A Checklist for Successful Quantitative Live Cell Imaging in Systems Biology

Abstract
:1. Introduction
2. Considerations for Artifact-Free Monitoring in Live Cell Imaging
2.1. Unnatural Activity of Fluorescent Fusion Proteins from the Transgene
2.1.1. Expression Level

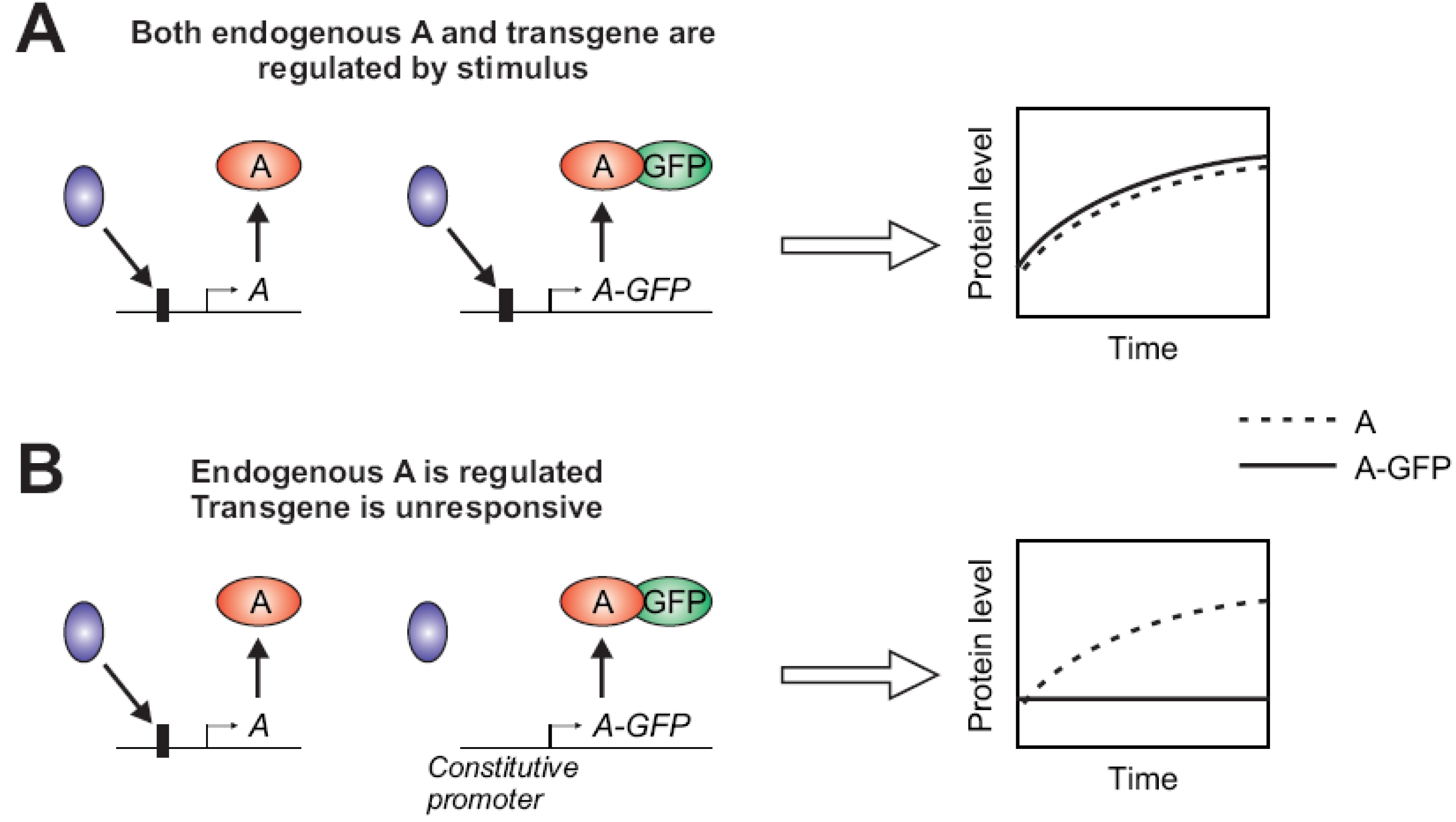
2.1.2. Stimulus-Dependent Regulation of Expression
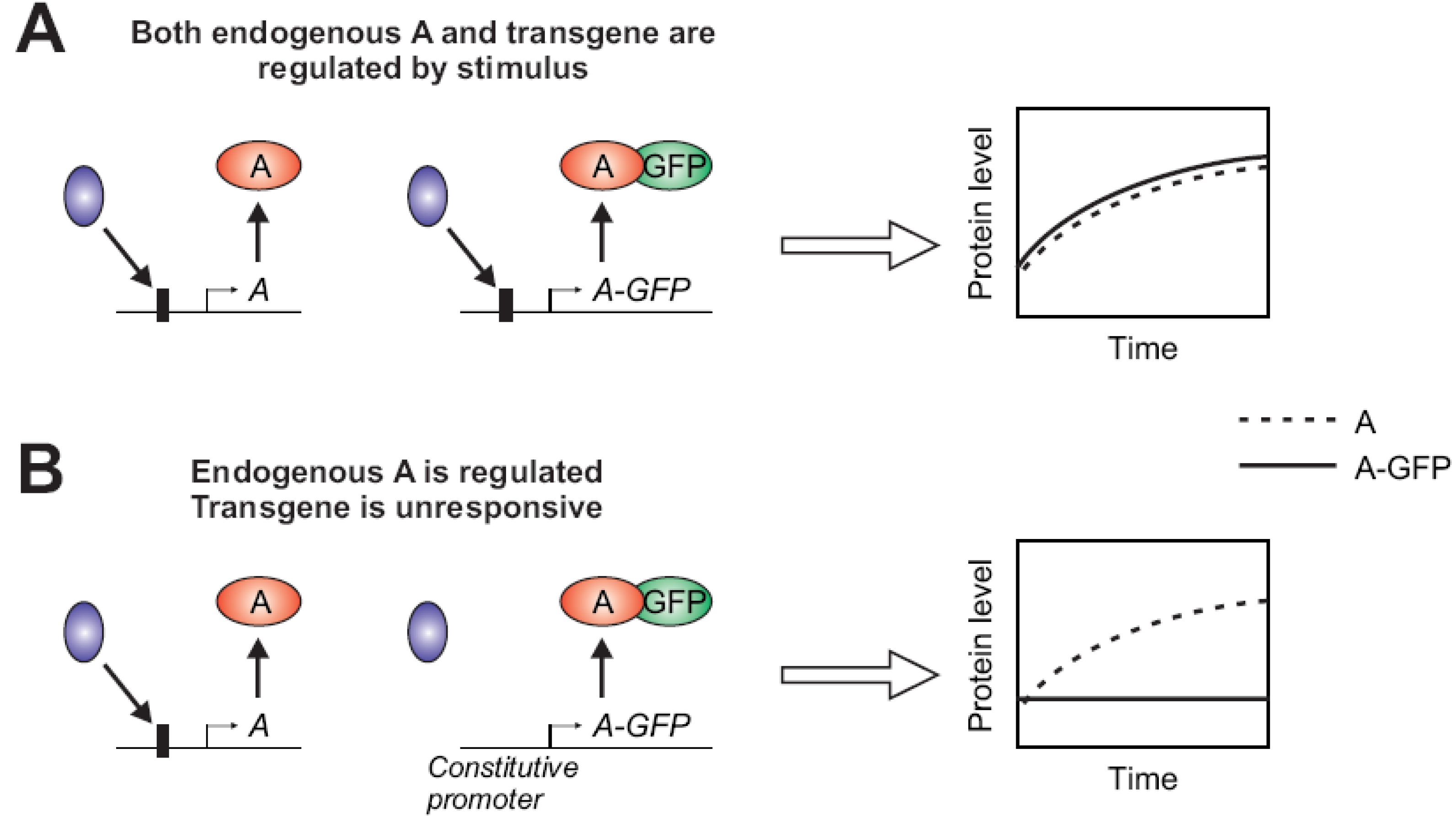
2.1.3. Fluorophore Interfering with Protein Function
2.2. Optimal Spatial Resolution
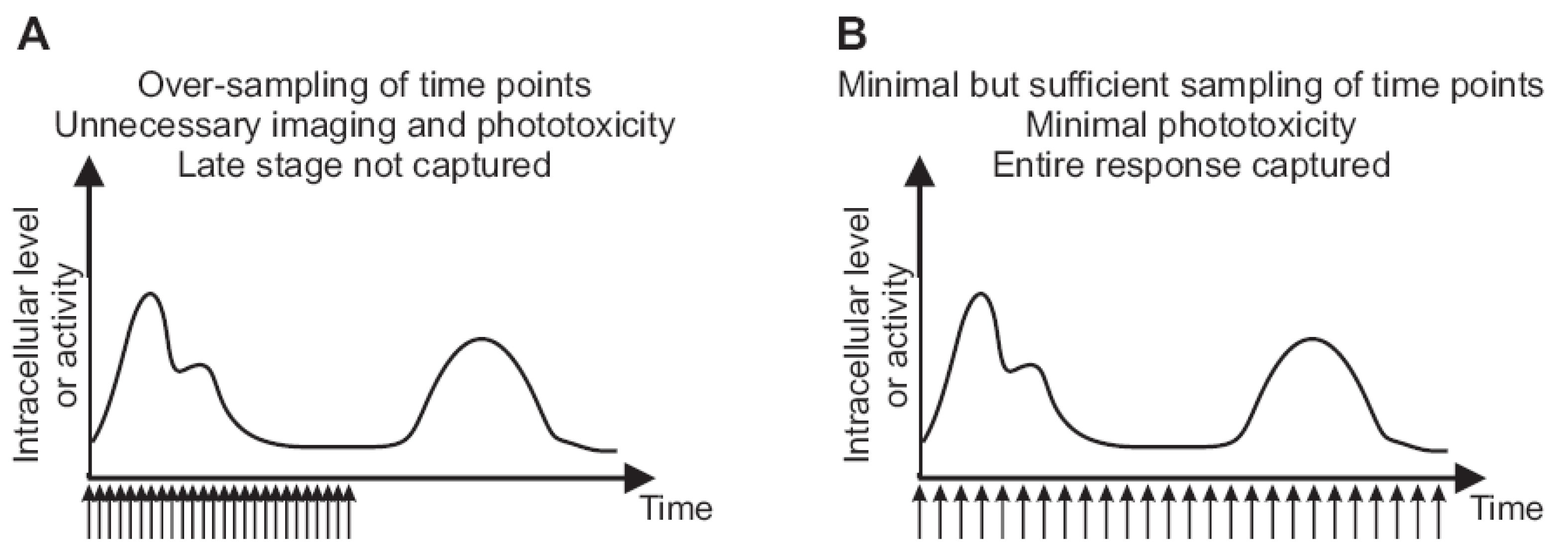
2.3. Imaging Duration and Temporal Resolution
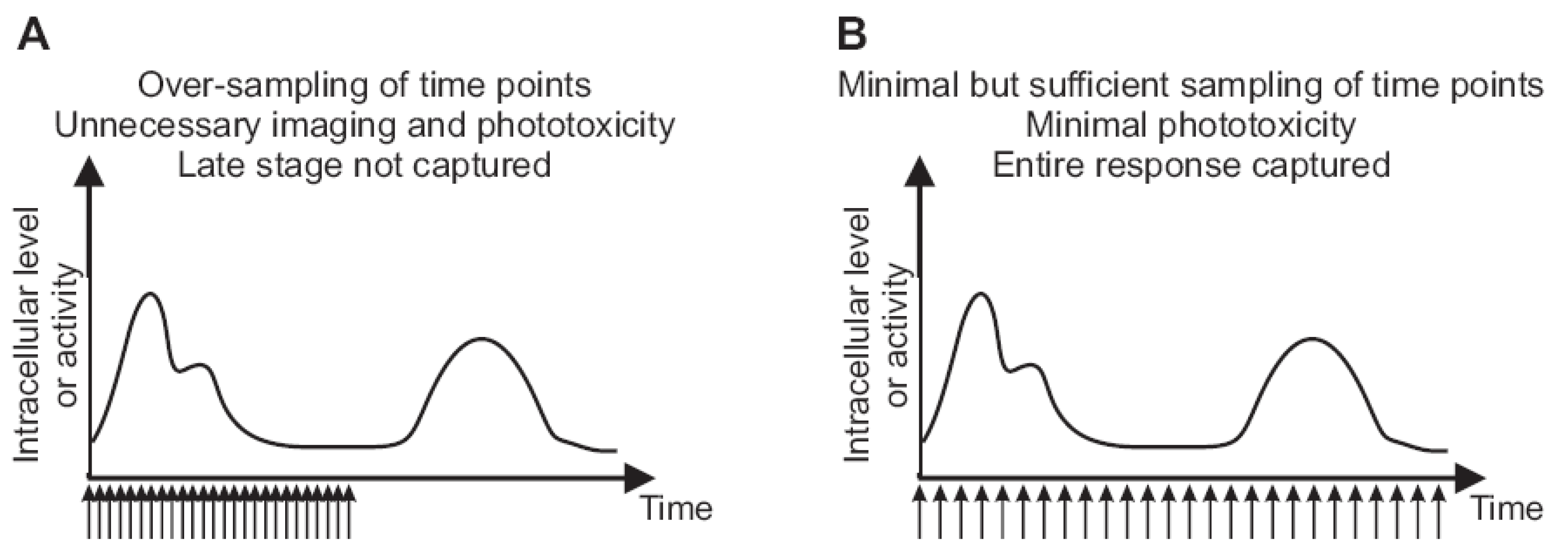
2.4. Autofocus
2.5. Importance of Cell Culture Conditions during Setup and Image Acquisition
2.6. Quantitative Analysis to Extract Relevant Information from Imaging Data
2.7. Sufficient Number of Single Cells for Statistically Significant Analysis Results
2.8. Comparison with a Computational Model of the Molecular Network
3. Conclusions

{kind=link}
{kind=link}
{kind=link}
| Sections | Multidisciplinary expertise | Hardware | Software |
|---|---|---|---|
| 2.1: Fluorescent protein behavior | Molecular biology, biochemistry | n.a. | n.a. |
| 2.2–2.4: Microscopy | Cell biology, biophysics, optics | Microscope control system | Time series, stage control, autofocus |
| 2.5: Cell physiology | Cell biology | Incubation control system | n.a. |
| 2.6: Image analysis | Math/physics | High performance computer | Image segmentation, tracking |
| 2.7: Population sampling | Statistics | High performance computer | Statistical analysis |
| 2.8: Network modeling | Math/physics/engineering | High performance computer | Mathematical modeling |
Acknowledgments
Conflict of Interest
References
- Sung, M.H.; McNally, J.G. Live cell imaging and systems biology. Wiley Interdiscip. Rev. Syst. Biol. Med. 2011, 3, 167–182. [Google Scholar] [CrossRef]
- Spiller, D.G.; Wood, C.D.; Rand, D.A.; White, M.R. Measurement of single-cell dynamics. Nature 2010, 465, 736–745. [Google Scholar]
- Lahav, G.; Rosenfeld, N.; Sigal, A.; Geva-Zatorsky, N.; Levine, A.J.; Elowitz, M.B.; Alon, U. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat. Genet. 2004, 36, 147–150. [Google Scholar]
- Yissachar, N.; Sharar Fischler, T.; Cohen, A.A.; Reich-Zeliger, S.; Russ, D.; Shifrut, E.; Porat, Z.; Friedman, N. Dynamic response diversity of NFAT isoforms in individual living cells. Mol. Cell 2013, 49, 1–9. [Google Scholar]
- Sung, M.H.; Salvatore, L.; de Lorenzi, R.; Indrawan, A.; Pasparakis, M.; Hager, G.L.; Bianchi, M.E.; Agresti, A. Sustained oscillations of NF-kappaB produce distinct genome scanning and gene expression profiles. PLoS One 2009, 4, e7163. [Google Scholar]
- Sung, M.H.; Lao, Q.; Ning, L.; Fraser, I.D.C.; Hager, G.L. National Institutes of Health: Bethesda, MD, USA, Unpublished work. 2013.
- Lee, T.K.; Denny, E.M.; Sanghvi, J.C.; Gaston, J.E.; Maynard, N.D.; Hughey, J.J.; Covert, M.W. A noisy paracrine signal determines the cellular NF-kappaB response to lipopolysaccharide. Sci. Signal. 2009, 2, ra65. [Google Scholar]
- Nelson, D.E.; Ihekwaba, A.E.; Elliott, M.; Johnson, J.R.; Gibney, C.A.; Foreman, B.E.; Nelson, G.; See, V.; Horton, C.A.; Spiller, D.G.; et al. Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science 2004, 306, 704–708. [Google Scholar] [CrossRef]
- Bartfeld, S.; Hess, S.; Bauer, B.; Machuy, N.; Ogilvie, L.A.; Schuchhardt, J.; Meyer, T.F. High-throughput and single-cell imaging of NF-kappaB oscillations using monoclonal cell lines. BMC Cell Biol. 2010, 11, 21. [Google Scholar]
- Harper, C.V.; Finkenstadt, B.; Woodcock, D.J.; Friedrichsen, S.; Semprini, S.; Ashall, L.; Spiller, D.G.; Mullins, J.J.; Rand, D.A.; Davis, J.R.; et al. Dynamic analysis of stochastic transcription cycles. PLoS Biol. 2011, 9, e1000607. [Google Scholar] [CrossRef]
- Rand, U.; Rinas, M.; Schwerk, J.; Nohren, G.; Linnes, M.; Kroger, A.; Flossdorf, M.; Kaly-Kullai, K.; Hauser, H.; Hofer, T.; et al. Multi-layered stochasticity and paracrine signal propagation shape the type-I interferon response. Mol. Syst. Biol. 2012, 8, 584. [Google Scholar]
- Bedell, V.M.; Wang, Y.; Campbell, J.M.; Poshusta, T.L.; Starker, C.G.; Krug, R.G., 2nd; Tan, W.; Penheiter, S.G.; Ma, A.C.; Leung, A.Y.; et al. In vivo genome editing using a high-efficiency TALEN system. Nature 2012, 491, 114–118. [Google Scholar]
- Reyon, D.; Tsai, S.Q.; Khayter, C.; Foden, J.A.; Sander, J.D.; Joung, J.K. FLASH assembly of TALENs for high-throughput genome editing. Nat. Biotechnol. 2012, 30, 460–465. [Google Scholar]
- microManager. Available online: https://valelab.ucsf.edu/~MM/MMwiki/ (accessed on 3 April 2013).
- ImageJ. Available online: https://rsb.info.nih.gov/ij/ (accessed on 3 April 2013).
- CellProfiler. Available online: https://www.cellprofiler.org/ (accessed on 3 April 2013).
- PhenoRipper. Available online: https://www4.utsouthwestern.edu/altschulerwulab/phenoripper/ (accessed on 3 April 2013).
- Cohen, A.A.; Geva-Zatorsky, N.; Eden, E.; Frenkel-Morgenstern, M.; Issaeva, I.; Sigal, A.; Milo, R.; Cohen-Saidon, C.; Liron, Y.; Kam, Z.; et al. Dynamic proteomics of individual cancer cells in response to a drug. Science 2008, 322, 1511–1516. [Google Scholar] [CrossRef]
- Cohen-Saidon, C.; Cohen, A.A.; Sigal, A.; Liron, Y.; Alon, U. Dynamics and variability of ERK2 response to EGF in individual living cells. Mol. Cell 2009, 36, 885–893. [Google Scholar] [CrossRef]
- Tay, S.; Hughey, J.J.; Lee, T.K.; Lipniacki, T.; Quake, S.R.; Covert, M.W. Single-cell NF-kappaB dynamics reveal digital activation and analogue information processing. Nature 2010, 466, 267–271. [Google Scholar]
- Awwad, Y.; Geng, T.; Baldwin, A.S.; Lu, C. Observing single cell NF-kappaB dynamics under stimulant concentration gradient. Anal. Chem. 2012, 84, 1224–1228. [Google Scholar] [CrossRef]
- James, C.D.; Moorman, M.W.; Carson, B.D.; Branda, C.S.; Lantz, J.W.; Manginell, R.P.; Martino, A.; Singh, A.K. Nuclear translocation kinetics of NF-kappaB in macrophages challenged with pathogens in a microfluidic platform. Biomed. Microdevices 2009, 11, 693–700. [Google Scholar] [CrossRef]
- Cai, L.; Dalal, C.K.; Elowitz, M.B. Frequency-modulated nuclear localization bursts coordinate gene regulation. Nature 2008, 455, 485–490. [Google Scholar]
- Purvis, J.E.; Karhohs, K.W.; Mock, C.; Batchelor, E.; Loewer, A.; Lahav, G. p53 dynamics control cell fate. Science 2012, 336, 1440–1444. [Google Scholar] [CrossRef]
- Sigal, A.; Milo, R.; Cohen, A.; Geva-Zatorsky, N.; Klein, Y.; Alaluf, I.; Swerdlin, N.; Perzov, N.; Danon, T.; Liron, Y.; et al. Dynamic proteomics in individual human cells uncovers widespread cell-cycle dependence of nuclear proteins. Nat. Methods 2006, 3, 525–531. [Google Scholar]
- Suel, G.M.; Garcia-Ojalvo, J.; Liberman, L.M.; Elowitz, M.B. An excitable gene regulatory circuit induces transient cellular differentiation. Nature 2006, 440, 545–550. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sung, M.-H. A Checklist for Successful Quantitative Live Cell Imaging in Systems Biology. Cells 2013, 2, 284-293. https://doi.org/10.3390/cells2020284
Sung M-H. A Checklist for Successful Quantitative Live Cell Imaging in Systems Biology. Cells. 2013; 2(2):284-293. https://doi.org/10.3390/cells2020284
Chicago/Turabian StyleSung, Myong-Hee. 2013. "A Checklist for Successful Quantitative Live Cell Imaging in Systems Biology" Cells 2, no. 2: 284-293. https://doi.org/10.3390/cells2020284
APA StyleSung, M.-H. (2013). A Checklist for Successful Quantitative Live Cell Imaging in Systems Biology. Cells, 2(2), 284-293. https://doi.org/10.3390/cells2020284