Antiproliferative Activity and Molecular Docking of Novel Double-Modified Colchicine Derivatives

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. General
2.2. Spectroscopic Measurements
2.3. Synthesis
2.3.1. Synthesis of 2
2.3.2. Synthesis of 3
2.3.3. Synthesis of 5
2.3.4. Synthesis of 7
2.4. Antiproliferative Activity of Colchicine and Its Derivatives
2.4.1. The Antiproliferative Assays In Vitro
2.4.2. SRB
2.5. Molecular Docking Simulations
3. Results
3.1. Chemistry
3.2. In Vitro Determination of Drug-Induced Inhibition of Human Cancer Cell Line Growth
3.3. Molecular Docking: in Silico Determination of Drug-Induced Inhibition of βI Tubulin
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hyams, J.; Stebbings, H. The mechanism of microtubule associated cytoplasmic transport. Cell Tissue Res. 1979, 196, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Dustin, P. Microtubules; Springer: Berlin, Germany, 1984; ISBN 978-3-642-69652-7. [Google Scholar]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Dutcher, S.K. The tubulin fraternity: alpha to eta. Curr. Opin. Cell Biol. 2001, 13, 49–54. [Google Scholar] [CrossRef]
- Vindya, N.G.; Sharma, N.; Yadav, M.; Ethiraj, K.R. Tubulins—The target for anticancer therapy. Curr. Top. Med. Chem. 2015, 15, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Seligmann, J.; Twelves, C. Tubulin: An example of targeted chemotherapy. Future Med. Chem. 2013, 5, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Katsetos, C.D.; Dráber, P. Tubulins as therapeutic targets in cancer: from bench to bedside. Curr. Pharm. Des. 2012, 18, 2778–2792. [Google Scholar] [CrossRef] [PubMed]
- Huzil, J.T.; Ludueña, R.F.; Tuszynski, J. Comparative modelling of human β tubulin isotypes and implications for drug binding. Nanotechnology 2006, 17, S90–S100. [Google Scholar] [CrossRef] [PubMed]
- Ravanbakhsh, S.; Gajewski, M.; Greiner, R.; Tuszynski, J.A. Determination of the optimal tubulin isotype target as a method for the development of individualized cancer chemotherapy. Theor. Biol. Med. Model. 2013, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Kumar, R.; Skvortsova, I.; Kumar, V. Mechanisms of tubulin binding ligands to target cancer cells: Updates on their therapeutic potential and clinical Trials. Curr. Cancer Drug Targets 2017, 17, 357–375. [Google Scholar] [CrossRef] [PubMed]
- Avendaño, C.; Menéndez, J.C. Medicinal Chemistry of Anticancer Drugs; Elsevier: Amsterdam, The Netherlands, 2008; ISBN 9780444528247. [Google Scholar]
- Slobodnick, A.; Shah, B.; Pillinger, M.H.; Krasnokutsky, S. Colchicine: Old and new. Am. J. Med. 2015, 128, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Nerlekar, N.; Beale, A.; Harper, R.W. Colchicine—A short history of an ancient drug. Med. J. Aust. 2014, 201, 687–688. [Google Scholar] [CrossRef] [PubMed]
- Grattagliano, I.; Bonfrate, L.; Ruggiero, V.; Scaccianoce, G.; Palasciano, G.; Portincasa, P. Novel therapeutics for the treatment of familial mediterranean fever: From colchicine to biologics. Clin. Pharmacol. Ther. 2013, 95, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Cocco, G.; Chu, D.C.C.; Pandolfi, S. Colchicine in clinical medicine. A guide for internists. Eur. J. Intern. Med. 2010, 21, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.P.H. Oral colchicine (Colcrys®). Drugs 2010, 70, 1603–1613. [Google Scholar] [CrossRef] [PubMed]
- Marangon, J.; Christodoulou, M.S.; Casagrande, F.V.M.; Tiana, G.; Dalla Via, L.; Aliverti, A.; Passarella, D.; Cappelletti, G.; Ricagno, S. Tools for the rational design of bivalent microtubule-targeting drugs. Biochem. Biophys. Res. Commun. 2016, 479, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Huczyński, A.; Rutkowski, J.; Popiel, K.; Maj, E.; Wietrzyk, J.; Stefańska, J.; Majcher, U.; Bartl, F. Synthesis, antiproliferative and antibacterial evaluation of C-ring modified colchicine analogues. Eur. J. Med. Chem. 2014, 90, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Huczyński, A.; Majcher, U.; Maj, E.; Wietrzyk, J.; Janczak, J.; Moshari, M.; Tuszynski, J.A.; Bartl, F. Synthesis, antiproliferative activity and molecular docking of Colchicine derivatives. Bioorg. Chem. 2016, 64, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kong, Y.; Zhang, J.; Su, M.; Zhou, Y.; Zang, Y.; Li, J.; Chen, Y.; Fang, Y.; Zhang, X.; et al. Design, synthesis and biological evaluation of colchicine derivatives as novel tubulin and histone deacetylase dual inhibitors. Eur. J. Med. Chem. 2015, 95, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Valiulin, R.A.; Pokorski, J.K.; Chang, V.; Chen, J.S. Bio-inspired synthesis and biological evaluation of a colchicine-related compound library. Bioorg. Med. Chem. Lett. 2012, 22, 3776–3780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, D.-J.; Yoon, E.-Y.; Lee, G.-B.; Kim, S.-O.; Kim, W.-J.; Kim, Y.-M.; Jung, J.-W.; An, H.; Suh, Y.-G. Design, synthesis and identification of novel colchicine-derived immunosuppressant. Bioorg. Med. Chem. Lett. 2009, 19, 4416–4420. [Google Scholar] [CrossRef] [PubMed]
- Marzo-Mas, A.; Barbier, P.; Breuzard, G.; Allegro, D.; Falomir, E.; Murga, J.; Carda, M.; Peyrot, V.; Marco, J.A. Interactions of long-chain homologues of colchicine with tubulin. Eur. J. Med. Chem. 2017, 126, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.; Goping, I.S.; Rieger, A.; Mane, J.Y.; Huzil, T.; Banerjee, A.; Luduena, R.; Hassani, B.; Winter, P.; Tuszynski, J.A. Novel Colchicine derivatives and their anti-cancer activity. Curr. Top. Med. Chem. 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Sharma, P.; Gupta, V.P.; Khullar, M.; Singh, S.; Dogra, N.; Kumar, V. Synthesis and biological evaluation of pyrimidine bridged combretastatin derivatives as potential anticancer agents and mechanistic studies. Bioorg. Chem. 2018, 78, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sharma, P.R.; Mondhe, D.M. Potential anticancer role of colchicine-based derivatives. Anticancer Drugs 2017, 28, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Shchegravina, E.S.; Maleev, A.A.; Ignatov, S.K.; Gracheva, I.A.; Stein, A.; Schmalz, H.G.; Gavryushin, A.E.; Zubareva, A.A.; Svirshchevskaya, E.V.; Fedorov, A.Y. Synthesis and biological evaluation of novel non-racemic indole-containing allocolchicinoids. Eur. J. Med. Chem. 2017, 141, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Bartusik, D.; Tomanek, B.; Lattová, E.; Perreault, H.; Tuszynski, J.; Fallone, G. Derivatives of thiocolchicine and its applications to CEM cells treatment using 19F Magnetic Resonance ex vivo. Bioorg. Chem. 2010, 38, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raspaglio, G.; Ferlini, C.; Mozzetti, S.; Prislei, S.; Gallo, D.; Das, N.; Scambia, G. Thiocolchicine dimers: A novel class of topoisomerase-I inhibitors. Biochem. Pharmacol. 2005, 69, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Kozaka, T.; Nakagawa-Goto, K.; Shi, Q.; Lai, C.Y.; Hamel, E.; Bastow, K.F.; Brossi, A.; Lee, K.H. Antitumor agents 273. Design and synthesis of N-alkyl-thiocolchicinoids as potential antitumor agents. Bioorg. Med. Chem. Lett. 2010, 20, 4091–4094. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa-Goto, K.; Chen, C.X.; Hamel, E.; Wu, C.-C.; Bastow, K.F.; Brossi, A.; Lee, K.-H. Antitumor agents. Part 236: Synthesis of water-soluble colchicine derivatives. Bioorg. Med. Chem. Lett. 2005, 15, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Yasobu, N.; Kitajima, M.; Kogure, N.; Shishido, Y.; Matsuzaki, T.; Nagaoka, M.; Takayama, H. Design, synthesis, and antitumor activity of 4-halocolchicines and their pro-drugs activated by cathepsin B. ACS Med. Chem. Lett. 2011, 2, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Shchegravina, E.S.; Knyazev, D.I.; Beletskaya, I.P.; Svirshchevskaya, E.V.; Schmalz, H.G.; Fedorov, A.Y. Synthesis of nonracemic pyrrolo-allocolchicinoids exhibiting potent cytotoxic activity. Eur. J. Org. Chem. 2016, 2016, 5620–5623. [Google Scholar] [CrossRef]
- Shi, Q.; Verdier-Pinard, P.; Brossi, A.; Hamel, E.; Lee, K.H. Antitumor agents-CLXXV. Anti-tubulin action of (+)-thiocolchicine prepared by partial synthesis. Bioorg. Med. Chem. 1997, 5, 2277–2282. [Google Scholar] [CrossRef]
- Banerjee, A.; Kasmala, L.T.; Hamel, E.; Sun, L.; Lee, K.-H. Interaction of novel thiocolchicine analogs with the tubulin isoforms from bovine brain. Biochem. Biophys. Res. Commun. 1999, 337, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, P.B.; Bodiwala, K.B.; Marolia, B.P. Oxidative degradation kinetic study of thiocolchicoside using stability indicating high performance thin layer chromatographic method. Pharm. Methods 2014, 5, 1–10. [Google Scholar] [CrossRef]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Nevozhay, D. Cheburator software for automatically calculating drug inhibitory concentrations from in vitro screening assays. PLoS ONE 2014, 9, e106186. [Google Scholar] [CrossRef] [PubMed]
- Majcher, U.; Urbaniak, A.; Maj, E.; Moshari, M.; Delgado, M.; Wietrzyk, J.; Bartl, F.; Chambers, T.C.; Tuszynski, J.A.; Huczyński, A. Synthesis, antiproliferative activity and molecular docking of thiocolchicine urethanes. Bioorg. Chem. 2018, 81, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Devalapally, H.; Chakilam, A.; Amiji, M.M. Role of nanotechnology in pharmaceutical product development. J. Pharm. Sci. 2007, 96, 2547–2565. [Google Scholar] [CrossRef] [PubMed]
- Ludueña, R.F. Multiple forms of tubulin: Different gene products and covalent modifications. Int. Rev. Cytol. 1997, 178, 207–275. [Google Scholar]
- Leandro-García, L.J.; Leskelä, S.; Landa, I.; Montero-Conde, C.; López-Jiménez, E.; Letón, R.; Cascón, A.; Robledo, M.; Rodríguez-Antona, C. Tumoral and tissue-specific expression of the major human β-tubulin isotypes. Cytoskeleton 2010, 67, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Mozzetti, S.; Ferlini, C.; Concolino, P.; Filippetti, F.; Raspaglio, G.; Prislei, S.; Gallo, D.; Martinelli, E.; Ranelletti, F.O.; Ferrandina, G.; et al. Class III β-tubulin overexpression is a prominent mechanism of paclitaxel resistance in ovarian cancer patients. Clin. Cancer Res. 2005, 11, 298–305. [Google Scholar] [PubMed]
- Sève, P.; Dumontet, C. Is class III β-tubulin a predictive factor in patients receiving tubulin-binding agents? Lancet Oncol. 2008, 9, 168–175. [Google Scholar] [CrossRef]
- Hiser, L.; Aggarwal, A.; Young, R.; Frankfurter, A.; Spano, A.; Correia, J.J.; Lobert, S. Comparison of β-tubulin mRNA and protein levels in 12 human cancer cell lines. Cell Motil. Cytoskeleton 2006, 63, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.; Martinez, S.; Nelson, D.; Middleton, K. A tubulin polymerization microassay used to compare ligand efficacy. In Methods in Cell Biology; Elsevier: Amsterdam, The Netherlands, 2010; Volume 95(C), pp. 331–351. ISBN 9780123748157. [Google Scholar]
- Tseng, C.Y.; Mane, J.Y.; Winter, P.; Johnson, L.; Huzil, T.; Izbicka, E.; Luduena, R.F.; Tuszynski, J.A. Quantitative analysis of the effect of tubulin isotype expression on sensitivity of cancer cell lines to a set of novel colchicine derivatives. Mol. Cancer 2010, 9, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.H.; Yu, A.-M. ABC transporters in multidrug resistance and pharmacokinetics, and strategies for drug development. Curr. Pharm. Des. 2014, 20, 793–807. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Compound | A549 | MCF-7 | LoVo | LoVo/DX | BALB/3T3 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (μM) | SI | IC50 (μM) | SI | IC50 (μM) | SI | IC50 (μM) | SI | RI | IC50 (μM) | |
| 1 | 0.149 ± 0.009 | 1.4 | 0.128 ± 0.135 | 1.6 | 0.108 ± 0.025 | 1.9 | 2.65 ± 0.96 | 0.1 | 24.5 | 0.208 ± 0.042 |
| 2 | 0.011 ± 0.001 | 10.1 | 0.010 ± 0.002 | 11.9 | 0.021 ± 0.006 | 5.5 | 0.398 ± 0.075 | 0.3 | 19.0 | 0.114 ± 0.072 |
| 3 | 0.046 ± 0.035 | 3.0 | 0.023 ± 0.005 | 6.0 | 0.069 ± 0.012 | 2.0 | 0.784 ± 0.28 | 0.2 | 11.4 | 0.138 ± 0.069 |
| 4 | 0.022 ± 0.002 | 1.0 | 0.022 ± 0.002 | 1.0 | 0.022 ± 0.002 | 1.0 | 0.111 ± 0.044 | 0.2 | 5.1 | 0.022 ± 0.002 |
| 5 | 0.105 ± 0.008 | 1.4 | 0.027 ± 0.008 | 5.3 | 0.084 ± 0.021 | 1.7 | 1.55 ± 0.17 | 0.1 | 18.5 | 0.142 ± 0.073 |
| 6 | 0.010 ± 0.0001 | 10.3 | 0.015 ± 0.002 | 6.9 | 0.014 ± 0.004 | 7.4 | 0.135 ± 0.012 | 0.8 | 9.6 | 0.103 ± 0.089 |
| 7 | 0.094 ± 0.006 | 1.4 | 0.098 ± 0.029 | 1.4 | 0.010 ± 0.002 | 13.5 | 2.78 ± 0.45 | 0.1 | 278.0 | 0.135 ± 0.056 |
| 8 | 0.011 ± 0.002 | 10.5 | 0.017 ± 0.006 | 6.8 | 0.007 ± 0.002 | 16.4 | 0.642 ± 0.084 | 0.2 | 91.7 | 0.115 ± 0.044 |
| Doxorubicin | 0.258 ± 0.044 | 0.6 | 0.386 ± 0.118 | 0.4 | 0.092 ± 0.018 | 1.8 | 4.75 ± 0.99 | <0.1 | 51.6 | 0.166 ± 0.074 |
| Cisplatin | 6.367 ± 1.413 | 0.6 | 10.70 ± 0.753 | 0.4 | 4.37 ± 0.73 | 0.9 | 5.70 ± 0.63 | 0.7 | 1.3 | 3.90 ± 1.50 |
| Structure | Interactions | Binding Energy (Kcal/mol) | MlogP | Active Residues |
|---|---|---|---|---|
1 |  | −8.09 | 1.37 | Asn258 |
2 |  | −8.13 | 1.56 | Met259 Asn258 Lys352 |
3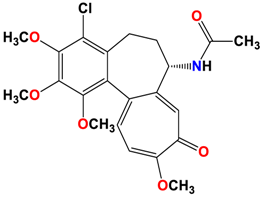 |  | −8.33 | 1.58 | Met259 Lys352 Val315 |
4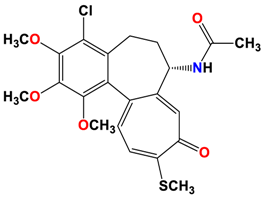 | 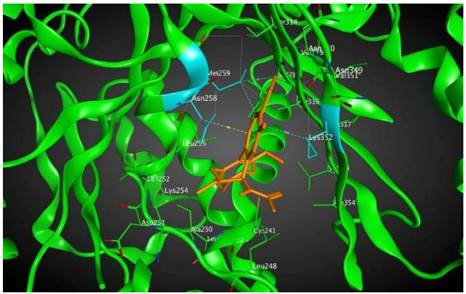 | −8.57 | 2.34 | Met259 Asn258 Lys352 |
5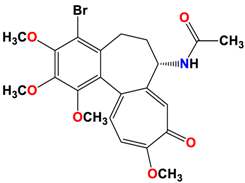 | 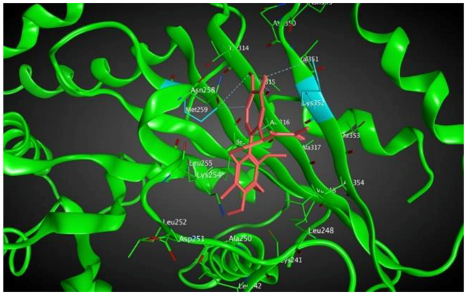 | −8.40 | 1.93 | Met259 Val315 Lys352 |
6 |  | −8.60 | 2.69 | Met259 Asn258 Lys352 |
7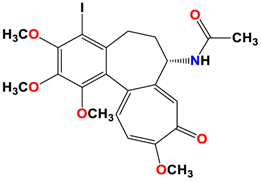 | 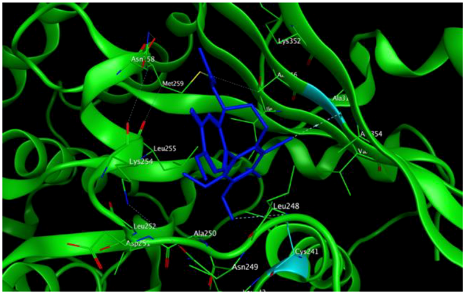 | −7.53 | 1.50 | Ala317 Cys241 |
8 | 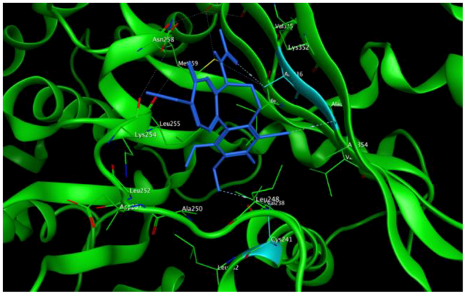 | −7.33 | 2.00 | Ala316 Ala317 Cys241 |
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
|---|---|---|---|---|---|---|---|---|
| βI | −8.090 | −8.130 | −8.330 | −8.570 | −8.400 | −8.600 | −7.530 | −7.330 |
| βIIa | −7.420 | −7.190 | −7.890 | −7.640 | −8.000 | −7.460 | −8.300 | −8.200 |
| βIIb | −7.050 | −6.680 | −6.800 | −6.900 | −6.890 | −7.050 | −6.430 | −7.040 |
| βIII | −7.490 | −7.150 | −7.470 | −7.620 | −7.850 | −7.780 | −8.200 | −7.710 |
| βIVa | −7.610 | −7.300 | −7.100 | −7.300 | −7.210 | −7.430 | −6.970 | −7.010 |
| βIVb | −7.260 | −7.210 | −7.420 | −7.610 | −7.370 | −7.620 | −7.180 | −6.340 |
| βV | −7.480 | −7.180 | −7.320 | −7.260 | −7.250 | −7.340 | −6.500 | −7.190 |
| βVI | −7.730 | −7.050 | −7.270 | −7.710 | −7.590 | −7.870 | −8.320 | −8.300 |
 | ||||||||
| Tubulin Isotype | Interactions | Binding Energy (Kcal/mol) | MlogP | Active Residues |
|---|---|---|---|---|
| βIIa | 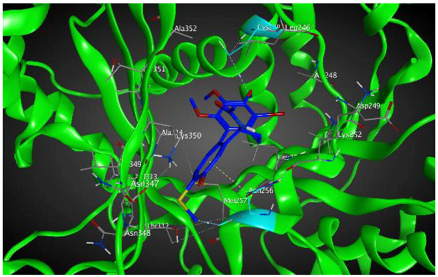 | −8.20 | 1.99 | Asn256 Cys239 |
| βVI | 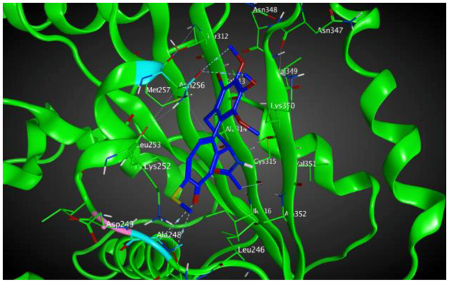 | −8.30 | 1.99 | Asn256 Asn247 Ala248 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majcher, U.; Klejborowska, G.; Moshari, M.; Maj, E.; Wietrzyk, J.; Bartl, F.; Tuszynski, J.A.; Huczyński, A. Antiproliferative Activity and Molecular Docking of Novel Double-Modified Colchicine Derivatives. Cells 2018, 7, 192. https://doi.org/10.3390/cells7110192
Majcher U, Klejborowska G, Moshari M, Maj E, Wietrzyk J, Bartl F, Tuszynski JA, Huczyński A. Antiproliferative Activity and Molecular Docking of Novel Double-Modified Colchicine Derivatives. Cells. 2018; 7(11):192. https://doi.org/10.3390/cells7110192
Chicago/Turabian StyleMajcher, Urszula, Greta Klejborowska, Mahshad Moshari, Ewa Maj, Joanna Wietrzyk, Franz Bartl, Jack A. Tuszynski, and Adam Huczyński. 2018. "Antiproliferative Activity and Molecular Docking of Novel Double-Modified Colchicine Derivatives" Cells 7, no. 11: 192. https://doi.org/10.3390/cells7110192
APA StyleMajcher, U., Klejborowska, G., Moshari, M., Maj, E., Wietrzyk, J., Bartl, F., Tuszynski, J. A., & Huczyński, A. (2018). Antiproliferative Activity and Molecular Docking of Novel Double-Modified Colchicine Derivatives. Cells, 7(11), 192. https://doi.org/10.3390/cells7110192