Assays to Monitor Autophagy in Saccharomyces cerevisiae


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Yeast as a Model Organism to Study Autophagy
3. End-Point Measurements
3.1. End-Point Measurements to Monitor Bulk Autophagy
3.1.1. The Pho8∆60 Assay (ALP Assay)
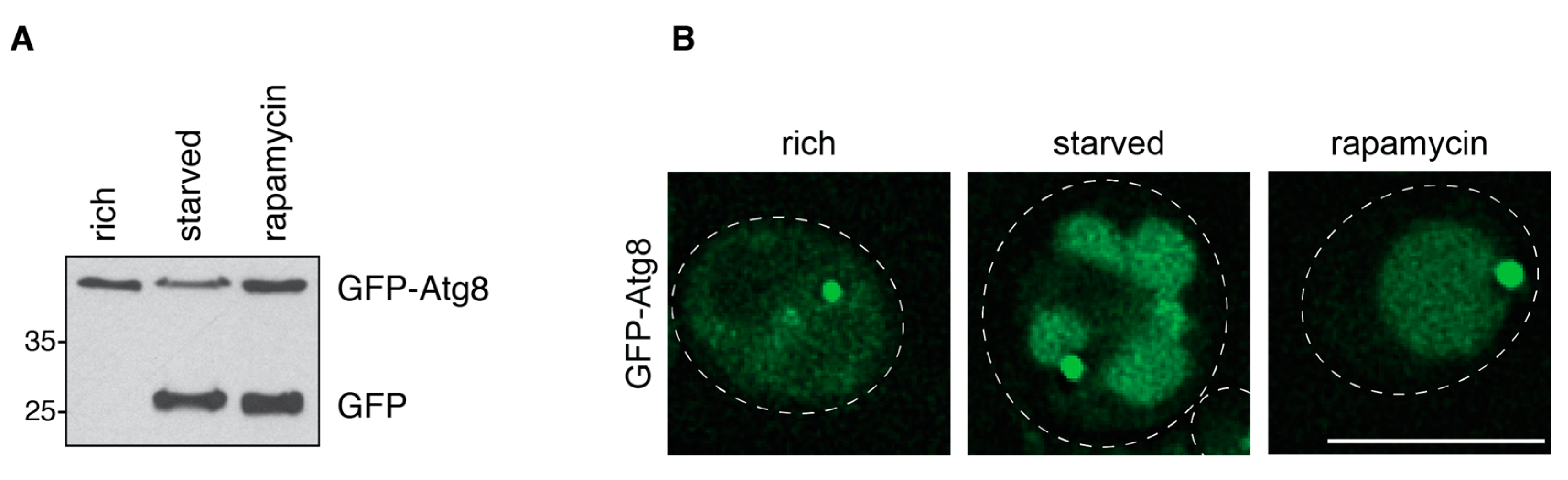
3.1.2. GFP-Atg8 Assays
3.1.3. Comparison of the Pho8∆60 and the GFP-Atg8 Assays
3.1.4. Cytoplasmic Proteins Tagged with GFP
3.1.5. Protein Degradation upon Starvation
3.2. End-Point Measurements to Monitor Selective Autophagy
3.2.1. The Cvt Pathway
3.2.2. Mitophagy
3.2.3. Pexophagy
3.2.4. Reticulophagy
3.2.5. Ribophagy
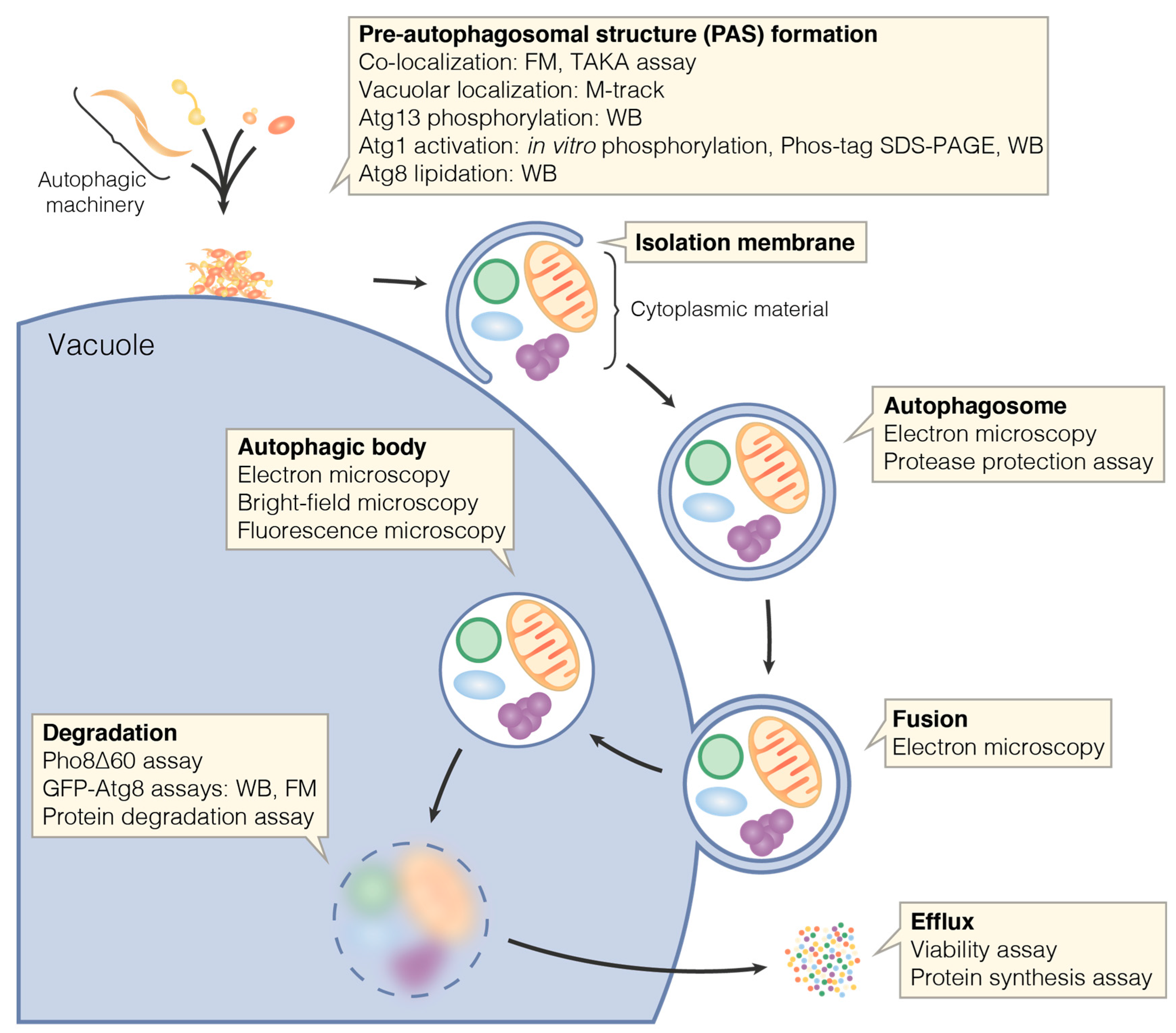
4. Monitoring Individual Steps during Autophagy
4.1. Autophagic Machinery Assembly
4.2. Protein Recruitment to Cellular Structures
4.3. Bypassing Protein Functions in Distinct Steps of Autophagy
4.4. Atg13 Phosphorylation
4.5. Atg1 Kinase Activation
4.6. Atg8 Lipidation
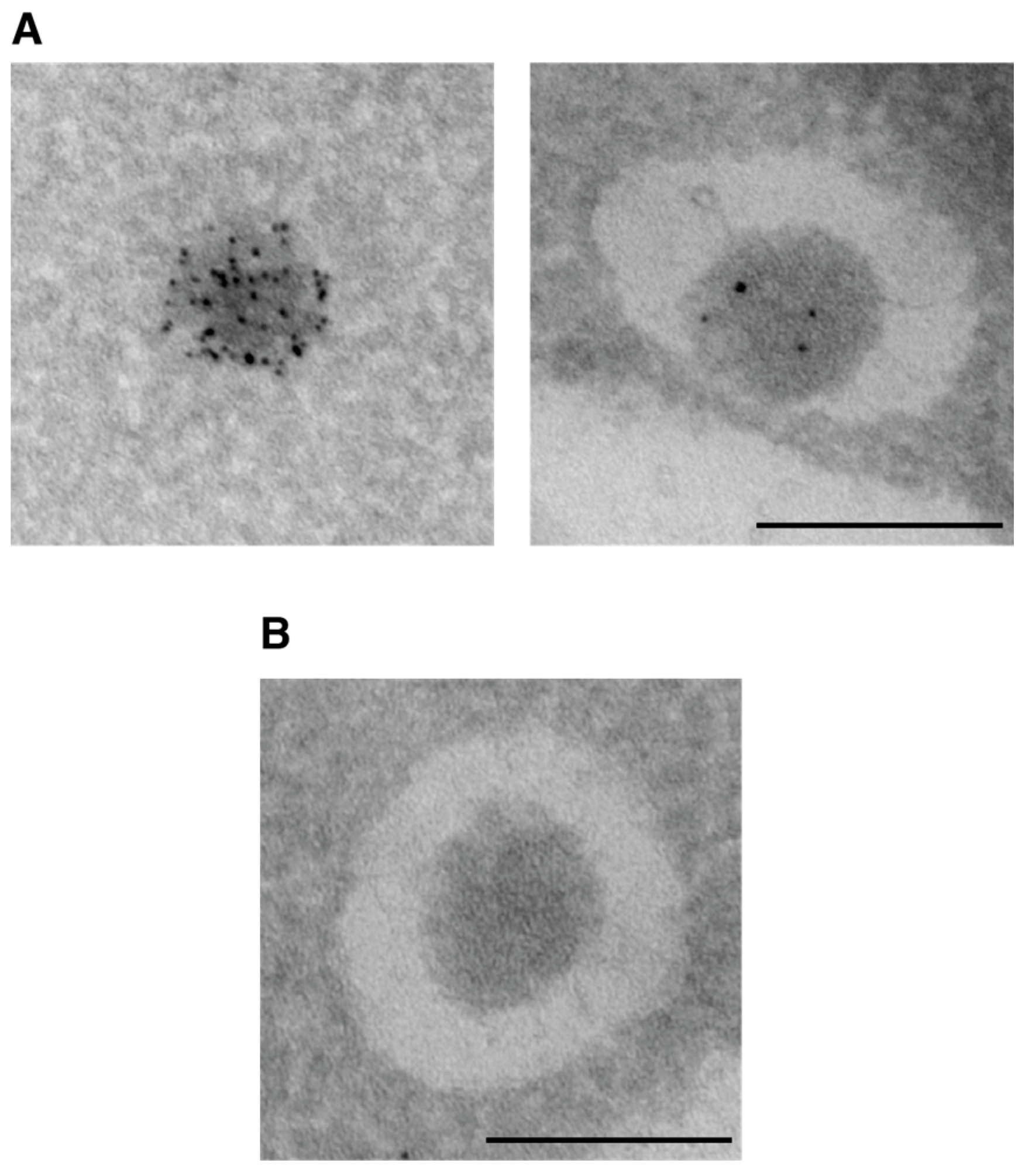
4.7. Autophagosome Formation
4.8. Vacuolar Delivery and Autophagic Body Degradation
4.9. Efflux of Catabolites from the Vacuole
4.10. Early Steps during the Cvt Pathway
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Kraft, C.; Martens, S. Mechanisms and regulation of autophagosome formation. Curr. Opin. Cell Biol. 2012, 24, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Kraft, C.; Reggiori, F.; Peter, M. Selective types of autophagy in yeast. Biochim. Biophys. Acta 2009, 1793, 1404–1412. [Google Scholar] [CrossRef] [PubMed]
- Baba, M.; Osumi, M.; Scott, S.V.; Klionsky, D.J.; Ohsumi, Y. Two distinct pathways for targeting proteins from the cytoplasm to the vacuole/lysosome. J. Cell Biol. 1997, 139, 1687–1695. [Google Scholar] [CrossRef] [PubMed]
- Sawa-Makarska, J.; Abert, C.; Romanov, J.; Zens, B.; Ibiricu, I.; Martens, S. Cargo binding to Atg19 unmasks additional Atg8 binding sites to mediate membrane-cargo apposition during selective autophagy. Nat. Cell Biol. 2014, 16, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Klionsky, D.J. Protein turnover via autophagy: Implications for metabolism. Annu. Rev. Nutr. 2007, 27, 19–40. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C. The Lysosome Concept. In Ciba Foundation Symposium—Lysosomes; de Reuck, A.V.S., Cameron, M.P., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 1963; pp. 1–35. [Google Scholar]
- De Duve, C.; Wattiaux, R. Functions of Lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, M.; Ohsumi, Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993, 333, 169–174. [Google Scholar] [CrossRef]
- Nakatogawa, H.; Suzuki, K.; Kamada, Y.; Ohsumi, Y. Dynamics and diversity in autophagy mechanisms: Lessons from yeast. Nat. Rev. Mol. Cell Biol. 2009, 10, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Thumm, M.; Egner, R.; Koch, B.; Schlumpberger, M.; Straub, M.; Veenhuis, M.; Wolf, D.H. Isolation of autophagocytosis mutants of Saccharomyces cerevisiae. FEBS Lett. 1994, 349, 275–280. [Google Scholar] [CrossRef]
- Harding, T.M.; Morano, K.A.; Scott, S.V.; Klionsky, D.J. Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. J. Cell Biol. 1995, 131, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Loewith, R.; Hall, M.N. Target of rapamycin (TOR) in nutrient signaling and growth control. Genetics 2011, 189, 1177–1201. [Google Scholar] [CrossRef] [PubMed]
- Noda, T.; Ohsumi, Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 1998, 273, 3963–3966. [Google Scholar] [CrossRef] [PubMed]
- Budovskaya, Y.V.; Stephan, J.S.; Reggiori, F.; Klionsky, D.J.; Herman, P.K. The Ras/cAMP-dependent protein kinase signaling pathway regulates an early step of the autophagy process in Saccharomyces cerevisiae. J. Biol. Chem. 2004, 279, 20663–20671. [Google Scholar] [CrossRef] [PubMed]
- Yorimitsu, T.; Zaman, S.; Broach, J.R.; Klionsky, D.J. Protein kinase A and Sch9 cooperatively regulate induction of autophagy in Saccharomyces cerevisiae. Mol. Biol. Cell 2007, 18, 4180–4189. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wilson, W.A.; Fujino, M.A.; Roach, P.J. Antagonistic controls of autophagy and glycogen accumulation by Snf1p, the yeast homolog of AMP-activated protein kinase, and the cyclin-dependent kinase Pho85p. Mol. Cell. Biol. 2001, 21, 5742–5752. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Kirisako, T.; Kamada, Y.; Mizushima, N.; Noda, T.; Ohsumi, Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 2001, 20, 5971–5981. [Google Scholar] [CrossRef] [PubMed]
- Papinski, D.; Schuschnig, M.; Reiter, W.; Wilhelm, L.; Barnes, C.A.; Maiolica, A.; Hansmann, I.; Pfaffenwimmer, T.; Kijanska, M.; Stoffel, I.; et al. Early steps in autophagy depend on direct phosphorylation of Atg9 by the Atg1 kinase. Mol. Cell 2014, 53, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Cueva, R.; Yaver, D.S. Aminopeptidase I of Saccharomyces cerevisiae is localized to the vacuole independent of the secretory pathway. J. Cell Biol. 1992, 119, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Hutchins, M.U.; Klionsky, D.J. Vacuolar localization of oligomeric alpha-mannosidase requires the cytoplasm to vacuole targeting and autophagy pathway components in Saccharomyces cerevisiae. J. Biol. Chem. 2001, 276, 20491–20498. [Google Scholar] [CrossRef] [PubMed]
- Yuga, M.; Gomi, K.; Klionsky, D.J.; Shintani, T. Aspartyl aminopeptidase is imported from the cytoplasm to the vacuole by selective autophagy in Saccharomyces cerevisiae. J. Biol. Chem. 2011, 286, 13704–13713. [Google Scholar] [CrossRef] [PubMed]
- Buschhorn, B.A.; Petzold, G.; Galova, M.; Dube, P.; Kraft, C.; Herzog, F.; Stark, H.; Peters, J.-M. Substrate binding on the APC/C occurs between the coactivator Cdh1 and the processivity factor Doc. Nat. Struct. Mol. Biol. 2011, 18, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Tong, A.H.; Evangelista, M.; Parsons, A.B.; Xu, H.; Bader, G.D.; Pagé, N.; Robinson, M.; Raghibizadeh, S.; Hogue, C.W.; Bussey, H.; et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 2001, 294, 2364–2368. [Google Scholar] [CrossRef] [PubMed]
- Longtine, M.S.; McKenzie, A.; Demarini, D.J.; Shah, N.G.; Wach, A.; Brachat, A.; Philippsen, P.; Pringle, J.R. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 1998, 14, 953–961. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.B.; Broach, J.R. Propagation and expression of cloned genes in yeast: 2-microns circle-based vectors. Meth. Enzymol. 1990, 185, 234–279. [Google Scholar] [CrossRef] [PubMed]
- Partow, S.; Siewers, V.; Bjørn, S.; Nielsen, J.; Maury, J. Characterization of different promoters for designing a new expression vector in Saccharomyces cerevisiae. Yeast 2010, 27, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Sikorski, R.S.; Hieter, P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 1989, 122, 19–27. [Google Scholar] [PubMed]
- Frazer, L.N.; O’Keefe, R.T. A new series of yeast shuttle vectors for the recovery and identification of multiple plasmids from Saccharomyces cerevisiae. Yeast 2007, 24, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Noda, T.; Matsuura, A.; Wada, Y.; Ohsumi, Y. Novel system for monitoring autophagy in the yeast Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 1995, 210, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J. Monitoring autophagy in yeast: The Pho8Delta60 assay. Methods Mol. Biol. 2007, 390, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Tuleva, B.; Vasileva-Tonkova, E.; Galabova, D. A specific alkaline phosphatase from Saccharomyces cerevisiae with protein phosphatase activity. FEMS Microbiol. Lett. 1998, 161, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Noda, T.; Klionsky, D.J. The quantitative Pho8Delta60 assay of nonspecific autophagy. Meth. Enzymol. 2008, 451, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Nakatogawa, H.; Ishii, J.; Asai, E.; Ohsumi, Y. Atg4 recycles inappropriately lipidated Atg8 to promote autophagosome biogenesis. Autophagy 2012, 8, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Araki, Y.; Kira, S.; Noda, T. Quantitative assay of macroautophagy using Pho8∆60 assay and GFP-cleavage assay in yeast. In Molecular Characterization of Autophagic Responses, Part B; Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2017; Volume 588, pp. 307–321. [Google Scholar]
- Kirisako, T.; Baba, M.; Ishihara, N.; Miyazawa, K.; Ohsumi, M.; Yoshimori, T.; Noda, T.; Ohsumi, Y. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J. Cell Biol. 1999, 147, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Kirisako, T.; Ichimura, Y.; Okada, H.; Kabeya, Y.; Mizushima, N.; Yoshimori, T.; Ohsumi, M.; Takao, T.; Noda, T.; Ohsumi, Y. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J. Cell Biol. 2000, 151, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.P.; Scott, S.V.; Kim, J.; Klionsky, D.J. The itinerary of a vesicle component, Aut7p/Cvt5p, terminates in the yeast vacuole via the autophagy/Cvt pathways. J. Biol. Chem. 2000, 275, 5845–5851. [Google Scholar] [CrossRef] [PubMed]
- Meiling-Wesse, K.; Barth, H.; Thumm, M. Ccz1p/Aut11p/Cvt16p is essential for autophagy and the cvt pathway. FEBS Lett. 2002, 526, 71–76. [Google Scholar] [CrossRef]
- Roberts, T.M.; Rudolf, F.; Meyer, A.; Pellaux, R.; Whitehead, E.; Panke, S.; Held, M. Identification and characterisation of a pH-stable GFP. Sci. Rep. 2016, 6, 28166. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Campbell, R.E.; Steinbach, P.A.; Giepmans, B.N.G.; Palmer, A.E.; Tsien, R.Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 2004, 22, 1567–1572. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Steinbach, P.A.; Tsien, R.Y. A guide to choosing fluorescent proteins. Nat. Meth. 2005, 2, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Vida, T.A.; Emr, S.D. A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. J. Cell Biol. 1995, 128, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Kraft, C.; Kijanska, M.; Kalie, E.; Siergiejuk, E.; Lee, S.S.; Semplicio, G.; Stoffel, I.; Brezovich, A.; Verma, M.; Hansmann, I.; et al. Binding of the Atg1/ULK1 kinase to the ubiquitin-like protein Atg8 regulates autophagy. EMBO J. 2012, 31, 3691–3703. [Google Scholar] [CrossRef] [PubMed]
- Welter, E.; Thumm, M.; Krick, R. Quantification of nonselective bulk autophagy in S. cerevisiae using Pgk1-GFP. Autophagy 2010, 6, 794–797. [Google Scholar] [CrossRef] [PubMed]
- Kraft, C.; Deplazes, A.; Sohrmann, M.; Peter, M. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat. Cell Biol. 2008, 10, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Gent, D.P.; Slaughter, J.C. Intracellular distribution of amino acids in an slp1 vacuole-deficient mutant of the yeast Saccharomyces cerevisiae. J. Appl. Microbiol. 1998, 84, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Rosado, C.; Mijaljica, D.; Hatzinisiriou, I.; Prescott, M.; Devenish, R.J. Rosella: A fluorescent pH-biosensor for reporting vacuolar turnover of cytosol and organelles in yeast. Autophagy 2008, 4, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Yorimitsu, T.; Klionsky, D.J. Atg11 links cargo to the vesicle-forming machinery in the cytoplasm to vacuole targeting pathway. Mol. Biol. Cell 2005, 16, 1593–1605. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.L.; Thorsness, P.E. Escape of mitochondrial DNA to the nucleus in yme1 yeast is mediated by vacuolar-dependent turnover of abnormal mitochondrial compartments. J. Cell Sci. 1998, 111, 2455. [Google Scholar] [PubMed]
- Scott, S.V.; Guan, J.; Hutchins, M.U.; Kim, J.; Klionsky, D.J. Cvt19 is a receptor for the cytoplasm-to-vacuole targeting pathway. Mol. Cell 2001, 7, 1131–1141. [Google Scholar] [CrossRef]
- Kabeya, Y.; Kamada, Y.; Baba, M.; Takikawa, H.; Sasaki, M.; Ohsumi, Y. Atg17 functions in cooperation with Atg1 and Atg13 in yeast autophagy. Mol. Biol. Cell 2005, 16, 2544–2553. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Kondo, C.; Morimoto, M.; Ohsumi, Y. Selective transport of alpha-mannosidase by autophagic pathways: Identification of a novel receptor, Atg34p. J. Biol. Chem. 2010, 285, 30019–30025. [Google Scholar] [CrossRef] [PubMed]
- Kanki, T.; Wang, K.; Cao, Y.; Baba, M.; Klionsky, D.J. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev. Cell 2009, 17, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Kondo-Okamoto, N.; Ohsumi, Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev. Cell 2009, 17, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Kiššová, I.; Deffieu, M.; Manon, S.; Camougrand, N. Uth1p is involved in the autophagic degradation of mitochondria. J. Biol. Chem. 2004, 279, 39068–39074. [Google Scholar] [CrossRef] [PubMed]
- Kanki, T.; Klionsky, D.J. Mitophagy in yeast occurs through a selective mechanism. J. Biol. Chem. 2008, 283, 32386–32393. [Google Scholar] [CrossRef] [PubMed]
- Eiyama, A.; Kondo-Okamoto, N.; Okamoto, K. Mitochondrial degradation during starvation is selective and temporally distinct from bulk autophagy in yeast. FEBS Lett. 2013, 587, 1787–1792. [Google Scholar] [CrossRef] [PubMed]
- Böckler, S.; Westermann, B. Mitochondrial ER contacts are crucial for mitophagy in yeast. Dev. Cell 2014, 28, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Tal, R.; Winter, G.; Ecker, N.; Klionsky, D.J.; Abeliovich, H. Aup1p, a yeast mitochondrial protein phosphatase homolog, is required for efficient stationary phase mitophagy and cell survival. J. Biol. Chem. 2007, 282, 5617–5624. [Google Scholar] [CrossRef] [PubMed]
- Nowikovsky, K.; Reipert, S.; Devenish, R.J.; Schweyen, R.J. Mdm38 protein depletion causes loss of mitochondrial K+/H+ exchange activity, osmotic swelling and mitophagy. Cell Death Differ. 2007, 14, 1647–1656. [Google Scholar] [CrossRef] [PubMed]
- Veenhuis, M.; Mateblowski, M.; Kunau, W.H.; Harder, W. Proliferation of microbodies in Saccharomyces cerevisiae. Yeast 1987, 3, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Sakai, Y.; Oku, M.; van der Klei, I.J.; Kiel, J.A.K.W. Pexophagy: Autophagic degradation of peroxisomes. Biochim. Biophys. Acta 2006, 1763, 1767–1775. [Google Scholar] [CrossRef] [PubMed]
- Motley, A.M.; Nuttall, J.M.; Hettema, E.H. Pex3-anchored Atg36 tags peroxisomes for degradation in Saccharomyces cerevisiae. EMBO J. 2012, 31, 2852–2868. [Google Scholar] [CrossRef] [PubMed]
- Chiang, H.L.; Schekman, R.; Hamamoto, S. Selective uptake of cytosolic, peroxisomal, and plasma membrane proteins into the yeast lysosome for degradation. J. Biol. Chem. 1996, 271, 9934–9941. [Google Scholar] [CrossRef] [PubMed]
- Hutchins, M.U.; Veenhuis, M.; Klionsky, D.J. Peroxisome degradation in Saccharomyces cerevisiae is dependent on machinery of macroautophagy and the Cvt pathway. J. Cell Sci. 1999, 112, 4079–4087. [Google Scholar] [PubMed]
- Torggler, R.; Papinski, D.; Brach, T.; Bas, L.; Schuschnig, M.; Pfaffenwimmer, T.; Rohringer, S.; Matzhold, T.; Schweida, D.; Brezovich, A.; et al. Two independent pathways within selective autophagy converge to activate Atg1 kinase at the vacuole. Mol. Cell 2016, 64, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Monastyrska, I.; Shintani, T.; Klionsky, D.J. The actin cytoskeleton is required for selective types of autophagy, but not nonspecific autophagy, in the yeast Saccharomyces cerevisiae. Mol. Biol. Cell 2005, 16, 5843–5856. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Stromhaug, P.E.; George, M.D.; Habibzadegah-Tari, P.; Bevan, A.; Dunn, W.A.; Klionsky, D.J. Cvt18/Gsa12 is required for cytoplasm-to-vacuole transport, pexophagy, and autophagy in Saccharomyces cerevisiae and Pichia pastoris. Mol. Biol. Cell 2001, 12, 3821–3838. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.A.; Cregg, J.M.; Kiel, J.A.K.W.; van der Klei, I.J.; Oku, M.; Sakai, Y.; Sibirny, A.A.; Stasyk, O.V.; Veenhuis, M. Pexophagy: The selective autophagy of peroxisomes. Autophagy 2005, 1, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Mochida, K.; Oikawa, Y.; Kimura, Y.; Kirisako, H.; Hirano, H.; Ohsumi, Y.; Nakatogawa, H. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 2015, 522, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, M.; Noda, T.; Baba, M.; Ohsumi, Y. Starvation triggers the delivery of the endoplasmic reticulum to the vacuole via autophagy in yeast. Traffic 2005, 6, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Bernales, S.; McDonald, K.L.; Walter, P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006, 4, e423. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Kubota, Y.; Sekito, T.; Ohsumi, Y. Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells 2007, 12, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Noda, T.; Kim, J.; Huang, W.P.; Baba, M.; Tokunaga, C.; Ohsumi, Y.; Klionsky, D.J. Apg9p/Cvt7p is an integral membrane protein required for transport vesicle formation in the Cvt and autophagy pathways. J. Cell Biol. 2000, 148, 465–480. [Google Scholar] [CrossRef] [PubMed]
- Tucker, K.A.; Reggiori, F.; Dunn, W.A.; Klionsky, D.J. Atg23 is essential for the cytoplasm to vacuole targeting pathway and efficient autophagy but not pexophagy. J. Biol. Chem. 2003, 278, 48445–48452. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Tucker, K.A.; Stromhaug, P.E.; Klionsky, D.J. The Atg1-Atg13 complex regulates Atg9 and Atg23 retrieval transport from the pre-autophagosomal structure. Dev. Cell 2004, 6, 79–90. [Google Scholar] [CrossRef]
- Yen, W.-L.; Legakis, J.E.; Nair, U.; Klionsky, D.J. Atg27 is required for autophagy-dependent cycling of Atg. Mol. Biol. Cell 2007, 18, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Kakuta, S.; Watanabe, T.M.; Kitamura, A.; Sekito, T.; Kondo-Kakuta, C.; Ichikawa, R.; Kinjo, M.; Ohsumi, Y. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J. Cell Biol. 2012, 198, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Sekito, T.; Kawamata, T.; Ichikawa, R.; Suzuki, K.; Ohsumi, Y. Atg17 recruits Atg9 to organize the pre-autophagosomal structure. Genes Cells 2009, 14, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Cheong, H.; Yorimitsu, T.; Reggiori, F.; Legakis, J.E.; Wang, C.-W.; Klionsky, D.J. Atg17 regulates the magnitude of the autophagic response. Mol. Biol. Cell 2005, 16, 3438–3453. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Baba, M.; Nair, U.; Klionsky, D.J. Quantitative analysis of autophagy-related protein stoichiometry by fluorescence microscopy. J. Cell Biol. 2008, 182, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Zuzuarregui, A.; Kupka, T.; Bhatt, B.; Dohnal, I.; Mudrak, I.; Friedmann, C.; Schüchner, S.; Frohner, I.E.; Ammerer, G.; Ogris, E. M-Track: Detecting short-lived protein-protein interactions in vivo. Nat. Methods 2012, 9, 594–596. [Google Scholar] [CrossRef] [PubMed]
- Brezovich, A.; Schuschnig, M.; Ammerer, G.; Kraft, C. An in vivo detection system for transient and low-abundant protein interactions and their kinetics in budding yeast. Yeast 2015, 32, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Kamada, Y.; Funakoshi, T.; Shintani, T.; Nagano, K.; Ohsumi, M.; Ohsumi, Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J. Cell Biol. 2000, 150, 1507–1513. [Google Scholar] [CrossRef] [PubMed]
- Kamada, Y.; Yoshino, K.-I.; Kondo, C.; Kawamata, T.; Oshiro, N.; Yonezawa, K.; Ohsumi, Y. Tor directly controls the Atg1 kinase complex to regulate autophagy. Mol. Cell. Biol. 2010, 30, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Stephan, J.S.; Yeh, Y.-Y.; Ramachandran, V.; Deminoff, S.J.; Herman, P.K. The Tor and PKA signaling pathways independently target the Atg1/Atg13 protein kinase complex to control autophagy. Proc. Natl. Acad. Sci. USA 2009, 106, 17049–17054. [Google Scholar] [CrossRef] [PubMed]
- Scott, S.V.; Nice, D.C.; Nau, J.J.; Weisman, L.S.; Kamada, Y.; Keizer-Gunnink, I.; Funakoshi, T.; Veenhuis, M.; Ohsumi, Y.; Klionsky, D.J. Apg13p and Vac8p are part of a complex of phosphoproteins that are required for cytoplasm to vacuole targeting. J. Biol. Chem. 2000, 275, 25840–25849. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, A.; Tsukada, M.; Wada, Y.; Ohsumi, Y. Apg1p, a novel protein kinase required for the autophagic process in Saccharomyces cerevisiae. Gene 1997, 192, 245–250. [Google Scholar] [CrossRef]
- Yeh, Y.-Y.; Wrasman, K.; Herman, P.K. Autophosphorylation within the Atg1 activation loop is required for both kinase activity and the induction of autophagy in Saccharomyces cerevisiae. Genetics 2010, 185, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Kijanska, M.; Dohnal, I.; Reiter, W.; Kaspar, S.; Stoffel, I.; Ammerer, G.; Kraft, C.; Peter, M. Activation of Atg1 kinase in autophagy by regulated phosphorylation. Autophagy 2010, 6, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, Y.; Suzuki, S.W.; Yamamoto, H.; Kondo-Kakuta, C.; Kimura, Y.; Hirano, H.; Akada, R.; Inagaki, F.; Ohsumi, Y.; Noda, N.N. Structural basis of starvation-induced assembly of the autophagy initiation complex. Nat. Struct. Mol. Biol. 2014, 21, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Pfaffenwimmer, T.; Reiter, W.; Brach, T.; Nogellova, V.; Papinski, D.; Schuschnig, M.; Abert, C.; Ammerer, G.; Martens, S.; Kraft, C. Hrr25 kinase promotes selective autophagy by phosphorylating the cargo receptor Atg. EMBO Rep. 2014, 15, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Kamber, R.A.; Shoemaker, C.J.; Denic, V. Receptor-bound targets of selective autophagy use a scaffold protein to activate the Atg1 kinase. Mol. Cell 2015, 59, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, E.; Kinoshita-Kikuta, E.; Takiyama, K.; Koike, T. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell Proteom. 2006, 5, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Fujioka, Y.; Suzuki, S.W.; Noshiro, D.; Suzuki, H.; Kondo-Kakuta, C.; Kimura, Y.; Hirano, H.; Ando, T.; Noda, N.N.; et al. The intrinsically disordered protein Atg13 mediates supramolecular assembly of autophagy initiation complexes. Dev. Cell 2016, 38, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Baba, M.; Takeshige, K.; Baba, N.; Ohsumi, Y. Ultrastructural analysis of the autophagic process in yeast: Detection of autophagosomes and their characterization. J. Cell Biol. 1994, 124, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Dalton, V.M.; Eggerton, K.P.; Scott, S.V.; Klionsky, D.J. Apg7p/Cvt2p is required for the cytoplasm-to-vacuole targeting, macroautophagy, and peroxisome degradation pathways. Mol. Biol. Cell 1999, 10, 1337–1351. [Google Scholar] [CrossRef] [PubMed]
- Darsow, T.; Rieder, S.E.; Emr, S.D. A multispecificity syntaxin homologue, Vam3p, essential for autophagic and biosynthetic protein transport to the vacuole. J. Cell Biol. 1997, 138, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.-L.; Reggiori, F.; Baba, M.; Kovács, A.L.; Seglen, P.O. Seeing is believing: The impact of electron microscopy on autophagy research. Autophagy 2011, 7, 935–956. [Google Scholar] [CrossRef] [PubMed]
- Baba, M.; Osumi, M.; Ohsumi, Y. Analysis of the membrane structures involved in autophagy in yeast by freeze-replica method. Cell Struct. Funct. 1995, 20, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Scott, S.V.; Baba, M.; Ohsumi, Y.; Klionsky, D.J. Aminopeptidase I is targeted to the vacuole by a nonclassical vesicular mechanism. J. Cell Biol. 1997, 138, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Nair, U.; Thumm, M.; Klionsky, D.J.; Krick, R. GFP-Atg8 protease protection as a tool to monitor autophagosome biogenesis. Autophagy 2011, 7, 1546–1550. [Google Scholar] [CrossRef] [PubMed]
- Abeliovich, H.; Dunn, W.A.; Kim, J.; Klionsky, D.J. Dissection of autophagosome biogenesis into distinct nucleation and expansion steps. J. Cell Biol. 2000, 151, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Nair, U.; Klionsky, D.J. Atg8 controls phagophore expansion during autophagosome formation. Mol. Biol. Cell 2008, 19, 3290–3298. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; He, D.; Backues, S.K.; Freeberg, M.A.; Liu, X.; Kim, J.K.; Klionsky, D.J. Transcriptional regulation by Pho23 modulates the frequency of autophagosome formation. Curr. Biol. 2014, 24, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- Epple, U.D.; Suriapranata, I.; Eskelinen, E.L.; Thumm, M. Aut5/Cvt17p, a putative lipase essential for disintegration of autophagic bodies inside the vacuole. J. Bacteriol. 2001, 183, 5942–5955. [Google Scholar] [CrossRef] [PubMed]
- Teter, S.A.; Eggerton, K.P.; Scott, S.V.; Kim, J.; Fischer, A.M.; Klionsky, D.J. Degradation of lipid vesicles in the yeast vacuole requires function of Cvt17, a putative lipase. J. Biol. Chem. 2001, 276, 2083–2087. [Google Scholar] [CrossRef] [PubMed]
- Onodera, J.; Ohsumi, Y. Autophagy is required for maintenance of amino acid levels and protein synthesis under nitrogen starvation. J. Biol. Chem. 2005, 280, 31582–31586. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Huang, J.; Geng, J.; Nair, U.; Klionsky, D.J. Atg22 recycles amino acids to link the degradative and recycling functions of autophagy. Mol. Biol. Cell 2006, 17, 5094–5104. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Akioka, M.; Kondo-Kakuta, C.; Yamamoto, H.; Ohsumi, Y. Fine mapping of autophagy-related proteins during autophagosome formation in Saccharomyces cerevisiae. J. Cell Sci. 2013, 126, 2534–2544. [Google Scholar] [CrossRef] [PubMed]








© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torggler, R.; Papinski, D.; Kraft, C. Assays to Monitor Autophagy in Saccharomyces cerevisiae. Cells 2017, 6, 23. https://doi.org/10.3390/cells6030023
Torggler R, Papinski D, Kraft C. Assays to Monitor Autophagy in Saccharomyces cerevisiae. Cells. 2017; 6(3):23. https://doi.org/10.3390/cells6030023
Chicago/Turabian StyleTorggler, Raffaela, Daniel Papinski, and Claudine Kraft. 2017. "Assays to Monitor Autophagy in Saccharomyces cerevisiae" Cells 6, no. 3: 23. https://doi.org/10.3390/cells6030023
APA StyleTorggler, R., Papinski, D., & Kraft, C. (2017). Assays to Monitor Autophagy in Saccharomyces cerevisiae. Cells, 6(3), 23. https://doi.org/10.3390/cells6030023