
Intermediate Filaments as Organizers of Cellular Space: How They Affect Mitochondrial Structure and Function



Abstract
:1. Introduction
1.1. Intermediate Filaments as Organizers of Cytoplasmic Space
1.2. Mitochondrial Trafficking and Shape Changes
2. Interactions of Intermediate Filaments and Mitochondria in Different Cell Types
2.1. Lessons Learned from Neurons: Intermediate Filaments Determine Mitochondrial Motility
2.2. Lessons Learned from Mesenchymal Cells: Intermediate Filaments Bind to Mitochondria
2.3. Lessons Learned from Muscle Cells: Intermediate Filaments Affect Mitochondrial Calcium Handling and Energy Production
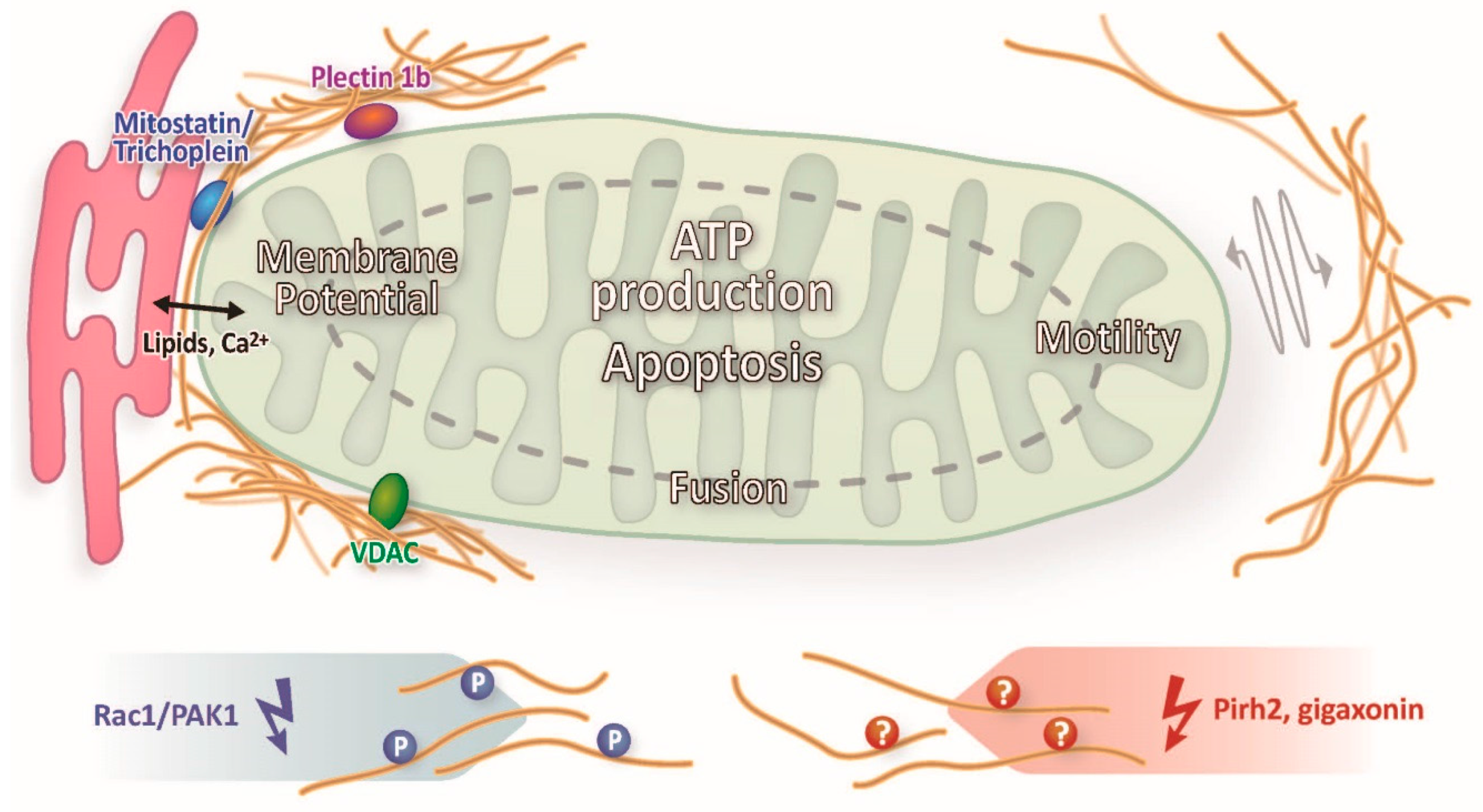
2.4. Lessons Learned in Epithelia: Intermediate Filaments Affect Mitochondrial Lipid Metabolism and Communicate through Signaling with Mitochondria
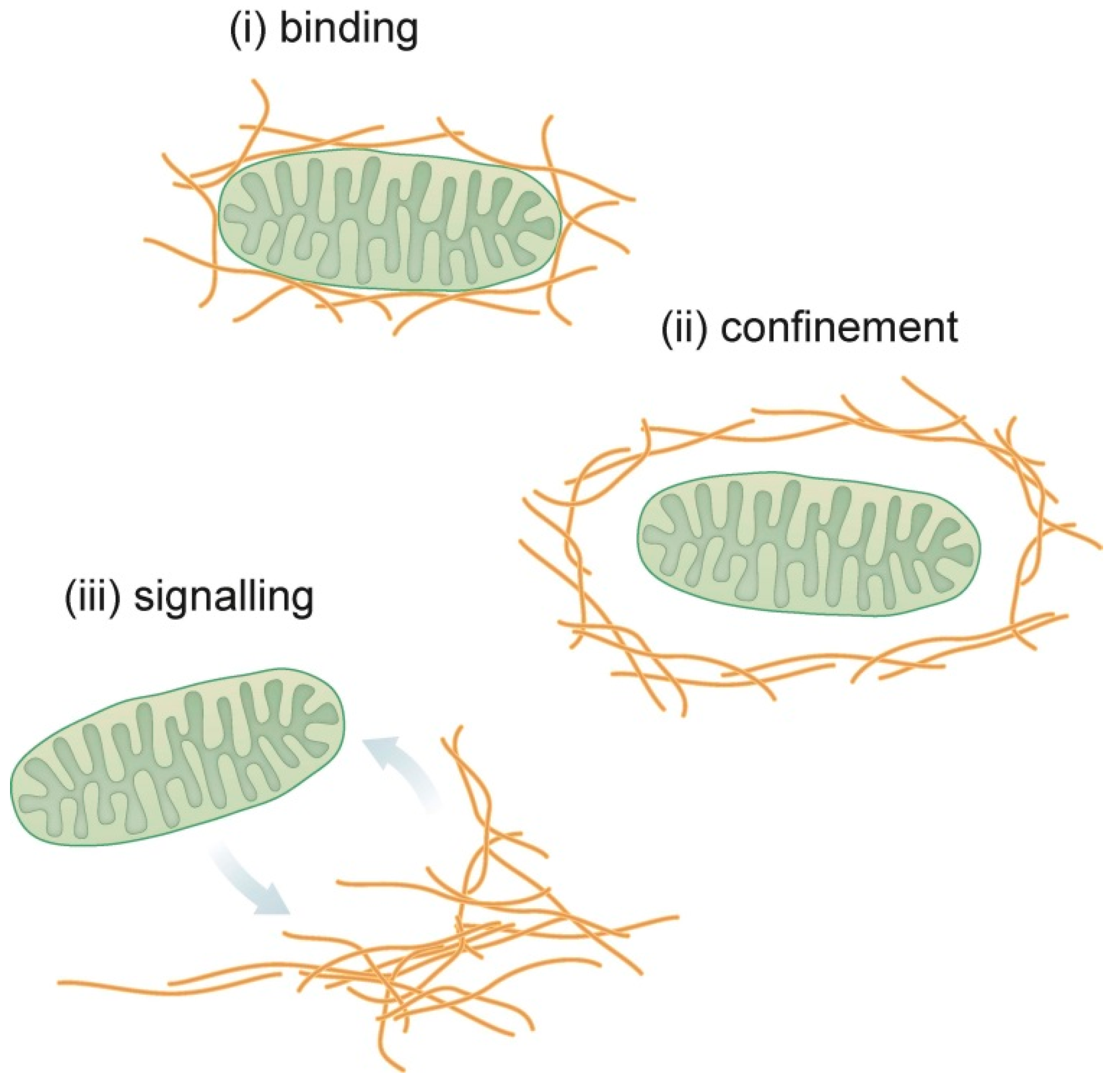
3. Conclusions and Future Directions
Acknowledgments
Conflicts of Interest
References
- Toivola, D.M.; Tao, G.-Z.; Habtezion, A.; Liao, J.; Omary, M.B. Cellular integrity plus: organelle-related and protein-targeting functions of intermediate filaments. Trends Cell Biol. 2005, 15, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.; Strelkov, S.V.; Burkhard, P.; Aebi, U. Intermediate filaments: Primary determinants of cell architecture and plasticity. J. Clin. Investig. 2009, 119, 1772–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coulombe, P.A.; Wong, P. Cytoplasmic intermediate filaments revealed as dynamic and multipurpose scaffolds. Nat. Cell Biol. 2004, 6, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Moll, R.; Divo, M.; Langbein, L. The human keratins: biology and pathology. Histochem. Cell Biol. 2008, 129, 705–733. [Google Scholar] [CrossRef] [PubMed]
- Bragulla, H.H.; Homberger, D.G. Structure and functions of keratin proteins in simple, stratified, keratinized and cornified epithelia. J. Anat. 2009, 214, 516–559. [Google Scholar] [CrossRef] [PubMed]
- Karantza, V. Keratins in health and cancer: More than mere epithelial cell markers. Oncogene 2011, 30, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Bauer, C.; Traub, P. Interaction of intermediate filaments with ribosomes in vitro. Eur. J. Cell Biol. 1995, 68, 288–296. [Google Scholar] [PubMed]
- Chang, L.; Barlan, K.; Chou, Y.-H.; Grin, B.; Lakonishok, M.; Serpinskaya, A.S.; Shumaker, D.K.; Herrmann, H.; Gelfand, V.I.; Goldman, R.D. The dynamic properties of intermediate filaments during organelle transport. J. Cell Sci. 2009, 122, 2914–2923. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zheng, Y. Lamins position the nuclear pores and centrosomes by modulating dynein. Mol. Biol. Cell 2015, 26, 3379–3389. [Google Scholar] [CrossRef] [PubMed]
- Kumemura, H.; Harada, M.; Omary, M.B.; Sakisaka, S.; Suganuma, T.; Namba, M.; Sata, M. Aggregation and loss of cytokeratin filament networks inhibit Golgi organization in liver-derived epithelial cell lines. Cell Motil. Cytoskeleton 2004, 57, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Mayerson, P.L.; Brumbaugh, J.A. Lavender, a chick melanocyte mutant with defective melanosome translocation: A possible role for 10 nm filaments and microfilaments but not microtubules. J. Cell Sci. 1981, 51, 25–51. [Google Scholar] [PubMed]
- Sarria, A.J.; Lieber, J.G.; Nordeen, S.K.; Evans, R.M. The presence or absence of a vimentin-type intermediate filament network affects the shape of the nucleus in human SW-13 cells. J. Cell Sci. 1994, 107, 1593–1607. [Google Scholar] [PubMed]
- Styers, M.L.; Salazar, G.; Love, R.; Peden, A.A.; Kowalczyk, A.P.; Faundez, V. The Endo-Lysosomal Sorting Machinery Interacts with the Intermediate Filament Cytoskeleton. Mol. Biol. Cell 2004, 15, 5369–5382. [Google Scholar] [CrossRef] [PubMed]
- Seltmann, K.; Roth, W.; Kröger, C.; Loschke, F.; Lederer, M.; Hüttelmaier, S.; Magin, T.M. Keratins Mediate Localization of Hemidesmosomes and Repress Cell Motility. J. Investig. Dermatol. 2013, 133, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Milner, D.J.; Weitzer, G.; Tran, D.; Bradley, A.; Capetanaki, Y. Disruption of muscle architecture and myocardial degeneration in mice lacking desmin. J. Cell Biol. 1996, 134, 1255–1270. [Google Scholar] [CrossRef] [PubMed]
- Lieber, J.G.; Evans, R.M. Disruption of the vimentin intermediate filament system during adipose conversion of 3T3-L1 cells inhibits lipid droplet accumulation. J. Cell Sci. 1996, 109, 3047–3058. [Google Scholar] [PubMed]
- Omary, M.B. “IF-pathies”: A broad spectrum of intermediate filament-associated diseases. J. Clin. Investig. 2009, 119, 1756–1762. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.L.; Hollenbeck, P.J. Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. J. Cell Biol. 1995, 131, 1315–1326. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.-H.; Cai, Q. Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.-H. Mitochondrial trafficking and anchoring in neurons: New insight and implications. J. Cell Biol. 2014, 204, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Stowers, R.S.; Megeath, L.J.; Górska-Andrzejak, J.; Meinertzhagen, I.A.; Schwarz, T.L. Axonal Transport of Mitochondria to Synapses Depends on Milton, a Novel Drosophila Protein. Neuron 2002, 36, 1063–1077. [Google Scholar] [CrossRef]
- Glater, E.E.; Megeath, L.J.; Stowers, R.S.; Schwarz, T.L. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J. Cell Biol. 2006, 173, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.; Aebi, U. Intermediate Filaments: Molecular Structure, Assembly Mechanism, and Integration into Functionally Distinct Intracellular Scaffolds. Annu. Rev. Biochem. 2004, 73, 749–789. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.-S.; Tian, J.-H.; Pan, P.-Y.; Zald, P.; Li, C.; Deng, C.; Sheng, Z.-H. Docking of Axonal Mitochondria by Syntaphilin Controls Their Mobility and Affects Short-Term Facilitation. Cell 2008, 132, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-M.; Gerwin, C.; Sheng, Z.-H. Dynein Light Chain LC8 Regulates Syntaphilin-Mediated Mitochondrial Docking in Axons. J. Neurosci. 2009, 29, 9429–9438. [Google Scholar] [CrossRef] [PubMed]
- Pathak, D.; Sepp, K.J.; Hollenbeck, P.J. Evidence That Myosin Activity Opposes Microtubule-Based Axonal Transport of Mitochondria. J. Neurosci. 2010, 30, 8984–8992. [Google Scholar] [CrossRef] [PubMed]
- Campello, S.; Scorrano, L. Mitochondrial shape changes: Orchestrating cell pathophysiology. EMBO Rep. 2010, 11, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.; Griparic, L.; Shurland, D.-L.; van der Bliek, A.M. Dynamin-related Protein Drp1 Is Required for Mitochondrial Division in Mammalian Cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed]
- James, D.I.; Parone, P.A.; Mattenberger, Y.; Martinou, J.-C. hFis1, a Novel Component of the Mammalian Mitochondrial Fission Machinery. J. Biol. Chem. 2003, 278, 36373–36379. [Google Scholar] [CrossRef] [PubMed]
- Gandre-Babbe, S.; van der Bliek, A.M. The Novel Tail-anchored Membrane Protein Mff Controls Mitochondrial and Peroxisomal Fission in Mammalian Cells. Mol. Biol. Cell 2008, 19, 2402–2412. [Google Scholar] [CrossRef] [PubMed]
- Santel, A.; Fuller, M.T. Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 2001, 114, 867–874. [Google Scholar] [PubMed]
- Cipolat, S.; de Brito, O.M.; Zilio, B.D.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, R.; Gilkerson, R.; Aggeler, R.; Yamagata, K.; Remington, S.J.; Capaldi, R.A. Energy Substrate Modulates Mitochondrial Structure and Oxidative Capacity in Cancer Cells. Cancer Res. 2004, 64, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Lehto, V.-P.; Virtanen, I. Association of intermediate filaments with other cell organelles in carcinoid tumor of the colon. Experientia 1979, 35, 35–36. [Google Scholar] [CrossRef] [PubMed]
- Toh, B.H.; Lolait, S.J.; Mathy, J.P.; Baum, R. Association of mitochondria with intermediate filaments and of polyribosomes with cytoplasmic actin. Cell Tissue Res. 1980, 211, 163–169. [Google Scholar] [CrossRef] [PubMed]
- David-Ferreira, K.L.; David-Ferreira, J.F. Association between intermediate-sized filaments and mitochondria in rat Leydig cells. Cell Biol. Int. Rep. 1980, 4, 655–662. [Google Scholar] [CrossRef]
- Hirokawa, N. Cross-linker system between neurofilaments, microtubules and membranous organelles in frog axons revealed by the quick-freeze, deep-etching method. J. Cell Biol. 1982, 94, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Mose-Larsen, P.; Bravo, R.; Fey, S.J.; Small, J.V.; Celis, J.E. Putative association of mitochondria with a subpopulation of intermediate-sized filaments in cultured human skin fibroblasts. Cell 1982, 31, 681–692. [Google Scholar] [CrossRef]
- Eckert, B.S. Alteration of the distribution of intermediate filaments in PtK1 cells by acrylamide II: Effect on the organization of cytoplasmic organelles. Cell Motil. Cytoskeleton 1986, 6, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Gentil, B.J.; Minotti, S.; Beange, M.; Baloh, R.H.; Julien, J.-P.; Durham, H.D. Normal role of the low-molecular-weight neurofilament protein in mitochondrial dynamics and disruption in Charcot-Marie-Tooth disease. FASEB J. 2012, 26, 1194–1203. [Google Scholar] [CrossRef] [PubMed]
- Perrot, R.; Julien, J.-P. Real-time imaging reveals defects of fast axonal transport induced by disorganization of intermediate filaments. FASEB J. 2009, 23, 3213–3225. [Google Scholar] [CrossRef] [PubMed]
- Nekrasova, O.E.; Mendez, M.G.; Chernoivanenko, I.S.; Tyurin-Kuzmin, P.A.; Kuczmarski, E.R.; Gelfand, V.I.; Goldman, R.D.; Minin, A.A. Vimentin intermediate filaments modulate the motility of mitochondria. Mol. Biol. Cell 2011, 22, 2282–2289. [Google Scholar] [CrossRef] [PubMed]
- Chernoivanenko, I.S.; Matveeva, E.A.; Gelfand, V.I.; Goldman, R.D.; Minin, A.A. Mitochondrial membrane potential is regulated by vimentin intermediate filaments. FASEB J. 2015, 29, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.L.; Lung, H.L.; Wu, K.C.; Le, A.-H.P.; Tang, H.M.; Fung, M.C. Vimentin supports mitochondrial morphology and organization. Biochem. J. 2008, 410, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Milner, D.J.; Mavroidis, M.; Weisleder, N.; Capetanaki, Y. Desmin Cytoskeleton Linked to Muscle Mitochondrial Distribution and Respiratory Function. J. Cell Biol. 2000, 150, 1283–1298. [Google Scholar] [CrossRef] [PubMed]
- Uttam, J.; Hutton, E.; Coulombe, P.A.; Anton-Lamprecht, I.; Yu, Q.C.; Gedde-Dahl, T.; Fine, J.D.; Fuchs, E. The genetic basis of epidermolysis bullosa simplex with mottled pigmentation. Proc. Natl. Acad. Sci. USA 1996, 93, 9079–9084. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Bouameur, J.-E.; Bär, J.; Rice, R.H.; Hornig-Do, H.-T.; Roop, D.R.; Schwarz, N.; Brodesser, S.; Thiering, S.; Leube, R.E.; et al. A keratin scaffold regulates epidermal barrier formation, mitochondrial lipid composition, and activity. J. Cell Biol. 2015, 211, 1057–1075. [Google Scholar] [CrossRef] [PubMed]
- Kumemura, H.; Harada, M.; Yanagimoto, C.; Koga, H.; Kawaguchi, T.; Hanada, S.; Taniguchi, E.; Ueno, T.; Sata, M. Mutation in keratin 18 induces mitochondrial fragmentation in liver-derived epithelial cells. Biochem. Biophys. Res. Commun. 2008, 367, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Stone, M.R.; O’Neill, A.; Lovering, R.M.; Strong, J.; Resneck, W.G.; Reed, P.W.; Toivola, D.M.; Ursitti, J.A.; Omary, M.B.; Bloch, R.J. Absence of keratin 19 in mice causes skeletal myopathy with mitochondrial and sarcolemmal reorganization. J. Cell Sci. 2007, 120, 3999–4008. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.; Smith, K.E.; O’Brien, B.J.; Angelides, K.J. Characterization of mammalian neurofilament triplet proteins. Subunit stoichiometry and morphology of native and reconstituted filaments. J. Biol. Chem. 1985, 260, 10736–10747. [Google Scholar] [PubMed]
- David, G.; Barrett, J.N.; Barrett, E.F. Evidence that mitochondria buffer physiological Ca2+ loads in lizard motor nerve terminals. J. Physiol. 1998, 509, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Straube-West, K.; Loomis, P.A.; Opal, P.; Goldman, R.D. Alterations in neural intermediate filament organization: functional implications and the induction of pathological changes related to motor neuron disease. J. Cell Sci. 1996, 109, 2319–2329. [Google Scholar] [PubMed]
- Rao, M.V.; Garcia, M.L.; Miyazaki, Y.; Gotow, T.; Yuan, A.; Mattina, S.; Ward, C.M.; Calcutt, N.A.; Uchiyama, Y.; Nixon, R.A.; et al. Gene replacement in mice reveals that the heavily phosphorylated tail of neurofilament heavy subunit does not affect axonal caliber or the transit of cargoes in slow axonal transport. J. Cell Biol. 2002, 158, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Szebenyi, G.; Smith, G.M.; Li, P.; Brady, S.T. Overexpression of neurofilament H disrupts normal cell structure and function. J. Neurosci. Res. 2002, 68, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Julien, J.P.; Mushynski, W.E. The distribution of phosphorylation sites among identified proteolytic fragments of mammalian neurofilaments. J. Biol. Chem. 1983, 258, 4019–4025. [Google Scholar] [PubMed]
- Hisanaga, S.; Hirokawa, N. Structure of the peripheral domains of neurofilaments revealed by low angle rotary shadowing. J. Mol. Biol. 1988, 202, 297–305. [Google Scholar] [CrossRef]
- Leterrier, J.F.; Rusakov, D.A.; Nelson, B.D.; Linden, M. Interactions between brain mitochondria and cytoskeleton: Evidence for specialized outer membrane domains involved in the association of cytoskeleton-associated proteins to mitochondria in situ and in vitro. Microsc. Res. Tech. 1994, 27, 233–261. [Google Scholar] [CrossRef] [PubMed]
- Overly, C.C.; Rieff, H.I.; Hollenbeck, P.J. Organelle motility and metabolism in axons vs. dendrites of cultured hippocampal neurons. J. Cell Sci. 1996, 109, 971–980. [Google Scholar] [PubMed]
- Wagner, O.I.; Lifshitz, J.; Janmey, P.A.; Linden, M.; McIntosh, T.K.; Leterrier, J.-F. Mechanisms of Mitochondria-Neurofilament Interactions. J. Neurosci. 2003, 23, 9046–9058. [Google Scholar] [PubMed]
- Brownlees, J. Charcot-Marie-Tooth disease neurofilament mutations disrupt neurofilament assembly and axonal transport. Hum. Mol. Genet. 2002, 11, 2837–2844. [Google Scholar] [CrossRef] [PubMed]
- Züchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Misko, A.; Jiang, S.; Wegorzewska, I.; Milbrandt, J.; Baloh, R.H. Mitofusin 2 Is Necessary for Transport of Axonal Mitochondria and Interacts with the Miro/Milton Complex. J. Neurosci. 2010, 30, 4232–4240. [Google Scholar] [CrossRef] [PubMed]
- Summerhayes, I.C.; Wong, D.; Chen, L.B. Effect of microtubules and intermediate filaments on mitochondrial distribution. J. Cell Sci. 1983, 61, 87–105. [Google Scholar] [PubMed]
- Almahbobi, G.; Williams, L.J.; Han, X.-G.; Hall, P.F. Binding of lipid droplets and mitochondria to intermediate filaments in rat Leydig cells. J. Reprod. Fertil. 1993, 98, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Collier, N.C.; Sheetz, M.P.; Schlesinger, M.J. Concomitant changes in mitochondria and intermediate filaments during heat shock and recovery of chicken embryo fibroblasts. J. Cell. Biochem. 1993, 52, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Tolstonog, G.V.; Belichenko-Weitzmann, I.V.; Lu, J.-P.; Hartig, R.; Shoeman, R.L.; Taub, U.; Traub, P. Spontaneously Immortalized Mouse Embryo Fibroblasts: Growth Behavior of Wild-Type and Vimentin-Deficient Cells in Relation to Mitochondrial Structure and Activity. DNA Cell Biol. 2005, 24, 680–709. [Google Scholar] [CrossRef] [PubMed]
- Wiche, G.; Krepler, R.; Artlieb, U.; Pytela, R.; Denk, H. Occurrence and immunolocalization of plectin in tissues. J. Cell Biol. 1983, 97, 887–901. [Google Scholar] [CrossRef] [PubMed]
- Rezniczek, G.A.; Abrahamsberg, C.; Fuchs, P.; Spazierer, D.; Wiche, G. Plectin 5′-transcript diversity: short alternative sequences determine stability of gene products, initiation of translation and subcellular localization of isoforms. Hum. Mol. Genet. 2003, 12, 3181–3194. [Google Scholar] [CrossRef] [PubMed]
- Winter, L.; Abrahamsberg, C.; Wiche, G. Plectin isoform 1b mediates mitochondrion-intermediate filament network linkage and controls organelle shape. J. Cell Biol. 2008, 181, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Rapaport, D. Finding the right organelle: Targeting signals in mitochondrial outer-membrane proteins. EMBO Rep. 2003, 4, 948–952. [Google Scholar] [CrossRef] [PubMed]
- Chernoivanenko, I.S.; Matveeva, E.A.; Minin, A.A. Vimentin intermediate filaments increase mitochondrial membrane potential. Biochem. Mosc. Suppl. Ser. Membr. Cell Biol. 2011, 5, 21–28. [Google Scholar] [CrossRef]
- Matveeva, E.A.; Venkova, L.S.; Chernoivanenko, I.S.; Minin, A.A. Vimentin is involved in regulation of mitochondrial motility and membrane potential by Rac1. Biol. Open 2015, 4, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Izawa, I.; Inagaki, M. Regulatory mechanisms and functions of intermediate filaments: A study using site- and phosphorylation state-specific antibodies. Cancer Sci. 2006, 97, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Lowery, J.; Jain, N.; Kuczmarski, E.R.; Mahammad, S.; Goldman, A.; Gelfand, V.I.; Opal, P.; Goldman, R.D. Abnormal Intermediate Filament Organization Alters Mitochondrial Motility in Giant Axonal Neuropathy Fibroblasts. Mol. Biol. Cell 2015. [Google Scholar] [CrossRef] [PubMed]
- Capetanaki, Y.; Bloch, R.J.; Kouloumenta, A.; Mavroidis, M.; Psarras, S. Muscle intermediate filaments and their links to membranes and membranous organelles. Exp. Cell Res. 2007, 313, 2063–2076. [Google Scholar] [CrossRef] [PubMed]
- Thornell, L.-E.; Carlsson, L.; Li, Z.; Mericskay, M.; Paulin, D. Null Mutation in the Desmin Gene Gives Rise to a Cardiomyopathy. J. Mol. Cell. Cardiol. 1997, 29, 2107–2124. [Google Scholar] [CrossRef] [PubMed]
- Stromer, M.H.; Bendayan, M. Immunocytochemical identification of cytoskeletal linkages to smooth muscle cell nuclei and mitochondria. Cell Motil. Cytoskeleton. 1990, 17, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Colucci-Guyon, E.; Pinçon-Raymond, M.; Mericskay, M.; Pournin, S.; Paulin, D.; Babinet, C. Cardiovascular Lesions and Skeletal Myopathy in Mice Lacking Desmin. Dev. Biol. 1996, 175, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Kostareva, A.; Sjöberg, G.; Bruton, J.; Zhang, S.-J.; Balogh, J.; Gudkova, A.; Hedberg, B.; Edström, L.; Westerblad, H.; Sejersen, T. Mice expressing L345P mutant desmin exhibit morphological and functional changes of skeletal and cardiac mitochondria. J. Muscle Res. Cell Motil. 2008, 29, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Schröder, R.; Goudeau, B.; Simon, M.C.; Fischer, D.; Eggermann, T.; Clemen, C.S.; Li, Z.; Reimann, J.; Xue, Z.; Rudnik-Schöneborn, S.; et al. On noxious desmin: functional effects of a novel heterozygous desmin insertion mutation on the extrasarcomeric desmin cytoskeleton and mitochondria. Hum. Mol. Genet. 2003, 12, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.; Waele, L.D.; Hudson, J.; Eagle, M.; Sewry, C.; Marsh, J.; Charlton, R.; He, L.; Blakely, E.L.; Horrocks, I.; et al. Recessive desmin-null muscular dystrophy with central nuclei and mitochondrial abnormalities. Acta Neuropathol. (Berl.) 2013, 125, 917–919. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. MAM (mitochondria-associated membranes) in mammalian cells: Lipids and beyond. Biochim. Biophys. Acta 2014, 1841, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Achleitner, G.; Gaigg, B.; Krasser, A.; Kainersdorfer, E.; Kohlwein, S.D.; Perktold, A.; Zellnig, G.; Daum, G. Association between the endoplasmic reticulum and mitochondria of yeast facilitates interorganelle transport of phospholipids through membrane contact. Eur. J. Biochem. 1999, 264, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Filippin, L.; Magalhães, P.J.; Benedetto, G.D.; Colella, M.; Pozzan, T. Stable Interactions between Mitochondria and Endoplasmic Reticulum Allow Rapid Accumulation of Calcium in a Subpopulation of Mitochondria. J. Biol. Chem. 2003, 278, 39224–39234. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; Stefani, D.D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Fountoulakis, M.; Soumaka, E.; Rapti, K.; Mavroidis, M.; Tsangaris, G.; Maris, A.; Weisleder, N.; Capetanaki, Y. Alterations in the heart mitochondrial proteome in a desmin null heart failure model. J. Mol. Cell. Cardiol. 2005, 38, 461–474. [Google Scholar] [CrossRef] [PubMed]
- Smolina, N.; Bruton, J.; Sjoberg, G.; Kostareva, A.; Sejersen, T. Aggregate-prone desmin mutations impair mitochondrial calcium uptake in primary myotubes. Cell Calcium 2014, 56, 269–275. [Google Scholar] [CrossRef] [PubMed]
- McCormack, J.G.; Denton, R.M. Intracellular calcium ions and intramitochondrial Ca in the regulation of energy metabolism in mammalian tissues. Proc. Nutr. Soc. 1990, 49, 57–75. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgó, J.; Müller, M.; Vais, H.; Cheung, K.-H.; Yang, J.; Parker, I.; et al. Essential Regulation of Cell Bioenergetics by Constitutive InsP3 Receptor Ca2+ Transfer to Mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, W.A.; Ahad, A.; Ahsan, H. The mystery of BCL2 family: Bcl-2 proteins and apoptosis: An update. Arch. Toxicol. 2015, 89, 289–317. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Narita, M.; Tsujimoto, Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 1999, 399, 483–487. [Google Scholar] [PubMed]
- Zhu, L.; Ling, S.; Yu, X.-D.; Venkatesh, L.K.; Subramanian, T.; Chinnadurai, G.; Kuo, T.H. Modulation of Mitochondrial Ca2+ Homeostasis by Bcl-2. J. Biol. Chem. 1999, 274, 33267–33273. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Ferrari, D.; Rapizzi, E.; Virgilio, F.D.; Pozzan, T.; Rizzuto, R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: Significance for the molecular mechanism of Bcl-2 action. EMBO J. 2001, 20, 2690–2701. [Google Scholar] [CrossRef] [PubMed]
- Weisleder, N.; Taffet, G.E.; Capetanaki, Y. Bcl-2 overexpression corrects mitochondrial defects and ameliorates inherited desmin null cardiomyopathy. Proc. Natl. Acad. Sci. USA 2004, 101, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Lindén, M.; Li, Z.; Paulin, D.; Gotow, T.; Leterrier, J.-F. Effects of Desmin Gene Knockout on Mice Heart Mitochondria. J. Bioenerg. Biomembr. 2001, 33, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Vendelin, M.; Lemba, M.; Saks, V.A. Analysis of Functional Coupling: Mitochondrial Creatine Kinase and Adenine Nucleotide Translocase. Biophys. J. 2004, 87, 696–713. [Google Scholar] [CrossRef] [PubMed]
- Kay, L.; Li, Z.; Mericskay, M.; Olivares, J.; Tranqui, L.; Fontaine, E.; Tiivel, T.; Sikk, P.; Kaambre, T.; Samuel, J.-L.; et al. Study of regulation of mitochondrial respiration in vivo: An analysis of influence of ADP diffusion and possible role of cytoskeleton. Biochim. Biophys. Acta BBA Bioenerg. 1997, 1322, 41–59. [Google Scholar] [CrossRef]
- Moll, R.; Franke, W.W.; Schiller, D.L.; Geiger, B.; Krepler, R. The catalog of human cytokeratins: Patterns of expression in normal epithelia, tumors and cultured cells. Cell 1982, 31, 11–24. [Google Scholar] [CrossRef]
- Coulombe, P.A.; Kerns, M.L.; Fuchs, E. Epidermolysis bullosa simplex: A paradigm for disorders of tissue fragility. J. Clin. Investig. 2009, 119, 1784–1793. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.M.; Yu, Q.C.; LeBlanc-Straceski, J.; Christiano, A.; Pulkkinen, L.; Kucherlapati, R.S.; Uitto, J.; Fuchs, E. Mutations in the non-helical linker segment L1–2 of keratin 5 in patients with Weber-Cockayne epidermolysis bullosa simplex. J. Cell Sci. 1994, 107, 765–774. [Google Scholar] [PubMed]
- Vance, J.E. Newly made phosphatidylserine and phosphatidylethanolamine are preferentially translocated between rat liver mitochondria and endoplasmic reticulum. J. Biol. Chem. 1991, 266, 89–97. [Google Scholar] [PubMed]
- Nishizawa, M.; Izawa, I.; Inoko, A.; Hayashi, Y.; Nagata, K.; Yokoyama, T.; Usukura, J.; Inagaki, M. Identification of trichoplein, a novel keratin filament-binding protein. J. Cell Sci. 2005, 118, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Cerqua, C.; Anesti, V.; Pyakurel, A.; Liu, D.; Naon, D.; Wiche, G.; Baffa, R.; Dimmer, K.S.; Scorrano, L. Trichoplein/mitostatin regulates endoplasmic reticulum-mitochondria juxtaposition. EMBO Rep. 2010, 11, 854–860. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Gohil, V.M.; Ma, L.; Greenberg, M.L. Absence of Cardiolipin Results in Temperature Sensitivity, Respiratory Defects, and Mitochondrial DNA Instability Independent of pet56. J. Biol. Chem. 2004, 279, 32294–32300. [Google Scholar] [CrossRef] [PubMed]
- DeVay, R.M.; Dominguez-Ramirez, L.; Lackner, L.L.; Hoppins, S.; Stahlberg, H.; Nunnari, J. Coassembly of Mgm1 isoforms requires cardiolipin and mediates mitochondrial inner membrane fusion. J. Cell Biol. 2009, 186, 793–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, G.-Z.; Looi, K.S.; Toivola, D.M.; Strnad, P.; Zhou, Q.; Liao, J.; Wei, Y.; Habtezion, A.; Omary, M.B. Keratins modulate the shape and function of hepatocyte mitochondria: A mechanism for protection from apoptosis. J. Cell Sci. 2009, 122, 3851–3855. [Google Scholar] [CrossRef] [PubMed]
- Leng, R.P.; Lin, Y.; Ma, W.; Wu, H.; Lemmers, B.; Chung, S.; Parant, J.M.; Lozano, G.; Hakem, R.; Benchimol, S. Pirh2, a p53-Induced Ubiquitin-Protein Ligase, Promotes p53 Degradation. Cell 2003, 112, 779–791. [Google Scholar] [CrossRef]
- Duan, S.; Yao, Z.; Zhu, Y.; Wang, G.; Hou, D.; Wen, L.; Wu, M. The Pirh2–keratin 8/18 interaction modulates the cellular distribution of mitochondria and UV-induced apoptosis. Cell Death Differ. 2009, 16, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial Fission, Fusion, and Stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Mathew, J.; Loranger, A.; Gilbert, S.; Faure, R.; Marceau, N. Keratin 8/18 regulation of glucose metabolism in normal versus cancerous hepatic cells through differential modulation of hexokinase status and insulin signaling. Exp. Cell Res. 2013, 319, 474–486. [Google Scholar] [CrossRef] [PubMed]
- Kröger, C.; Loschke, F.; Schwarz, N.; Windoffer, R.; Leube, R.E.; Magin, T.M. Keratins control intercellular adhesion involving PKC-α-mediated desmoplakin phosphorylation. J. Cell Biol. 2013, 201, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Baines, C.P.; Song, C.-X.; Zheng, Y.-T.; Wang, G.-W.; Zhang, J.; Wang, O.-L.; Guo, Y.; Bolli, R.; Cardwell, E.M.; Ping, P. Protein Kinase Cε Interacts With and Inhibits the Permeability Transition Pore in Cardiac Mitochondria. Circ. Res. 2003, 92, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Krols, M.; van Isterdael, G.; Asselbergh, B.; Kremer, A.; Lippens, S.; Timmerman, V.; Janssens, S. Mitochondria-associated membranes as hubs for neurodegeneration. Acta Neuropathol. (Berl.) 2016, 131, 505–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snider, N.T.; Omary, M.B. Post-translational modifications of intermediate filament proteins: Mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2014, 15, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Parysek, L.M.; Eckert, B.S. Vimentin filaments in spreading, randomly locomoting, and f-met-leu-phe-treated neutrophils. Cell Tissue Res. 1984, 235, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Campello, S.; Lacalle, R.A.; Bettella, M.; Mañes, S.; Scorrano, L.; Viola, A. Orchestration of lymphocyte chemotaxis by mitochondrial dynamics. J. Exp. Med. 2006, 203, 2879–2886. [Google Scholar] [CrossRef] [PubMed]
- Helfand, B.T.; Mendez, M.G.; Murthy, S.N.P.; Shumaker, D.K.; Grin, B.; Mahammad, S.; Aebi, U.; Wedig, T.; Wu, Y.I.; Hahn, K.M.; et al. Vimentin organization modulates the formation of lamellipodia. Mol. Biol. Cell 2011, 22, 1274–1289. [Google Scholar] [CrossRef] [PubMed]
- Desai, S.P.; Bhatia, S.N.; Toner, M.; Irimia, D. Mitochondrial Localization and the Persistent Migration of Epithelial Cancer cells. Biophys. J. 2013, 104, 2077–2088. [Google Scholar] [CrossRef] [PubMed]
- Kidd, M.E.; Shumaker, D.K.; Ridge, K.M. The Role of Vimentin Intermediate Filaments in the Progression of Lung Cancer. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1–6. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Intermediate Filament Protein | Modification | Cell Type | Mitochondrial Phenotype | Reference |
|---|---|---|---|---|
| NF-L | Knock-Out | Neurons | Decreased length and fusion rate, increased motility | [40] |
| Peripherin | Overexpression | NFL knock-out neurons | Increased retrograde transport of mitochondria | [41] |
| Vimentin | Knock-Out | Fibroblasts | Decreased membrane potential, altered distribution, increased motility | [42,43] |
| Vimentin | Knock-Down | Cos7 cell line | Fragmentation, altered distribution | [44] |
| Desmin | Knock-Out | Cardiac and skeletal muscle | Abnormal shape and positioning, decreased maximal respiration rate, decreased oxygen consumption | [45] |
| Keratin 5 | P24L mutation | Epidermis | Intracellular clustering | [46] |
| Keratin | Knock-Out | Epidermis | Altered lipid composition and activity | [47] |
| Keratin 18 | R89C mutation | Liver-derived cell lines | Fragmentation | [48] |
| Keratin 19 | Knock-Out | Muscle | Mitochondrial disorganization | [49] |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schwarz, N.; Leube, R.E. Intermediate Filaments as Organizers of Cellular Space: How They Affect Mitochondrial Structure and Function. Cells 2016, 5, 30. https://doi.org/10.3390/cells5030030
Schwarz N, Leube RE. Intermediate Filaments as Organizers of Cellular Space: How They Affect Mitochondrial Structure and Function. Cells. 2016; 5(3):30. https://doi.org/10.3390/cells5030030
Chicago/Turabian StyleSchwarz, Nicole, and Rudolf E. Leube. 2016. "Intermediate Filaments as Organizers of Cellular Space: How They Affect Mitochondrial Structure and Function" Cells 5, no. 3: 30. https://doi.org/10.3390/cells5030030
APA StyleSchwarz, N., & Leube, R. E. (2016). Intermediate Filaments as Organizers of Cellular Space: How They Affect Mitochondrial Structure and Function. Cells, 5(3), 30. https://doi.org/10.3390/cells5030030