
The Role of G-Protein-Coupled Receptor Proteolysis Site Cleavage of Polycystin-1 in Renal Physiology and Polycystic Kidney Disease

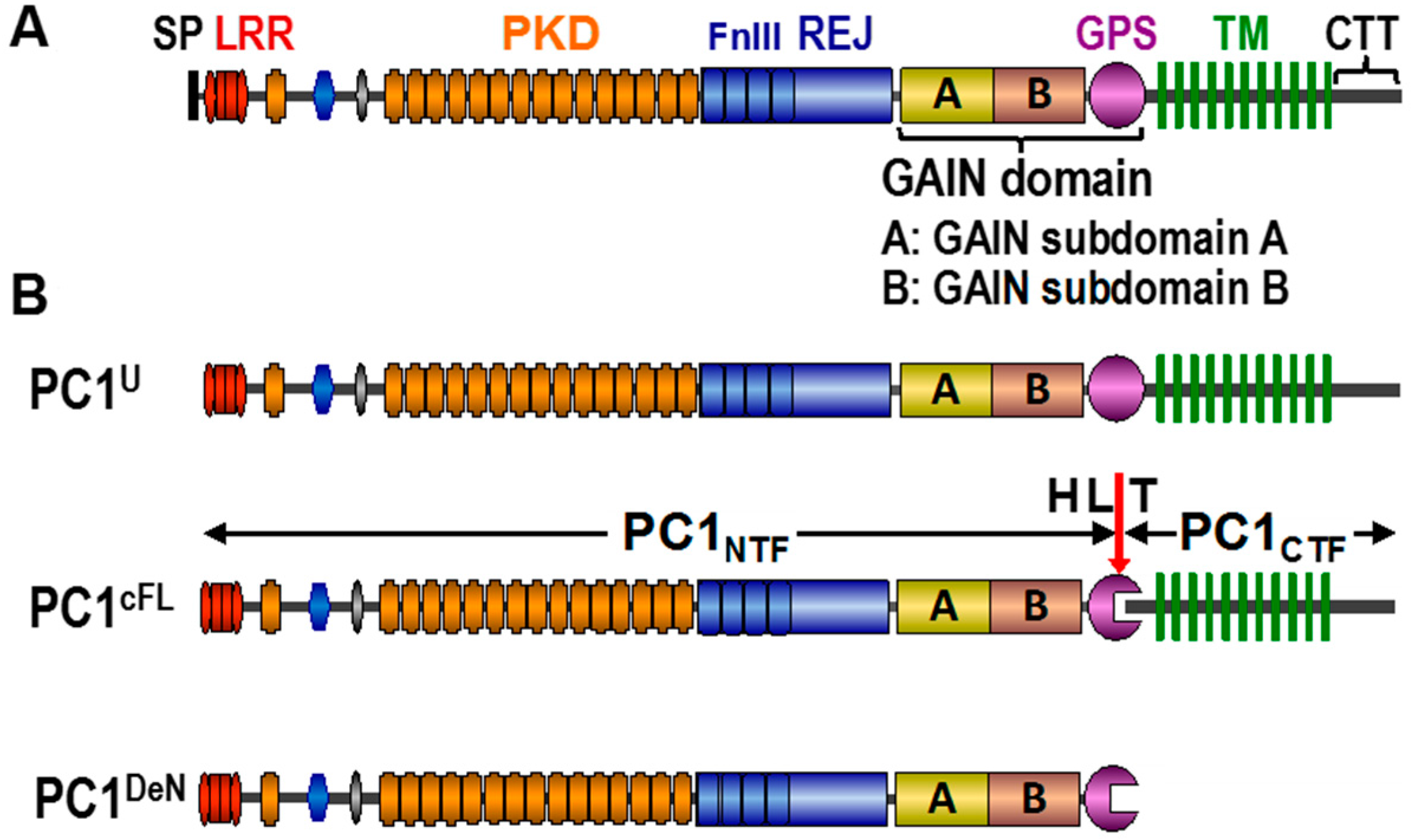
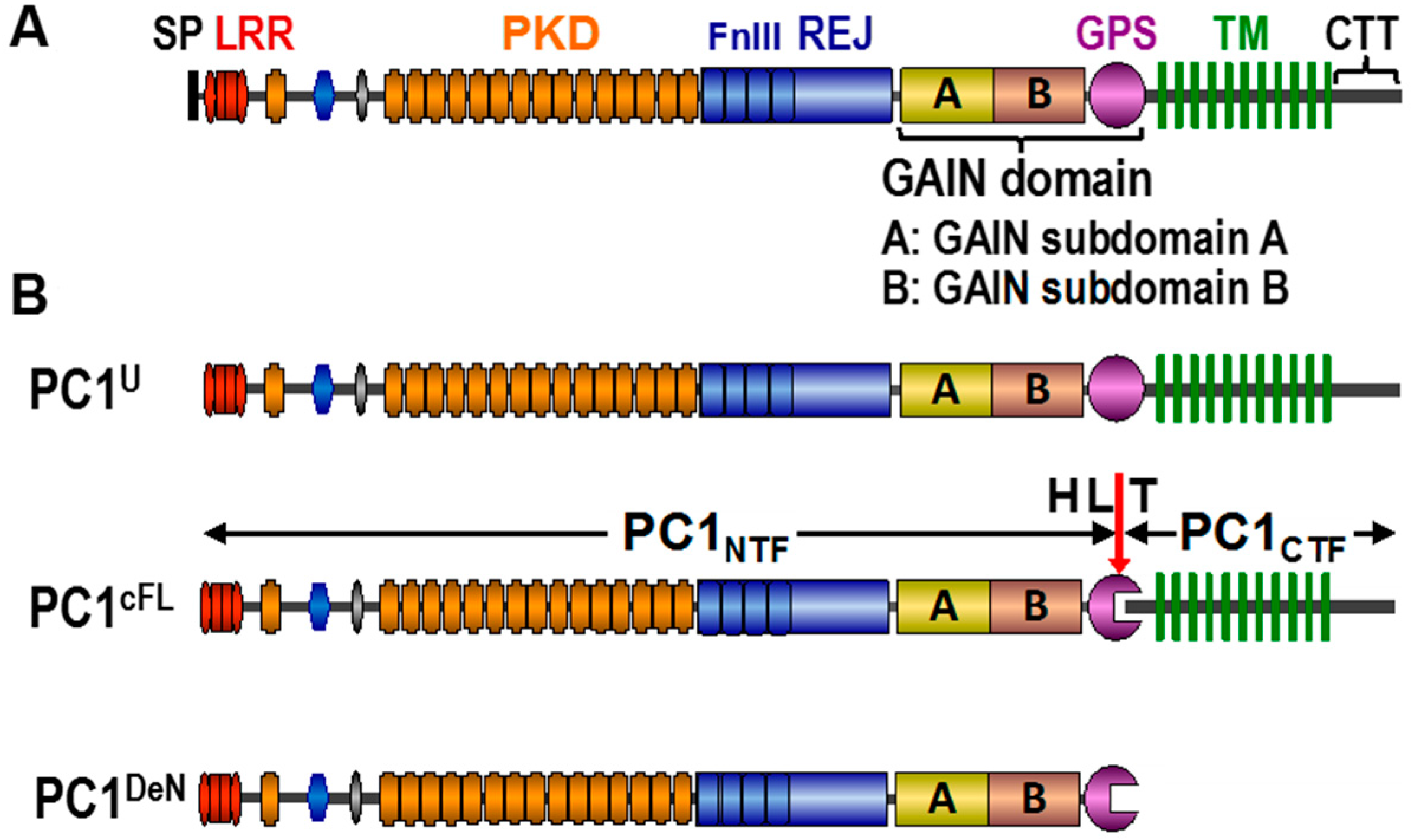{kind=link}
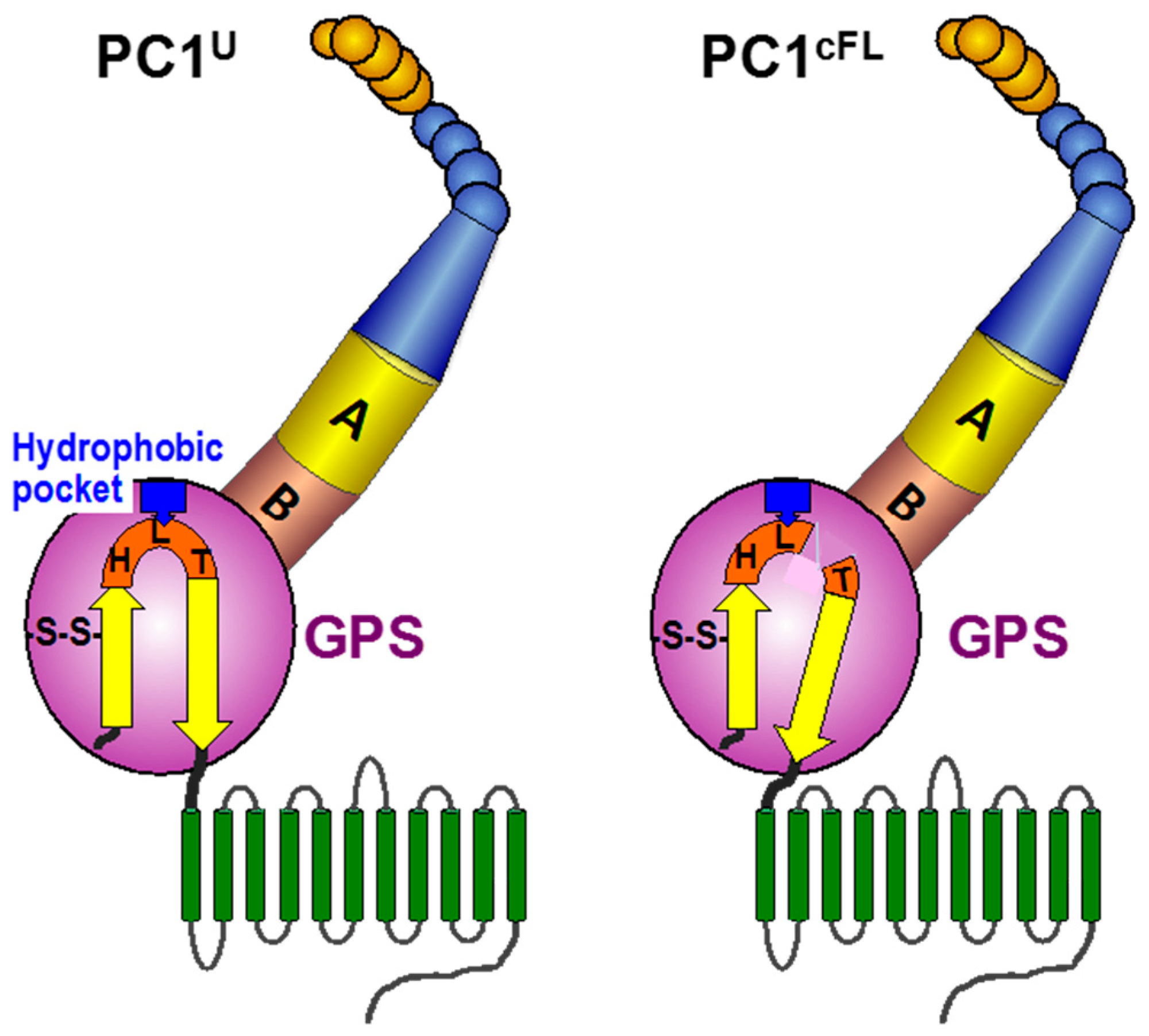{kind=link}
Abstract
:1. Introduction
2. Specific Role of GPS Cleavage of Polycystin-1 in the Kidney
2.1. Potential Role of PC1U in Kidney Development and Proximal Nephron Segments
2.2. Essential Role of PC1cFL for Distal Nephron Segments in the Postnatal Period
3. Structural Basis of GPS Cleavage and the Heterodimeric Association

4. Role of GPS Cleavage for PC1 Trafficking
5. Significance of Individual Mutations at the Critical HL*T Tripeptide for PC1 Trafficking and Function
6. Defective GPS Cleavage of PC1 in Polycystic Kidney Disease
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Harris, P.C.; Torres, V.E. Polycystic kidney disease. Annu. Rev. Med. 2009, 60, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T.E.P.K.D. The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. Cell 1994, 77, 881–894. [Google Scholar] [Green Version]
- Mochizuki, T.; Wu, G.; Hayashi, T.; Xenophontos, S.L.; Veldhuisen, B.; Saris, J.J.; Reynolds, D.M.; Cai, Y.; Gabow, P.A.; Pierides, A.; et al. Pkd2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 1996, 272, 1339–1342. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.; Ward, C.J.; Peral, B.; Aspinwall, R.; Clark, K.; San Millan, J.L.; Gamble, V.; Harris, P.C. The polycystic kidney disease 1 (pkd1) gene encodes a novel protein with multiple cell recognition domains. Nat. Genet. 1995, 10, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, P.; Somlo, S. Genetics and pathogenesis of polycystic kidney disease. J. Am. Soc. Nephrol 2002, 13, 2384–2398. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.E. New insights into polycystic kidney disease and its treatment. Curr. Opinion Nephrol. Hypertens. 1998, 7, 159–169. [Google Scholar] [CrossRef]
- Boyer, O.; Gagnadoux, M.F.; Guest, G.; Biebuyck, N.; Charbit, M.; Salomon, R.; Niaudet, P. Prognosis of autosomal dominant polycystic kidney disease diagnosed in utero or at birth. Pediatr. Nephrol. 2007, 22, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Fick, G.M.; Johnson, A.M.; Strain, J.D.; Kimberling, W.J.; Kumar, S.; Manco-Johnson, M.L.; Duley, I.T.; Gabow, P.A. Characteristics of very early onset autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 1993, 3, 1863–1870. [Google Scholar] [CrossRef] [PubMed]
- Grantham, J.J.; Cook, L.T.; Wetzel, L.H.; Cadnapaphornchai, M.A.; Bae, K.T. Evidence of extraordinary growth in the progressive enlargement of renal cysts. Clin. J. Am. Soc. Nephrol. 2010, 5, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Schroder, S.; Fraternali, F.; Quan, X.; Scott, D.; Qian, F.; Pfuhl, M. When a module is not a domain: The case of the rej module and the redefinition of the architecture of polycystin-1. Biochem. J. 2011, 435, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.; Wei, W.; Germino, G.; Oberhauser, A. The nanomechanics of polycystin-1 extracellular region. J. Biol. Chem. 2005, 280, 40723–40730. [Google Scholar] [CrossRef] [PubMed]
- Ponting, C.P.; Hofmann, K.; Bork, P. A latrophilin/cl-1-like gps domain in polycystin-1. Curr. Biol. 1999, 9, R585–588. [Google Scholar] [CrossRef]
- Qian, F.; Boletta, A.; Bhunia, A.K.; Xu, H.; Liu, L.; Ahrabi, A.K.; Watnick, T.J.; Zhou, F.; Germino, G.G. Cleavage of polycystin-1 requires the receptor for egg jelly domain and is disrupted by human autosomal-dominant polycystic kidney disease 1-associated mutations. Proc. Natl. Acad. Sci. USA 2002, 99, 16981–16986. [Google Scholar] [CrossRef] [PubMed]
- Krasnoperov, V.G.; Bittner, M.A.; Beavis, R.; Kuang, Y.; Salnikow, K.V.; Chepurny, O.G.; Little, A.R.; Plotnikov, A.N.; Wu, D.; Holz, R.W.; et al. Alpha-latrotoxin stimulates exocytosis by the interaction with a neuronal g-protein-coupled receptor. Neuron 1997, 18, 925–937. [Google Scholar] [CrossRef]
- Arac, D.; Boucard, A.A.; Bolliger, M.F.; Nguyen, J.; Soltis, S.M.; Sudhof, T.C.; Brunger, A.T. A novel evolutionarily conserved domain of cell-adhesion gpcrs mediates autoproteolysis. EMBO J. 2012, 31, 1364–1378. [Google Scholar] [CrossRef] [PubMed]
- Bjarnadottir, T.K.; Fredriksson, R.; Hoglund, P.J.; Gloriam, D.E.; Lagerstrom, M.C.; Schioth, H.B. The human and mouse repertoire of the adhesion family of g-protein-coupled receptors. Genomics 2004, 84, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Bjarnadottir, T.K.; Fredriksson, R.; Schioth, H.B. The adhesion gpcrs: A unique family of g protein-coupled receptors with important roles in both central and peripheral tissues. Cell. Mol. Life Sci. 2007, 64, 2104–2119. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Santoso, N.G.; Yu, S.; Woodward, O.M.; Qian, F.; Guggino, W.B. Polycystin-1 interacts with inositol 1,4,5-trisphosphate receptor to modulate intracellular Ca2+ signaling with implications for polycystic kidney disease. J. Biol. Chem. 2009, 284, 36431–36441. [Google Scholar] [CrossRef] [PubMed]
- Manzati, E.; Aguiari, G.; Banzi, M.; Manzati, M.; Selvatici, R.; Falzarano, S.; Maestri, I.; Pinton, P.; Rizzuto, R.; del Senno, L. The cytoplasmic c-terminus of polycystin-1 increases cell proliferation in kidney epithelial cells through serum-activated and Ca2+-dependent pathway(s). Exp. Cell Res. 2005, 304, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Arnould, T.; Sellin, L.K.; Benzing, T.; Fan, M.J.; Gruning, W.; Sokol, S.Y.; Drummond, I.; Walz, G. The polycystic kidney disease 1 gene product modulates wnt signaling. J. Biol. Chem. 1999, 274, 4947–4953. [Google Scholar] [CrossRef] [PubMed]
- Shillingford, J.M.; Murcia, N.S.; Larson, C.H.; Low, S.H.; Hedgepeth, R.; Brown, N.; Flask, C.A.; Novick, A.C.; Goldfarb, D.A.; Kramer-Zucker, A.; et al. The mtor pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5466–5471. [Google Scholar] [CrossRef] [PubMed]
- Rowe, I.; Chiaravalli, M.; Mannella, V.; Ulisse, V.; Quilici, G.; Pema, M.; Song, X.W.; Xu, H.; Mari, S.; Qian, F.; et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat. Med. 2013, 19, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Menezes, L.F.; Zhou, F.; Patterson, A.D.; Piontek, K.B.; Krausz, K.W.; Gonzalez, F.J.; Germino, G.G. Network analysis of a pkd1-mouse model of autosomal dominant polycystic kidney disease identifies hnf4alpha as a disease modifier. PLoS Genetics 2012, 8, e1003053. [Google Scholar] [CrossRef] [PubMed]
- Parnell, S.C.; Magenheimer, B.S.; Maser, R.L.; Rankin, C.A.; Smine, A.; Okamoto, T.; Calvet, J.P. The polycystic kidney disease-1 protein, polycystin-1, binds and activates heterotrimeric g-proteins in vitro. Biochem. Biophys. Res. Commun. 1998, 251, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Parnell, S.C.; Magenheimer, B.S.; Maser, R.L.; Zien, C.A.; Frischauf, A.M.; Calvet, J.P. Polycystin-1 activation of c-jun n-terminal kinase and ap-1 is mediated by heterotrimeric g proteins. J. Biol. Chem. 2002, 277, 19566–19572. [Google Scholar] [CrossRef] [PubMed]
- Chauvet, V.; Tian, X.; Husson, H.; Grimm, D.H.; Wang, T.; Hiesberger, T.; Igarashi, P.; Bennett, A.M.; Ibraghimov-Beskrovnaya, O.; Somlo, S.; et al. Mechanical stimuli induce cleavage and nuclear translocation of the polycystin-1 c terminus. J. Clin. Invest. 2004, 114, 1433–1443. [Google Scholar] [CrossRef] [PubMed]
- Merrick, D.; Chapin, H.; Baggs, J.E.; Yu, Z.; Somlo, S.; Sun, Z.; Hogenesch, J.B.; Caplan, M.J. The gamma-secretase cleavage product of polycystin-1 regulates tcf and chop-mediated transcriptional activation through a p300-dependent mechanism. Dev. Cell 2012, 22, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Hanaoka, K.; Qian, F.; Boletta, A.; Bhunia, A.K.; Piontek, K.; Tsiokas, L.; Sukhatme, V.P.; Guggino, W.B.; Germino, G.G. Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature 2000, 408, 990–994. [Google Scholar] [PubMed]
- Qian, F.; Germino, F.J.; Cai, Y.; Zhang, X.; Somlo, S.; Germino, G.G. Pkd1 interacts with pkd2 through a probable coiled-coil domain. Nat. Genet. 1997, 16, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Tsiokas, L.; Kim, E.; Arnould, T.; Sukhatme, V.P.; Walz, G. Homo- and heterodimeric interactions between the gene products of pkd1 and pkd2. Proc. Natl. Acad. Sci. USA 1997, 94, 6965–6970. [Google Scholar] [CrossRef] [PubMed]
- Nauli, S.M.; Alenghat, F.J.; Luo, Y.; Williams, E.; Vassilev, P.; Li, X.; Elia, A.E.; Lu, W.; Brown, E.M.; Quinn, S.J.; et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 2003, 33, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Pazour, G.J. Intraflagellar transport and cilia-dependent renal disease: The ciliary hypothesis of polycystic kidney disease. J. Am. Soc. Nephrol. 2004, 15, 2528–2536. [Google Scholar] [CrossRef] [PubMed]
- Yoder, B.K. Role of primary cilia in the pathogenesis of polycystic kidney disease. J. Am. Soc. Nephrol. 2007, 18, 1381–1388. [Google Scholar] [CrossRef] [PubMed]
- Yoder, B.K.; Hou, X.; Guay-Woodford, L.M. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J. Am. Soc. Nephrol. 2002, 13, 2508–2516. [Google Scholar] [CrossRef] [PubMed]
- Delling, M.; DeCaen, P.G.; Doerner, J.F.; Febvay, S.; Clapham, D.E. Primary cilia are specialized calcium signalling organelles. Nature 2013, 504, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Qian, F. Polycystin-1, The Handbook of Proteolytic Enzymes, 3rd ed.; Elsevier: Oxford, UK, 2013; Volume 3, pp. 3728–3736. [Google Scholar]
- Wei, W.; Hackmann, K.; Xu, H.; Germino, G.; Qian, F. Characterization of cis-autoproteolysis of polycystin-1, the product of human polycystic kidney disease 1 gene. J. Biol. Chem. 2007, 30, 21729–21737. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.H.; Chang, G.W.; Davies, J.Q.; Stacey, M.; Harris, J.; Gordon, S. Autocatalytic cleavage of the emr2 receptor occurs at a conserved g protein-coupled receptor proteolytic site motif. J. Biol. Chem. 2004, 279, 31823–31832. [Google Scholar] [CrossRef] [PubMed]
- Kurbegovic, A.; Kim, H.; Xu, H.; Yu, S.; Cruanes, J.; Maser, R.L.; Boletta, A.; Trudel, M.; Qian, F. Novel functional complexity of polycystin-1 by gps cleavage in vivo: Role in polycystic kidney disease. Mol. Cell. Biol. 2014, 34, 3341–3353. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Hackmann, K.; Gao, J.; He, X.; Piontek, K.; Garcia-Gonzalez, M.A.; Menezes, L.F.; Xu, H.; Germino, G.G.; Zuo, J.; et al. Essential role of cleavage of polycystin-1 at g protein-coupled receptor proteolytic site for kidney tubular structure. Proc. Natl. Acad. Sci. USA 2007, 104, 18688–18693. [Google Scholar] [CrossRef] [PubMed]
- Castelli, M.; Boca, M.; Chiaravalli, M.; Ramalingam, H.; Rowe, I.; Distefano, G.; Carroll, T.; Boletta, A. Polycystin-1 binds par3/apkc and controls convergent extension during renal tubular morphogenesis. Nat. Commun. 2013, 4, 2658. [Google Scholar] [CrossRef] [PubMed]
- Ahrabi, A.K.; Jouret, F.; Marbaix, E.; Delporte, C.; Horie, S.; Mulroy, S.; Boulter, C.; Sandford, R.; Devuyst, O. Glomerular and proximal tubule cysts as early manifestations of pkd1 deletion. Nephrol. Dial. Transpl. 2010, 25, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Shen, X.; Pavlova, A.; Lakkis, M.; Ward, C.J.; Pritchard, L.; Harris, P.C.; Genest, D.R.; Perez-Atayde, A.R.; Zhou, J. Comparison of pkd1-targeted mutants reveals that loss of polycystin-1 causes cystogenesis and bone defects. Hum. Mol. Genet. 2001, 10, 2385–2396. [Google Scholar] [CrossRef] [PubMed]
- Piontek, K.B.; Huso, D.L.; Grinberg, A.; Liu, L.; Bedja, D.; Zhao, H.; Gabrielson, K.; Qian, F.; Mei, C.; Westphal, H.; et al. A functional floxed allele of pkd1 that can be conditionally inactivated in vivo. J. Am. Soc. Nephrol. 2004, 15, 3035–3043. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.T.; Chiou, Y.Y.; Wang, E.; Lin, H.K.; Lin, Y.T.; Chi, Y.C.; Wang, C.K.; Tang, M.J.; Li, H. Defining a link with autosomal-dominant polycystic kidney disease in mice with congenitally low expression of pkd1. Am. J. Pathol. 2006, 168, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Hiesberger, T.; Cordes, K.; Sinclair, A.M.; Goldstein, L.S.; Somlo, S.; Igarashi, P. Kidney-specific inactivation of the kif3a subunit of kinesin-ii inhibits renal ciliogenesis and produces polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2003, 100, 5286–5291. [Google Scholar] [CrossRef] [PubMed]
- Jonassen, J.A.; San Agustin, J.; Follit, J.A.; Pazour, G.J. Deletion of ift20 in the mouse kidney causes misorientation of the mitotic spindle and cystic kidney disease. J. Cell. Biol. 2008, 183, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Lantinga-van Leeuwen, I.S.; Dauwerse, J.G.; Baelde, H.J.; Leonhard, W.N.; van de Wal, A.; Ward, C.J.; Verbeek, S.; Deruiter, M.C.; Breuning, M.H.; de Heer, E.; et al. Lowering of pkd1 expression is sufficient to cause polycystic kidney disease. Hum. Mol. Genet. 2004, 13, 3069–3077. [Google Scholar] [CrossRef] [PubMed]
- Tesmer, J.J. A gain in understanding autoproteolytic g protein-coupled receptors and polycystic kidney disease proteins. EMBO J. 2012, 31, 1334–1335. [Google Scholar] [CrossRef] [PubMed]
- Paavola, K.J.; Stephenson, J.R.; Ritter, S.L.; Alter, S.P.; Hall, R.A. The n terminus of the adhesion g protein-coupled receptor gpr56 controls receptor signaling activity. J. Biol. Chem. 2011, 286, 28914–28921. [Google Scholar] [CrossRef] [PubMed]
- Ward, Y.; Lake, R.; Yin, J.J.; Heger, C.D.; Raffeld, M.; Goldsmith, P.K.; Merino, M.; Kelly, K. Lpa receptor heterodimerizes with cd97 to amplify lpa-initiated rho-dependent signaling and invasion in prostate cancer cells. Cancer Res. 2011, 71, 7301–7311. [Google Scholar] [CrossRef] [PubMed]
- Paavola, K.J.; Hall, R.A. Adhesion g protein-coupled receptors: Signaling, pharmacology, and mechanisms of activation. Mol. Pharmacol. 2012, 82, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Xu, H.; Yao, Q.; Li, W.; Huang, Q.; Outeda, P.; Cebotaru, V.; Chiaravalli, M.; Boletta, A.; Piontek, K.; et al. Ciliary membrane proteins traffic through the golgi via a rabep1/gga1/arl3-dependent mechanism. Nat. Commun. 2014, 5, 5482. [Google Scholar] [CrossRef] [PubMed]
- Puertollano, R.; Randazzo, P.A.; Presley, J.F.; Hartnell, L.M.; Bonifacino, J.S. The ggas promote arf-dependent recruitment of clathrin to the tgn. Cell 2001, 105, 93–102. [Google Scholar] [CrossRef]
- Schrick, J.J.; Vogel, P.; Abuin, A.; Hampton, B.; Rice, D.S. Adp-ribosylation factor-like 3 is involved in kidney and photoreceptor development. Am. J. Pathol. 2006, 168, 1288–1298. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Fedeles, S.V.; Dong, K.; Anyatonwu, G.; Onoe, T.; Mitobe, M.; Gao, J.D.; Okuhara, D.; Tian, X.; Gallagher, A.R.; et al. Altered trafficking and stability of polycystins underlie polycystic kidney disease. J. Clin. Invest. 2014, 124, 5129–5144. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Buckley, D.; Guan, C.; Guo, H.C. Structural insights into the mechanism of intramolecular proteolysis. Cell 1999, 98, 651–661. [Google Scholar] [CrossRef]
- Garcia-Gonzalez, M.A.; Jones, J.G.; Allen, S.K.; Palatucci, C.M.; Batish, S.D.; Seltzer, W.K.; Lan, Z.; Allen, E.; Qian, F.; Lens, X.M.; et al. Evaluating the clinical utility of a molecular genetic test for polycystic kidney disease. Mol. Genet. Metab 2007, 92, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Hopp, K.; Ward, C.J.; Hommerding, C.J.; Nasr, S.H.; Tuan, H.F.; Gainullin, V.G.; Rossetti, S.; Torres, V.E.; Harris, P.C. Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J. Clin. Invest. 2012, 122, 4257–4273. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.C.; Torres, V.E. Understanding pathogenic mechanisms in polycystic kidney disease provides clues for therapy. Curr. Opin. Nephrol. Hy. 2006, 15, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.E.; Wang, X.; Qian, Q.; Somlo, S.; Harris, P.C.; Gattone, V.H., 2nd. Effective treatment of an orthologous model of autosomal dominant polycystic kidney disease. Nat. Med. 2004, 10, 363–364. [Google Scholar] [CrossRef] [PubMed]
- Czarnecki, P.G.; Steinman, T.I. Polycystic kidney disease: New horizons and therapeutic frontiers. Minerva Urol. Nefrol. 2013, 65, 61–68. [Google Scholar] [PubMed]
- Baur, B.P.; Meaney, C.J. Review of tolvaptan for autosomal dominant polycystic kidney disease. Pharmacotherapy 2014, 34, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Watnick, T.; Germino, G.G. Mtor inhibitors in polycystic kidney disease. N. Engl. J. Med. 2010, 363, 879–881. [Google Scholar] [CrossRef] [PubMed]
- Holleran, J.P.; Zeng, J.; Frizzell, R.A.; Watkins, S.C. Regulated recycling of mutant cftr is partially restored by pharmacological treatment. J. Cell Sci. 2013, 126 Pt. 12, 2692–2703. [Google Scholar] [CrossRef] [PubMed]
- Morello, J.P.; Salahpour, A.; Laperriere, A.; Bernier, V.; Arthus, M.F.; Lonergan, M.; Petaja-Repo, U.; Angers, S.; Morin, D.; Bichet, D.G.; et al. Pharmacological chaperones rescue cell-surface expression and function of misfolded v2 vasopressin receptor mutants. J. Clin. Invest. 2000, 105, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Virant-Young, D.; Thomas, J.; Woiderski, S.; Powers, M.; Carlier, J.; McCarty, J.; Kupchick, T.; Larder, A. Cystic fibrosis: A novel pharmacologic approach to cystic fibrosis transmembrane regulator modulation therapy. J. Am. Osteopath. Assoc. 2015, 115, 546–555. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trudel, M.; Yao, Q.; Qian, F. The Role of G-Protein-Coupled Receptor Proteolysis Site Cleavage of Polycystin-1 in Renal Physiology and Polycystic Kidney Disease. Cells 2016, 5, 3. https://doi.org/10.3390/cells5010003
Trudel M, Yao Q, Qian F. The Role of G-Protein-Coupled Receptor Proteolysis Site Cleavage of Polycystin-1 in Renal Physiology and Polycystic Kidney Disease. Cells. 2016; 5(1):3. https://doi.org/10.3390/cells5010003
Chicago/Turabian StyleTrudel, Marie, Qin Yao, and Feng Qian. 2016. "The Role of G-Protein-Coupled Receptor Proteolysis Site Cleavage of Polycystin-1 in Renal Physiology and Polycystic Kidney Disease" Cells 5, no. 1: 3. https://doi.org/10.3390/cells5010003
APA StyleTrudel, M., Yao, Q., & Qian, F. (2016). The Role of G-Protein-Coupled Receptor Proteolysis Site Cleavage of Polycystin-1 in Renal Physiology and Polycystic Kidney Disease. Cells, 5(1), 3. https://doi.org/10.3390/cells5010003