Assessing Autophagy in Mouse Models and Patients with Systemic Autoimmune Diseases



Abstract
:1. Autophagy in Immunity and Autoimmune Diseases
2. MRL/lpr Mice as a Model for SLE
3. Methods and Notes
- We highly recommend, whenever possible, to analyze specific cell subtypes rather than studying whole organ homogenates or unfractionated peripheral blood samples that contain mixed cell subsets, as the latter may exhibit very different autophagy activation status that can affect the detection of events.
- As recommended in authoritative reviews on autophagy, several different autophagic assays need to be applied to make reliable conclusions [67,68,69,70]. One single assay is by far not sufficient to determine whether the autophagy activity is abnormally increased or decreased. The number of individual samples analyzed also has to be sufficient to allow robust statistic interpretation of data.
3.1. Obtaining Cell Homogenates from Organs
3.1.1. Obtaining Homogenates from the Spleen
- Some fat tissues are commonly found in the splenic cell suspension prepared from MRL/lpr mice (rarely observed in the case of control CBA/J or C57BL/6 mice). They can be removed by filtering cell fraction through a 40-µm cell strainer, according to our experiences.
- The spleen of MRL/lpr mice is usually 4–6 times the size of that of CBA/J mice at the same age (Figure 2A). It is important to take into consideration this huge difference, as for certain experiments pooling 2–3 spleens from CBA/J or C57BL/6 control mice will be required to have enough control splenocytes.
- The final purity of B or T cells should always be checked as it has been sometimes observed, for example, that depending on the isolation kits, 5–10% DN T cells can remain in the MRL/lpr B cell fraction in our experiences.
3.1.2. Obtaining Homogenates from Lymph Nodes
3.1.3. Obtaining Homogenates from Salivary Glands
- The size of SGs from MRL/lpr mice is usually 1.5–2 times larger compared to that of SGs from CBA/J or C57BL/6 mice at the same age, depending on the severity of the disease (Figure 2B).
- The concentration of collagenase D and DNase needs to be optimized, as an excessive enzyme concentration can induce a loss of cell viability and a too low enzyme dose will obviously lead to insufficient digestion.
- When SG cell suspensions are prepared for FC measurement, it is essential to add EDTA in the FC buffer (such as PBS supplemented with 2% v/v FBS) to avoid cell aggregation and ensure single cell suspension. We have determined that 0.3 mM EDTA is the optimal concentration as higher concentration causes toxicity and a lower concentration is insufficient to separate cells.
3.1.4. Isolating Peripheral Blood Mononuclear Cells from Patient’s Blood
- In the case of blood samples taken from lupus patients, we have occasionally observed a large amount of red blood cells remaining in the PBMC layer after centrifugation in Ficoll. In this case, an additional step of ACK lysis has been included after the Ficoll step for lysing these red blood cells.
- In the case of CD4+ T cell isolation from blood, instead of performing a Ficoll density gradient centrifugation, RosetteSepTM Human CD4+ T Cell Enrichment Cocktail (Stemcell Technologies, Vancouver, Canada, 15062) can be added directly to the whole blood to isolate CD4+ T cells by negative selection.
- Note that the number of B cells recovered from the blood of lupus patients is usually very low (2–5 × 106 cells from around 40 mL of blood). This is probably related to the immunosuppressive treatments given to patients with lupus. This considerably reduces the number of assays that can be performed. Therefore, it is important to miniaturize the assays to maximum (without losing too much sensitivity and specificity) and prioritize the tests that will be carried out for the autophagy analysis.
3.2. Measurement of Macroautophagy by Electron Microscopy
- For statistical reasons, a minimum of 50 cell sections per condition should be examined.
- It is recommended to examine grids prepared from different resin blocks to avoid counting the same cells several times.
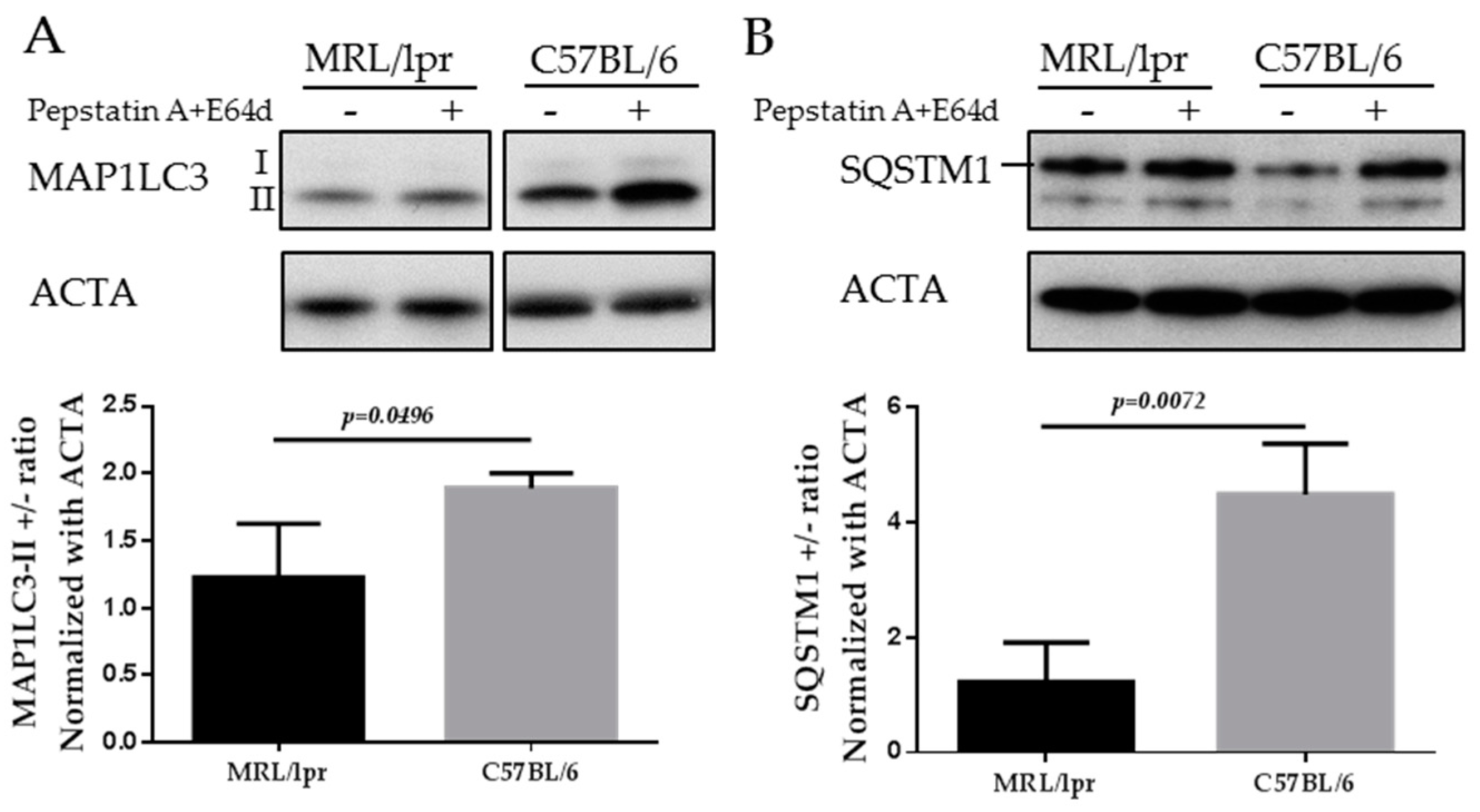
3.3. Measurement of Macroautophagy by Western Blot
- Serum deprivation is widely used as a stimulus to induce autophagy. It is important to keep in mind that B cells are much more sensitive to serum withdrawal compared to T cells and many other cells. Therefore, we do not recommend incubating B cells in serum free media for more than 4 h, while more than 12 h serum withdrawal can be used for T cells and SG cells.
- The limitation of using Western blotting for B cells isolated from the blood of lupus patients relies to the number of recovered cells, which is generally too low to perform several conditions (including the controls run in the presence or absence of lysosomal inhibitors). In our experience, it is optimal to use one million cells (or around 20 µg protein) per condition/lane to obtain good signals, while commonly only 2–5 million B cells are recovered from lupus patients. It is therefore more feasible to use alternative approaches such as FC-based methods, as in the latter, much fewer cells are needed per condition.
- Loading controls are essential for proper interpretation of Western blots. They are important to assess the total proteins that have been loaded in each lane across the gel, thus allowing a more accurate blot calibration. Loading controls also permit gel conditions to be checked and compared from gel to gel.
- Regarding the type of proteins used as loading controls, we noticed that actin-α, but not actin-β, can be readily used as a loading control of SG extracts, whereas actin-β, but not actin-α, can be used when splenocytes are studied. Among usual loading controls, attention should be paid not to use a marker that is affected by autophagy alteration; for example, in certain cases, glial fibrillary acidic protein (GFAP) is not an appropriate control.
3.4. Measurement of Macroautophagy by Flow Cytometry
3.4.1. Measurement of MAP1LC3 by Flow Cytometry
- The first advantage of measuring autophagosomes by MAP1LC3 staining with FC is that a limited number of cells is needed (around 0.1 million per condition, except in the imaging cytometer), compared with the number of cells required for Western blot (0.5–1 million cells per condition).
- Secondly, surface staining to distinguish cell populations (Figure 5A) can be done before permeabilization and immunostaining of MAP1LC3; therefore, no prior cell isolation is needed. We highly recommend FC measurement of MAP1LC3 staining in combination of surface cell markers when the number of cells that are available is low, such as it is the case when B cells from lupus patients are analyzed.
- It is very important to avoid high background staining, which we have observed when some MAP1LC3 staining kits are used. We strongly advise the users to include the following controls when calibrating the MAP1LC3 assay:
- −
- positive controls: cells treated with lysosomal inhibitors that block degradation of autophagosomes and therefore increase the amount of MAP1LC3-II;
- −
- negative controls: cells in which the initiation of autophagy has been blocked, e.g., cells from autophagy deficient mice, and unstained cells.
3.4.2. Measurement of SQSTM1 with Flow Cytometry
- Autophagic flux can also be evaluated by measuring the difference of SQSTM1 level in the presence and absence of lysosomal inhibitors [31].
3.5. Measurement of CMA by Western Blot
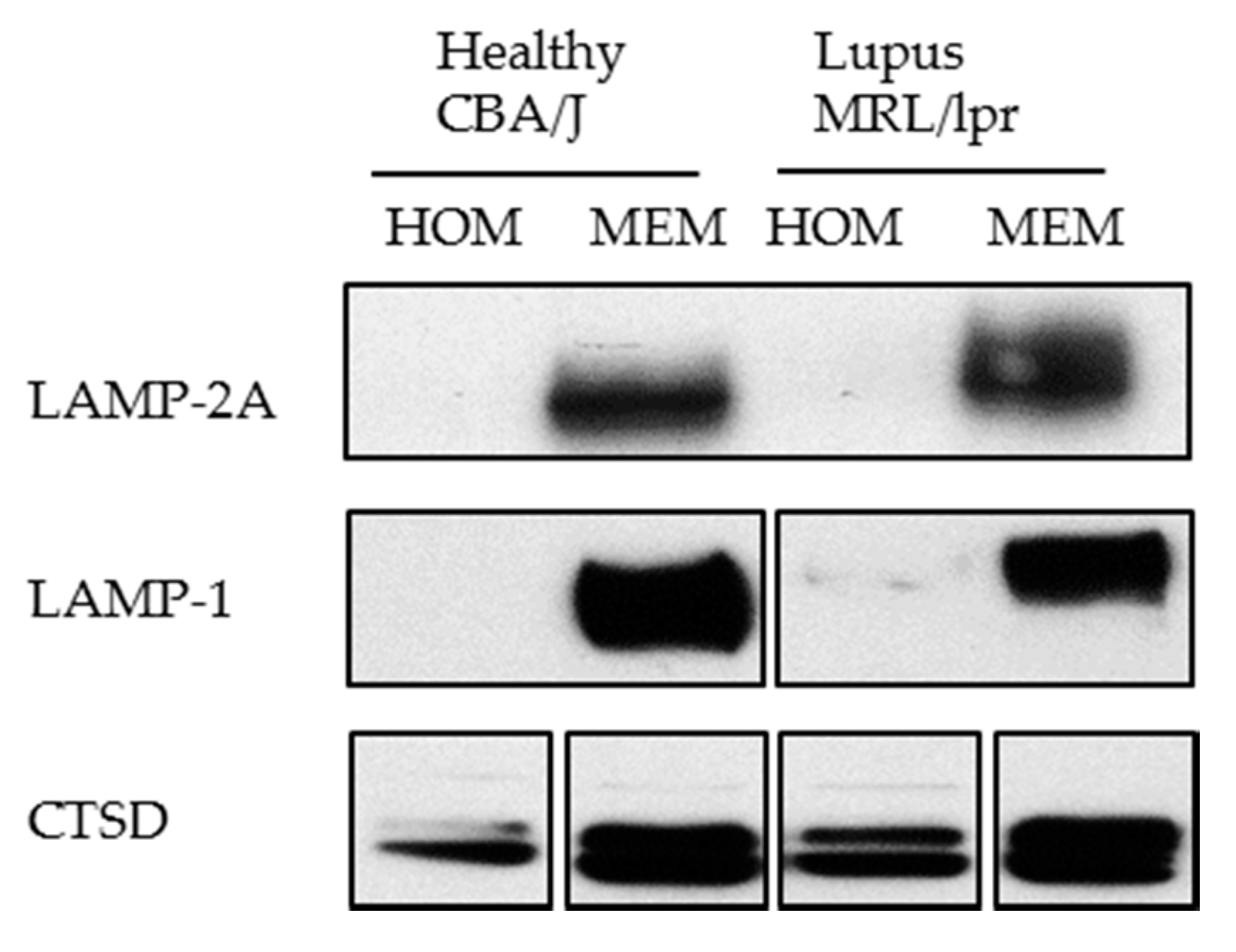
- It should be kept in mind that three variants of LAMP-2 exist and share the same lysosomal lumen region [90]. Therefore, for detecting LAMP-2A only, it is important to use antibodies that specifically detect the cytosolic tail of LAMP-2A. Specific antibodies can be raised as the 12 amino acid residues on the cytosolic side of LAMP-2A significantly differ from those encompassed in its variants LAMP-2B and C.
- We have experienced that sometimes LAMP-2A cannot be detected using whole cell lysate (homogenate). In this case, for studying LAMP-2A, we recommend preparing a membranous cell fraction with enriched lysosomes and mitochondria through classical step-wise centrifugation. This membrane cell fraction can be prepared by following the protocol published in Kaushik and Cuervo, 2008 (Section 3.1, Step 1–4) [91], however without going through the long and challenging process of preparing the highly purified lysosomes. Figure 6 illustrates our own data: LAMP-2A level is not detectable in cell homogenates (HOM), while there is a strong signal in the membrane fraction (MEM).
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
References
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Eskelinen, E.-L.; Deretic, V. Autophagosomes, phagosomes, autolysosomes, phagolysosomes, autophagolysosomes… Wait, I’m confused. Autophagy 2014, 10, 549–551. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, J.; Bao, J. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V. Autophagy: An emerging immunological paradigm. J. Immunol. 2012, 189, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Galluzzi, L.; Zitvogel, L.; Kroemer, G. Autophagy and cellular immune responses. Immunity 2013, 39, 211–227. [Google Scholar] [CrossRef] [PubMed]
- Mintern, J.D.; Harris, J. Autophagy and immunity. Immunol. Cell Biol. 2015, 93, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Gros, F.; Muller, S. Pharmacological regulators of autophagy and their link with modulators of lupus disease. Br. J. Pharmacol. 2014, 171, 4337–4359. [Google Scholar] [CrossRef] [PubMed]
- Münz, C. Autophagy beyond intracellular MHC Class II antigen presentation. Trends Immunol. 2016, 37, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Crotzer, V.L.; Blum, J.S. Autophagy and its role in MHC-mediated antigen presentation. J. Immunol. 2009, 182, 3335–3341. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, F.; Milosevic, S.; Behrends, U.; Jaffee, E.M.; Pardoll, D.M.; Bornkamm, G.W.; Mautner, J. Major histocompatibility complex class II-restricted presentation of a cytosolic antigen by autophagy. Eur. J. Immunol. 2003, 33, 1250–1259. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Mattei, L.M.; Steinberg, B.E.; Alberts, P.; Lee, Y.H.; Chervonsky, A.; Mizushima, N.; Grinstein, S.; Iwasaki, A. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity 2010, 32, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Schmid, D.; Pypaert, M.; Münz, C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 2007, 26, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Saini, N.K.; Baena, A.; Ng, T.W.; Venkataswamy, M.M.; Kennedy, S.C.; Kunnath-Velayudhan, S.; Carreño, L.J.; Xu, J.; Chan, J.; Larsen, M.H.; et al. Suppression of autophagy and antigen presentation by Mycobacterium tuberculosis PE_PGRS47. Nat. Microbiol. 2016, 1, 16133. [Google Scholar] [CrossRef] [PubMed]
- Blanchet, F.P.; Moris, A.; Nikolic, D.S.; Lehmann, M.; Cardinaud, S.; Stalder, R.; Garcia, E.; Dinkins, C.; Leuba, F.; Wu, L.; et al. Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity 2010, 32, 654–669. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Li, P.; Lin, Y.; Lott, J.M.; Hislop, A.D.; Canaday, D.H.; Brutkiewicz, R.R.; Blum, J.S. Lamp-2a facilitates MHC class II presentation of cytoplasmic antigens. Immunity 2005, 22, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Netea-Maier, R.T.; Plantinga, T.S.; van de Veerdonk, F.L.; Smit, J.W.; Netea, M.G. Modulation of inflammation by autophagy: Consequences for human disease. Autophagy 2016, 12, 245–260. [Google Scholar] [CrossRef] [PubMed]
- McLeod, I.X.; Jia, W.; He, Y.-W. The contribution of autophagy to lymphocyte survival and homeostasis. Immunol. Rev. 2012, 249, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Patterson, N.L.; Mintern, J.D. Intersection of autophagy with pathways of antigen presentation. Protein Cell 2012, 3, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Isenberg, D.A. Systemic lupus erythematosus. N. Engl. J. Med. 2008, 358, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, M.; Muller, S. Target autophagy as a novel therapeutic strategy in autoimmune diseases. In Autophagy Networks in Inflammation; Maiuri, M.C., Stefano, D.D., Eds.; Progress in Inflammation Research Series; Springer: Cham, Switzerland, 2016; pp. 267–295. ISBN 978-3-319-30077-1. [Google Scholar]
- Jeltsch-David, H.; Muller, S. Neuropsychiatric systemic lupus erythematosus: Pathogenesis and biomarkers. Nat. Rev. Neurol. 2014, 10, 579–596. [Google Scholar] [CrossRef] [PubMed]
- International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN); Harley, J.B.; Alarcón-Riquelme, M.E.; Criswell, L.A.; Jacob, C.O.; Kimberly, R.P.; Moser, K.L.; Tsao, B.P.; Vyse, T.J.; Langefeld, C.D. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat. Genet. 2008, 40, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Tang, H.; Zhang, Y.; Tang, X.; Zhang, J.; Sun, L.; Yang, J.; Cui, Y.; Zhang, L.; Hirankarn, N.; et al. Meta-analysis followed by replication identifies loci in or near CDKN1B, TET3, CD80, DRAM1, and ARID5B as associated with systemic lupus erythematosus in asians. Am. J. Hum. Genet. 2013, 92, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Lu, X.; Lv, J.; Yang, H.; Qin, L.; Zhao, M.; Su, Y.; Li, Z.; Zhang, H. Genetic association of PRDM1-ATG5 intergenic region and autophagy with systemic lupus erythematosus in a Chinese population. Ann. Rheum. Dis. 2011, 70, 1330–1337. [Google Scholar] [CrossRef] [PubMed]
- Dang, J.; Li, J.; Xin, Q.; Shan, S.; Bian, X.; Yuan, Q.; Liu, N.; Ma, X.; Li, Y.; Liu, Q. Gene-gene interaction of ATG5, ATG7, BLK and BANK1 in systemic lupus erythematosus. Int. J. Rheum. Dis. 2016, 19, 1284–1293. [Google Scholar] [CrossRef] [PubMed]
- Gros, F.; Arnold, J.; Page, N.; Décossas, M.; Korganow, A.-S.; Martin, T.; Muller, S. Macroautophagy is deregulated in murine and human lupus T lymphocytes. Autophagy 2012, 8, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Macri, C.; Wang, F.; Tasset, I.; Schall, N.; Page, N.; Briand, J.-P.; Cuervo, A.M.; Muller, S. Modulation of deregulated chaperone-mediated autophagy by a phosphopeptide. Autophagy 2015, 11, 472–486. [Google Scholar] [CrossRef] [PubMed]
- Clarke, A.J.; Ellinghaus, U.; Cortini, A.; Stranks, A.; Simon, A.K.; Botto, M.; Vyse, T.J. Autophagy is activated in systemic lupus erythematosus and required for plasmablast development. Ann. Rheum. Dis. 2015, 74, 912–920. [Google Scholar] [CrossRef] [PubMed]
- Alessandri, C.; Barbati, C.; Vacirca, D.; Piscopo, P.; Confaloni, A.; Sanchez, M.; Maselli, A.; Colasanti, T.; Conti, F.; Truglia, S.; et al. T lymphocytes from patients with systemic lupus erythematosus are resistant to induction of autophagy. FASEB J. 2012, 26, 4722–4732. [Google Scholar] [CrossRef] [PubMed]
- Nambiar, M.P.; Juang, Y.-T.; Krishnan, S.; Tsokos, G.C. Dissecting the molecular mechanisms of TCR ζ chain downregulation and T cell signaling abnormalities in human systemic lupus erythematosus. Int. Rev. Immunol. 2004, 23, 245–263. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.H. Autoimmunity: Antigen presentation by B cells contributes to murine lupus. Nat. Rev. Rheumatol. 2015, 11, 564. [Google Scholar] [CrossRef] [PubMed]
- Nashi, E.; Wang, Y.; Diamond, B. The role of B cells in lupus pathogenesis. Int. J. Biochem. Cell Biol. 2010, 42, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Muller, S. Manipulating autophagic processes in autoimmune diseases: A special focus on modulating chaperone-mediated autophagy, an emerging therapeutic target. Front. Immunol. 2015, 6, 252. [Google Scholar] [CrossRef] [PubMed]
- Weindel, C.G.; Richey, L.J.; Bolland, S.; Mehta, A.J.; Kearney, J.F.; Huber, B.T. B cell autophagy mediates TLR7-dependent autoimmunity and inflammation. Autophagy 2015, 11, 1010–1024. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.; Brun, S.; René, F.; de Sèze, J.; Loeffler, J.-P.; Jeltsch-David, H. Autophagy in neuroinflammatory diseases. Autoimmun. Rev. in press, available online 29 May 2017. [CrossRef] [PubMed]
- Wu, Z.-Z.; Zhang, J.-J.; Gao, C.-C.; Zhao, M.; Liu, S.-Y.; Gao, G.-M.; Zheng, Z.-H. Expression of autophagy related genes mTOR, Becline-1, LC3 and p62 in the peripheral blood mononuclear cells of systemic lupus erythematosus. Am. J. Clin. Exp. Immunol. 2017, 6, 1–8. [Google Scholar] [PubMed]
- Li, B.; Yue, Y.; Dong, C.; Shi, Y.; Xiong, S. Blockade of macrophage autophagy ameliorates activated lymphocytes-derived DNA induced murine lupus possibly via inhibition of proinflammatory cytokine production. Clin. Exp. Rheumatol. 2014, 32, 705–714. [Google Scholar] [PubMed]
- Page, N.; Gros, F.; Schall, N.; Décossas, M.; Bagnard, D.; Briand, J.-P.; Muller, S. HSC70 blockade by the therapeutic peptide P140 affects autophagic processes and endogenous MHCII presentation in murine lupus. Ann. Rheum. Dis. 2011, 70, 837–843. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, F.; Zhang, X.; Shi, G.; Ren, J.; Ji, J.; Ding, L.; Fan, H.; Dou, H.; Hou, Y. Notch-Hes-1 axis controls TLR7-mediated autophagic death of macrophage via induction of P62 in mice with lupus. Cell Death Dis. 2016, 7, e2341. [Google Scholar] [CrossRef] [PubMed]
- Hampe, J.; Franke, A.; Rosenstiel, P.; Till, A.; Teuber, M.; Huse, K.; Albrecht, M.; Mayr, G.; De La Vega, F.M.; Briggs, J.; et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 2007, 39, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Rioux, J.D.; Xavier, R.J.; Taylor, K.D.; Silverberg, M.S.; Goyette, P.; Huett, A.; Green, T.; Kuballa, P.; Barmada, M.M.; Datta, L.W.; et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 2007, 39, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Glas, J.; Seiderer, J.; Bues, S.; Stallhofer, J.; Fries, C.; Olszak, T.; Tsekeri, E.; Wetzke, M.; Beigel, F.; Steib, C.; et al. IRGM variants and susceptibility to inflammatory bowel disease in the german population. PLoS ONE 2013, 8, e54338. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.C.; Tao, Y.; Wu, C.; Zhao, P.L.; Li, K.; Zheng, J.Y.; Li, L.X. Association between variants of the autophagy related gene—IRGM and susceptibility to Crohn’s disease and ulcerative colitis: A meta-analysis. PLoS ONE 2013, 8, e80602. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.-Y.; Beyer, C.; Giessl, A.; Kireva, T.; Scholtysek, C.; Uderhardt, S.; Munoz, L.E.; Dees, C.; Distler, A.; Wirtz, S.; et al. Autophagy regulates TNFα-mediated joint destruction in experimental arthritis. Ann. Rheum. Dis. 2013, 72, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Orozco, G.; Eyre, S.; Hinks, A.; Bowes, J.; Morgan, A.W.; Wilson, A.G.; Wordsworth, P.; Steer, S.; Hocking, L.; UKRAG consortium; et al. Study of the common genetic background for rheumatoid arthritis and systemic lupus erythematosus. Ann. Rheum. Dis. 2011, 70, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Wang, H.; Wu, Y.; He, Z.; Qin, Y.; Shen, Q. The autophagy level is increased in the synovial tissues of patients with active rheumatoid arthritis and is correlated with disease severity. Mediators Inflamm. 2017, 2017, e7623145. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Ospelt, C.; Gay, R.E.; Gay, S.; Klein, K. Dual role of autophagy in stress-induced cell death in rheumatoid arthritis synovial fibroblasts. Arthritis Rheumatol. Hoboken NJ 2014, 66, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Nogalska, A.; D’Agostino, C.; Terracciano, C.; Engel, W.K.; Askanas, V. Impaired autophagy in sporadic inclusion-body myositis and in endoplasmic reticulum stress-provoked cultured human muscle fibers. Am. J. Pathol. 2010, 177, 1377–1387. [Google Scholar] [CrossRef] [PubMed]
- Alirezaei, M.; Fox, H.S.; Flynn, C.T.; Moore, C.S.; Hebb, A.L.O.; Frausto, R.F.; Bhan, V.; Kiosses, W.B.; Whitton, J.L.; Robertson, G.S.; et al. Elevated ATG5 expression in autoimmune demyelination and multiple sclerosis. Autophagy 2009, 5, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Igci, M.; Baysan, M.; Yigiter, R.; Ulasli, M.; Geyik, S.; Bayraktar, R.; Bozgeyik, İ.; Bozgeyik, E.; Bayram, A.; Cakmak, E.A. Gene expression profiles of autophagy-related genes in multiple sclerosis. Gene 2016, 588, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Kobayashi, S.; Chen, K.; Timm, D.; Volden, P.; Huang, Y.; Gulick, J.; Yue, Z.; Robbins, J.; Epstein, P.N.; et al. Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J. Biol. Chem. 2013, 288, 18077–18092. [Google Scholar] [CrossRef] [PubMed]
- Andrews, B.S.; Eisenberg, R.A.; Theofilopoulos, A.N.; Izui, S.; Wilson, C.B.; McConahey, P.J.; Murphy, E.D.; Roths, J.B.; Dixon, F.J. Spontaneous murine lupus-like syndromes. Clinical and immunopathological manifestations in several strains. J. Exp. Med. 1978, 148, 1198–1215. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zeumer, L.; Reeves, W.H.; Morel, L. Induced murine models of systemic lupus erythematosus. Methods Mol. Biol. Clifton NJ 2014, 1134, 103–130. [Google Scholar] [CrossRef]
- Kono, D.H.; Theofilopoulos, A.N. Genetics of SLE in mice. Springer Semin. Immunopathol. 2006, 28, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Morel, L. Genetics of SLE: Evidence from mouse models. Nat. Rev. Rheumatol. 2010, 6, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Perry, D.; Sang, A.; Yin, Y.; Zheng, Y.-Y.; Morel, L. Murine models of systemic lupus erythematosus. BioMed Res. Int. 2011, 2011, e271694. [Google Scholar] [CrossRef] [PubMed]
- Theofilopoulos, A.N.; Dixon, F.J. Murine models of systemic lupus erythematosus. Adv. Immunol. 1985, 37, 269–390. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.J. CD95 and the MRL-lpr Mouse Model. Methods Mol. Biol. Clifton NJ 2017, 1557, 219–228. [Google Scholar] [CrossRef]
- Martina, M.N.; Noel, S.; Saxena, A.; Rabb, H.; Hamad, A.R.A. Double negative (DN) αβ T cells: Misperception and overdue recognition. Immunol. Cell Biol. 2015, 93, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Hammond, D.M.; Nagarkatti, P.S.; Goté, L.R.; Seth, A.; Hassuneh, M.R.; Nagarkatti, M. Double-negative T cells from MRL-lpr/lpr mice mediate cytolytic activity when triggered through adhesion molecules and constitutively express perforin gene. J. Exp. Med. 1993, 178, 2225–2230. [Google Scholar] [CrossRef] [PubMed]
- Jeltsch-David, H.; Muller, S. Neuropsychiatric systemic lupus erythematosus and cognitive dysfunction: The MRL-lpr mouse strain as a model. Autoimmun. Rev. 2014, 13, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Haneji, N.; Hamano, H. Pathogenesis of Sjögren’s syndrome-like autoimmune lesions in MRL/lpr mice. Pathol. Int. 1994, 44, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Gulinello, M.; Putterman, C. The MRL/lpr mouse strain as a model for neuropsychiatric systemic lupus erythematosus. J. Biomed. Biotechnol. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, E.; Galluzzi, L.; Maiuri, M.C.; Criollo, A.; Vitale, I.; Hangen, E.; Modjtahedi, N.; Kroemer, G. Methods for asssessing autophagy and autophagic cell death. In Autophagosome and Phagosome; Deretic, V., Ed.; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2008; pp. 29–76. ISBN 978-1-58829-853-9. [Google Scholar]
- Rubinsztein, D.C.; Cuervo, A.M.; Ravikumar, B.; Sarkar, S.; Korolchuk, V.; Kaushik, S.; Klionsky, D.J. In search of an “autophagomometer”. Autophagy 2009, 5, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed]
- Reeves, J.P.; Reeves, P.A. Removal of lymphoid organs. Curr. Protoc. Immunol. 2001. [Google Scholar] [CrossRef]
- Monneaux, F.; Muller, S. Laboratory protocols for the identification of Th cell epitopes on self-antigens in mice with systemic autoimmune diseases. J. Immunol. Methods 2000, 244, 195–204. [Google Scholar] [CrossRef]
- Monneaux, F.; Briand, J.P.; Muller, S. B and T cell immune response to small nuclear ribonucleoprotein particles in lupus mice: Autoreactive CD4(+) T cells recognize a T cell epitope located within the RNP80 motif of the 70K protein. Eur. J. Immunol. 2000, 30, 2191–2200. [Google Scholar] [CrossRef]
- Henri, S.; Vremec, D.; Kamath, A.; Waithman, J.; Williams, S.; Benoist, C.; Burnham, K.; Saeland, S.; Handman, E.; Shortman, K. The Dendritic Cell Populations of Mouse Lymph Nodes. J. Immunol. 2001, 167, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Broggi, M.A.S.; Schmaler, M.; Lagarde, N.; Rossi, S.W. Isolation of Murine Lymph Node Stromal Cells. J. Vis. Exp. JoVE 2014. [Google Scholar] [CrossRef] [PubMed]
- Amano, O.; Mizobe, K.; Bando, Y.; Sakiyama, K. Anatomy and histology of rodent and human major salivary glands. Acta Histochem. Cytochem. 2012, 45, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Fuss, I.J.; Kanof, M.E.; Smith, P.D.; Zola, H. Isolation of whole mononuclear cells from peripheral blood and cord blood. Curr. Protoc. Immunol. 2009. [Google Scholar] [CrossRef]
- Graham, L.; Orenstein, J.M. Processing tissue and cells for transmission electron microscopy in diagnostic pathology and research. Nat. Protoc. 2007, 2, 2439–2450. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.-L. To be or not to be? Examples of incorrect identification of autophagic compartments in conventional transmission electron microscopy of mammalian cells. Autophagy 2008, 4, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and autophagy. Methods Mol. Biol. Clifton NJ 2008, 445, 77–88. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Sánchez, R.; Yakhine-Diop, S.M.S.; Rodríguez-Arribas, M.; Bravo-San Pedro, J.M.; Martínez-Chacón, G.; Uribe-Carretero, E.; Pinheiro de Castro, D.C.J.; Pizarro-Estrella, E.; Fuentes, J.M.; González-Polo, R.A. mRNA and protein dataset of autophagy markers (LC3 and p62) in several cell lines. Data Brief 2016, 7, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Sánchez, R.; Pizarro-Estrella, E.; Yakhine-Diop, S.M.S.; Rodríguez-Arribas, M.; Bravo-San Pedro, J.M.; Fuentes, J.M.; González-Polo, R.A. Routine Western blot to check autophagic flux: Cautions and recommendations. Anal. Biochem. 2015, 477, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Rajan, R.; Karbowniczek, M.; Pugsley, H.R.; Sabnani, M.K.; Astrinidis, A.; La-Beck, N.M. Quantifying autophagosomes and autolysosomes in cells using imaging flow cytometry. Cytom. Part J. Int. Soc. Anal. Cytol. 2015, 87, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-W.; Lo, S.; Hsu, C.; Hsieh, C.-H.; Chang, Y.-F.; Hou, B.-S.; Kao, Y.-H.; Lin, C.-C.; Yu, M.-L.; Yuan, S.-S.; et al. T-cell autophagy deficiency increases mortality and suppresses immune responses after sepsis. PLoS ONE 2014, 9, e102066. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Liang, Y.; Murphy, S.F.; Huang, A.; Shen, H.; Kelly, D.F.; Sobrado, P.; Sheng, Z. A rapid and high content assay that measures cyto-ID-stained autophagic compartments and estimates autophagy flux with potential clinical applications. Autophagy 2015, 11, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Shvets, E.; Elazar, Z. Chapter 9 Flow cytometric analysis of autophagy in living mammalian cells. In Autophagy in Mammalian Systems, Part B; Klionsky, D.J., Ed.; Academic Press: Cambridge, MA, USA, 2009; Volume 452, pp. 131–141. [Google Scholar]
- Gump, J.M.; Thorburn, A. Sorting cells for basal and induced autophagic flux by quantitative ratiometric flow cytometry. Autophagy 2014, 10, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. Methods to monitor chaperone-mediated autophagy. Methods Enzymol. 2009, 452, 297–324. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.; Cuervo, A.M. Methods to study chaperone-mediated autophagy. Methods San Diego Calif. 2015, 75, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy. Methods Mol. Biol. Clifton NJ 2008, 445, 227–244. [Google Scholar] [CrossRef]
- Koga, H.; Martinez-Vicente, M.; Macian, F.; Verkhusha, V.V.; Cuervo, A.M. A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nat. Commun. 2011, 2, 386. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Dice, J.F.; Knecht, E. A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. J. Biol. Chem. 1997, 272, 5606–5615. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Autoimmune Diseases | Autophagy Abnormalities | Methods | Model Systems or Patient Samples Tested | Ref. |
|---|---|---|---|---|
| Systemic lupus erythematosus | Associated genes: ATG5, ATG7, PRDM1, DRAM1, IRGM | N/A | N/A | [25,26,27,28] |
| Accumulation of autophagosomes and increased MaA flux in T splenocytes | WB, EM | MRL/lpr and (NZB/W)F1 mice (thymus, spleen) | [29] | |
| Increased amount of autophagosomes in T cells | EM | Patients (blood) | ||
| Increased MAP1LC3 puncta, decreased SQSTM1/p62, and increased MaA flux in B cells | MIFC, FC | NZB/W F1 mice (spleen, bone marrow) | [31] | |
| Increased MAP1LC3 puncta and increased MaA flux in B cells | MIFC | Patients (blood) | ||
| Increased mRNA of Beclin-1, MAP1LC3 and SQSTM1 in PBMCs | qPCR | Patients (blood) | [39] | |
| Increased expression of ATG5, ATG12 and BECN1 in macrophages | pPCR | Induced lupus mice (spleen, kidneys) and patients (blood) | [40] | |
| Increased HSPA8 expression in B cells | WB, FC, qPCR | MRL/lpr mice (spleen) | [41] | |
| Increased LAMP-2A and CTSD expression in B cells; defective lysosomes in B cells | WB, FC | MRL/lpr mice (spleen) | [21] | |
| Increased MAP1LC3-II protein level | FC | MRL/lpr mice (spleen) | [42] | |
| Secondary Sjögren’s syndrome | Defective autophagy in salivary glands | WB, EM | MRL/lpr mice (salivary glands) | Li & Muller, unpublished |
| Crohn’s disease | Associated genes: ATG16L1, IRGM | N/A | N/A | [43,44,45,46] |
| Rheumatoid arthritis | Associated genes: ATG5, ATG7, BECN1 | N/A | N/A | [47,48] |
| Increased protein expression of ATG7 and BECN1 | WB, IHC | Patients (bones) | [47] | |
| Increased BECN1, ATG5, MAP1LC3 mRNA expression; increased MAP1LC3-II protein level | qPCR, IH, WB | Patients (synovial tissues) | [49] | |
| Decreased SQSTM1 protein expression | WB | Patients (synovial tissues) | [50] | |
| Polymyositis | Increased levels of MAP1LC3-II and decreased level of p70S6 kinase | WB | Patients (muscle) | [51] |
| Multiple sclerosis | Associated gene: ATG5 | N/A | N/A | [52] |
| Increased mRNA and protein level of ATG5 | qPCR, WB | EAE mice (blood) and patient (blood and brain) | [52] | |
| Decreased expression of ATG16L2 and ATG9A genes and increased expression of ULK1 gene | qPCR | Patient (blood) | [53] | |
| Type 1 diabetes | Decreased MAP1LC3 and ATG5/12 protein level | WB | Induced diabetic mice (heart) | [54] |
| Mice | Human | |||
|---|---|---|---|---|
| Spleen | Lymph Node | Salivary Gland | Blood | |
| EM | Yes | No | Yes | Yes |
| WB | Yes | Yes | Yes | Yes |
| FC | Yes | No | No | Yes |
| Antibodies | Company, References | Organs or Tissues Tested | |
|---|---|---|---|
| WB | MAP1LC3 | MBL International Corporation, M186-3 | Mice (spleen, LN, SG), human (blood) |
| SQSTM1 | Abcam, ab109012 | Mice (spleen, SG), human (blood) | |
| ATG5/ATG12 | Abcam, ab155589 | Mice (spleen, SG), human (blood) | |
| LAMP-2A | Abcam, ab18528, polyclonal; Abcam, ab125068 monoclonal | Mice (spleen, LN, SG), human (blood) | |
| HSPA8 | Abcam, ab51052 | Mice (spleen, SG), human (blood) | |
| HSP90 | ENZO, ADI-SPA-831 | Mice (spleen, SG), human (blood) | |
| Actin-β HRP | Abcam, ab49900 | Mice (spleen), human (blood) | |
| Actin-α HRP | Abcam, ab203696 | Mice (SG) | |
| FC | MAP1LC3-FITC (FlowCellectTM) | Millipore, FCCH10071 | Mice (spleen), human (blood) |
| SQSTM1 AlexaFluor 647 | MBL International Corporation, M162-A64 | Mice (spleen), human (blood) | |
| HSPA8-PE | Abcam, ab65170 | Mice (spleen), human (blood) | |
| HSP90-PE | Abcam, ab65171 | Mice (spleen), human (blood) | |
| Pros | Cons | |
|---|---|---|
| EM |
|
|
| WB |
|
|
| FC |
|
|
| MIFC |
|
|
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, F.; Li, B.; Schall, N.; Wilhelm, M.; Muller, S. Assessing Autophagy in Mouse Models and Patients with Systemic Autoimmune Diseases. Cells 2017, 6, 16. https://doi.org/10.3390/cells6030016
Wang F, Li B, Schall N, Wilhelm M, Muller S. Assessing Autophagy in Mouse Models and Patients with Systemic Autoimmune Diseases. Cells. 2017; 6(3):16. https://doi.org/10.3390/cells6030016
Chicago/Turabian StyleWang, Fengjuan, Baihui Li, Nicolas Schall, Maud Wilhelm, and Sylviane Muller. 2017. "Assessing Autophagy in Mouse Models and Patients with Systemic Autoimmune Diseases" Cells 6, no. 3: 16. https://doi.org/10.3390/cells6030016
APA StyleWang, F., Li, B., Schall, N., Wilhelm, M., & Muller, S. (2017). Assessing Autophagy in Mouse Models and Patients with Systemic Autoimmune Diseases. Cells, 6(3), 16. https://doi.org/10.3390/cells6030016