MicroRNA-Regulated Rickettsial Invasion into Host Endothelium via Fibroblast Growth Factor 2 and Its Receptor FGFR1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Endothelial Cell Culture
2.2. Cell Infection and Transfection
2.3. RNA Preparation
2.4. Quantitative Real-Time PCR
2.5. In Vivo Model of Infection
2.6. Statistical Analysis
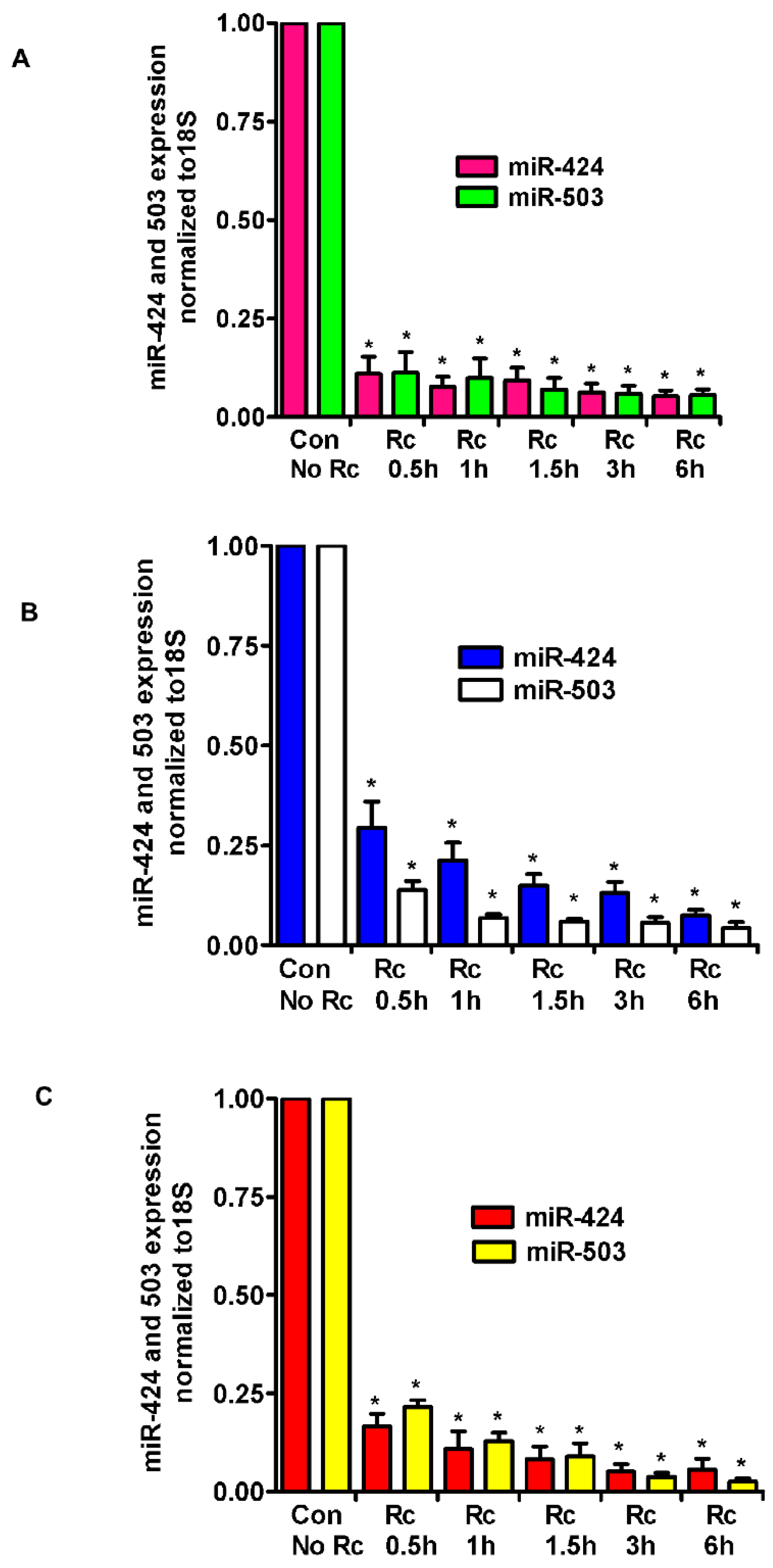
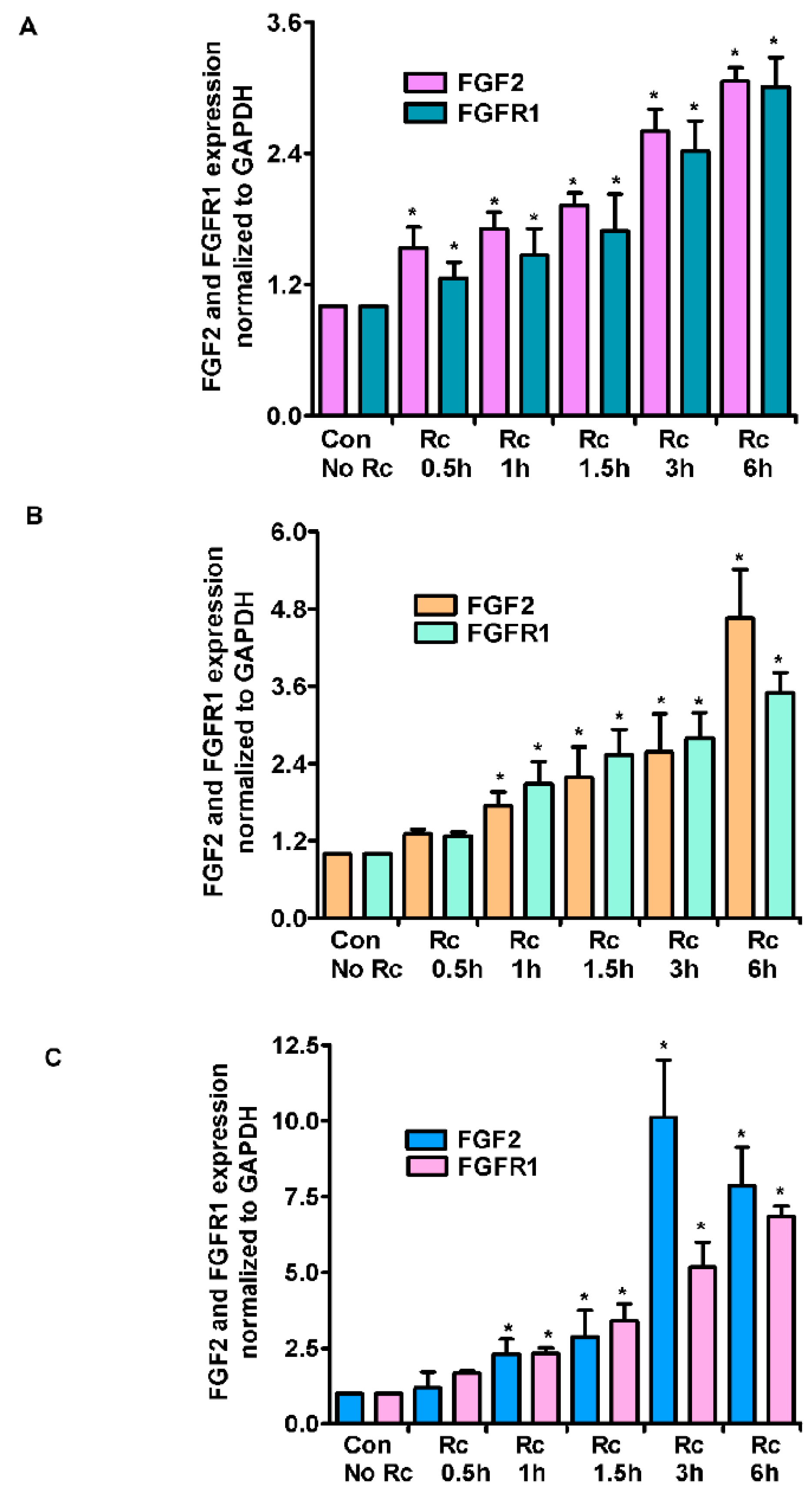
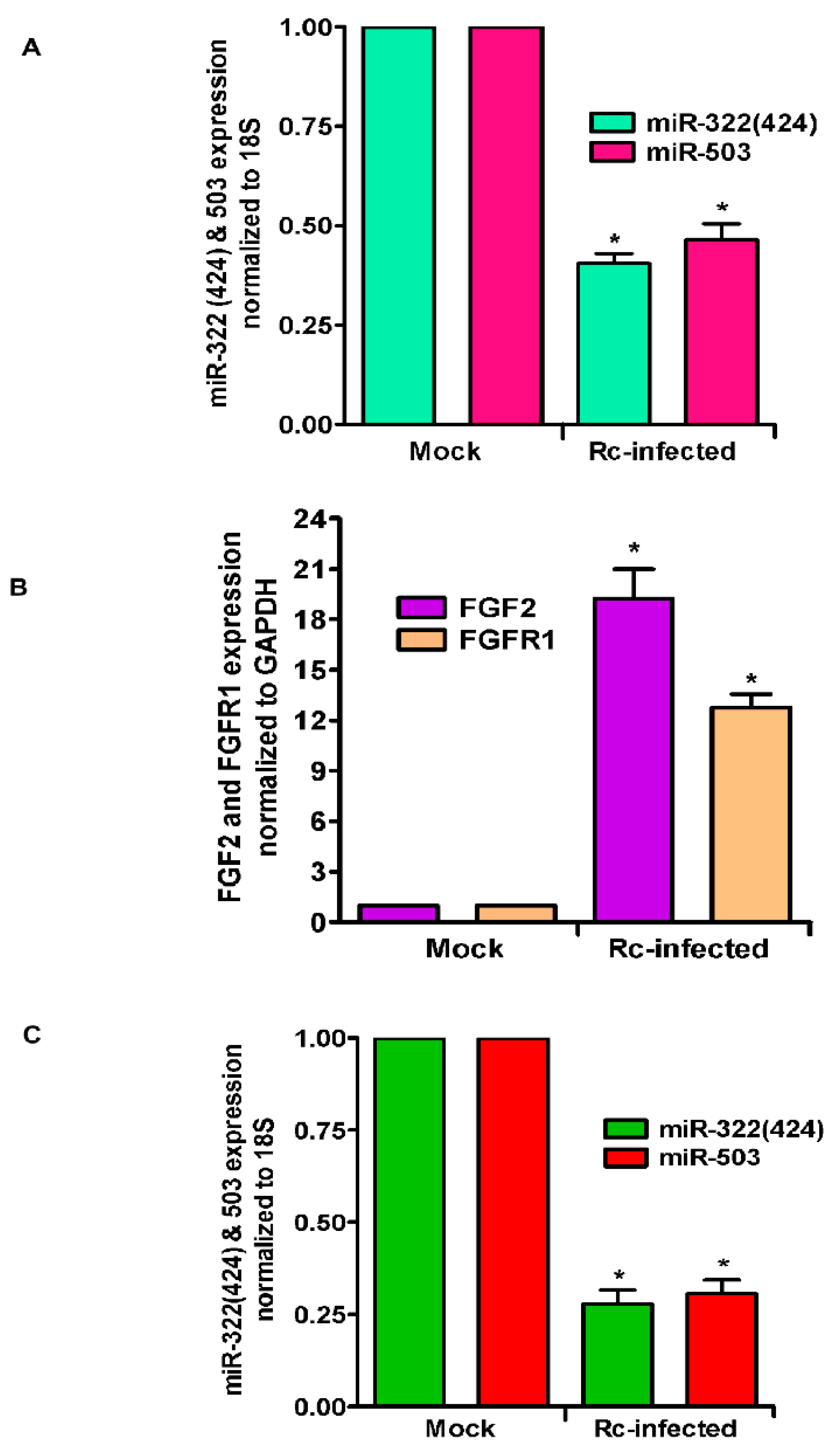
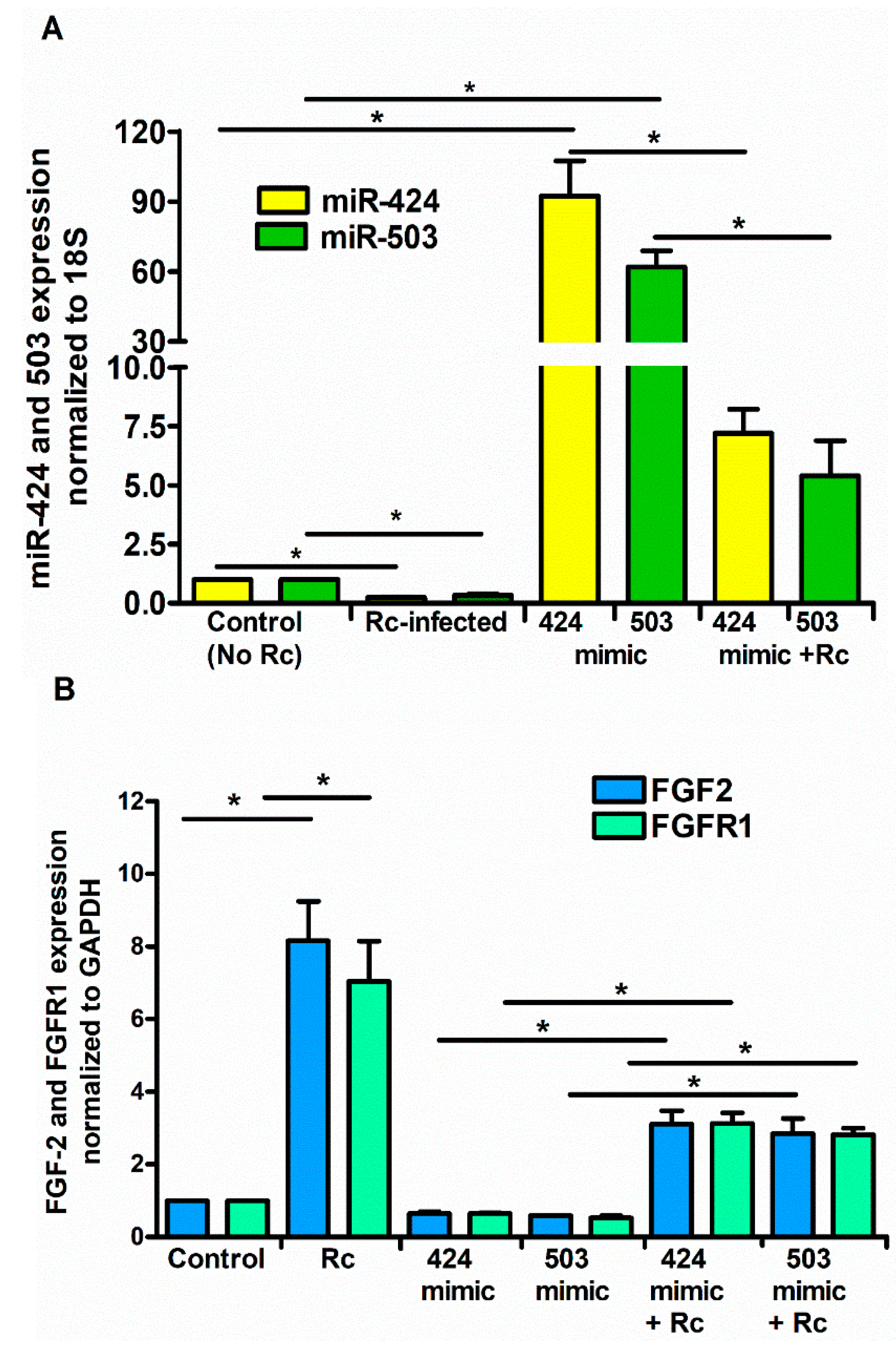
3. Results
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Sahni, A.; Narra, H.P.; Walker, D.H.; Sahni, S.K. Endothelial activation and injury: The mechanisms of rickettsial vasculitis. In Vascular Responses to Pathogens; Gavins, F., Stokes, K.Y., Eds.; Elsevier: Atlanta, GA, USA, 2016; pp. 111–122. [Google Scholar]
- Valbuena, G.; Walker, D.H. The endothelium as a target for infections. Annu. Rev. Pathol. 2006, 1, 171–198. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Yu, X.; Hu, S.; Yu, J. A brief review on the mechanisms of miRNA regulation. Genom. Proteom. Bioinform. 2009, 7, 147–154. [Google Scholar] [CrossRef]
- Catalanotto, C.; Cogoni, C.; Zardo, G. MicroRNA in Control of Gene Expression: An Overview of Nuclear Functions. Int. J. Mol. Sci. 2016, 17, 1712. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, F.; Rossato, M.; Fabbri, M.; Gaudiosi, D.; Mirolo, M.; Mori, L.; Tamassia, N.; Mantovani, A.; Cassatella, M.A.; Locati, M. Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc. Natl. Acad. Sci. USA 2009, 106, 5282–5287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtale, G.; Mirolo, M.; Renzi, T.A.; Rossato, M.; Bazzoni, F.; Locati, M. Negative regulation of Toll-like receptor 4 signaling by IL-10-dependent microRNA-146b. Proc. Natl. Acad. Sci. USA 2013, 110, 11499–11504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koralov, S.B.; Muljo, S.A.; Galler, G.R.; Krek, A.; Chakraborty, T.; Kanellopoulou, C.; Jensen, K.; Cobb, B.S.; Merkenschlager, M.; Rajewsky, N.; et al. Dicer ablation affects antibody diversity and cell survival in the B lymphocyte lineage. Cell 2008, 132, 860–874. [Google Scholar] [CrossRef]
- Mehta, A.; Baltimore, D. MicroRNAs as regulatory elements in immune system logic. Nat. Rev. Immunol. 2016, 16, 279–294. [Google Scholar] [CrossRef]
- Rossato, M.; Curtale, G.; Tamassia, N.; Castellucci, M.; Mori, L.; Gasperini, S.; Mariotti, B.; De Luca, M.; Mirolo, M.; Cassatella, M.A.; et al. IL-10-induced microRNA-187 negatively regulates TNF-alpha, IL-6, and IL-12p40 production in TLR4-stimulated monocytes. Proc. Natl. Acad. Sci. USA 2012, 109, E3101–E3110. [Google Scholar] [CrossRef]
- Krek, A.; Grun, D.; Poy, M.N.; Wolf, R.; Rosenberg, L.; Epstein, E.J.; MacMenamin, P.; da Piedade, I.; Gunsalus, K.C.; Stoffel, M.; et al. Combinatorial microRNA target predictions. Nat. Genet. 2005, 37, 495–500. [Google Scholar] [CrossRef]
- Cullen, B.R. Viruses and microRNAs: RISCy interactions with serious consequences. Genes Dev. 2011, 25, 1881–1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottwein, E.; Cullen, B.R. Viral and cellular microRNAs as determinants of viral pathogenesis and immunity. Cell Host Microbe 2008, 3, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 2008, 454, 780–783. [Google Scholar] [CrossRef] [Green Version]
- Eulalio, A.; Schulte, L.; Vogel, J. The mammalian microRNA response to bacterial infections. RNA Biol. 2012, 9, 742–750. [Google Scholar] [CrossRef] [Green Version]
- Harapan, H.; Fitra, F.; Ichsan, I.; Mulyadi, M.; Miotto, P.; Hasan, N.A.; Calado, M.; Cirillo, D.M. The roles of microRNAs on tuberculosis infection: Meaning or myth? Tuberculosis (Edinb) 2013, 93, 596–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staedel, C.; Darfeuille, F. MicroRNAs and bacterial infection. Cell Microbiol. 2013, 15, 1496–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maudet, C.; Mano, M.; Eulalio, A. MicroRNAs in the interaction between host and bacterial pathogens. FEBS Lett. 2014, 588, 4140–4147. [Google Scholar] [CrossRef] [Green Version]
- Maudet, C.; Mano, M.; Sunkavalli, U.; Sharan, M.; Giacca, M.; Forstner, K.U.; Eulalio, A. Functional high–throughput screening identifies the miR-15 microRNA family as cellular restriction factors for Salmonella infection. Nat. Commun. 2014, 5, 4718. [Google Scholar] [CrossRef]
- Sahni, A.; Narra, H.P.; Patel, J.; Sahni, S.K. MicroRNA signature of human microvascular endothelium infected with Rickettsia rickettsii. Int. J. Mol. Sci. 2017, 18, 1471. [Google Scholar] [CrossRef]
- Sahni, A.; Patel, J.; Narra, H.P.; Schroeder, C.L.C.; Walker, D.H.; Sahni, S.K. Fibroblast growth factor receptor-1 mediates internalization of pathogenic spotted fever rickettsiae into host endothelium. PLoS ONE 2017, 12, e0183181. [Google Scholar] [CrossRef]
- Rydkina, E.; Turpin, L.C.; Sahni, S.K. Rickettsia rickettsii infection of human macrovascular and microvascular endothelial cells reveals activation of both common and cell type-specific host response mechanisms. Infect. Immun. 2010, 78, 2599–2606. [Google Scholar] [CrossRef] [PubMed]
- Ades, E.W.; Candal, F.J.; Swerlick, R.A.; George, V.G.; Summers, S.; Bosse, D.C.; Lawley, T.J. HMEC-1: Establishment of an immortalized human microvascular endothelial cell line. J. Invest. Dermatol. 1992, 99, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Grab, D.J.; Nyarko, E.; Barat, N.C.; Nikolskaia, O.V.; Dumler, J.S. Anaplasma phagocytophilum-Borrelia burgdorferi coinfection enhances chemokine, cytokine, and matrix metalloprotease expression by human brain microvascular endothelial cells. Clin. Vaccine Immunol. 2007, 14, 1420–1424. [Google Scholar] [CrossRef] [PubMed]
- Rydkina, E.; Sahni, S.K.; Santucci, L.A.; Turpin, L.C.; Baggs, R.B.; Silverman, D.J. Selective modulation of antioxidant enzyme activities in host tissues during Rickettsia conorii infection. Microb. Pathog. 2004, 36, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Labruna, M.B.; Whitworth, T.; Horta, M.C.; Bouyer, D.H.; McBride, J.W.; Pinter, A.; Popov, V.; Gennari, S.M.; Walker, D.H. Rickettsia species infecting Amblyomma cooperi ticks from an area in the state of Sao Paulo, Brazil, where Brazilian spotted fever is endemic. J. Clin. Microbiol. 2004, 42, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, A.; Mueller, O.; Stocker, S.; Salowsky, R.; Leiber, M.; Gassmann, M.; Lightfoot, S.; Menzel, W.; Granzow, M.; Ragg, T. The RIN: An RNA integrity number for assigning integrity values to RNA measurements. BMC Mol. Biol. 2006, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Eremeeva, M.E.; Dasch, G.A.; Silverman, D.J. Evaluation of a PCR assay for quantitation of Rickettsia rickettsii and closely related spotted fever group rickettsiae. J. Clin. Microbiol. 2003, 41, 5466–5472. [Google Scholar] [CrossRef]
- Kim, J.; Kang, Y.; Kojima, Y.; Lighthouse, J.K.; Hu, X.; Aldred, M.A.; McLean, D.L.; Park, H.; Comhair, S.A.; Greif, D.M.; et al. An endothelial apelin-FGF link mediated by miR-424 and miR-503 is disrupted in pulmonary arterial hypertension. Nat. Med. 2013, 19, 74–82. [Google Scholar] [CrossRef]
- Di Leva, G.; Garofalo, M.; Croce, C.M. MicroRNAs in cancer. Annu. Rev. Pathol. 2014, 9, 287–314. [Google Scholar] [CrossRef]
- Chang, T.C.; Mendell, J.T. MicroRNAs in vertebrate physiology and human disease. Annu. Rev. Genomics Hum. Genet. 2007, 8, 215–239. [Google Scholar] [CrossRef]
- Zhang, X.; Guo, J.; Fan, S.; Li, Y.; Wei, L.; Yang, X.; Jiang, T.; Chen, Z.; Wang, C.; Liu, J.; et al. Screening and identification of six serum microRNAs as novel potential combination biomarkers for pulmonary tuberculosis diagnosis. PLoS ONE 2013, 8, e81076. [Google Scholar] [CrossRef] [PubMed]
- Shiota, S.; Yamaoka, Y. Biomarkers for Helicobacter pylori infection and gastroduodenal diseases. Biomark. Med. 2014, 8, 1127–1137. [Google Scholar] [CrossRef] [PubMed]
- Eswarakumar, V.P.; Lax, I.; Schlessinger, J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005, 16, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, M.; Trueb, B. Characterization of a novel protein (FGFRL1) from human cartilage related to FGF receptors. Genomics 2000, 69, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Florkiewicz, R.Z.; Baird, A.; Gonzalez, A.M. Multiple forms of bFGF: Differential nuclear and cell surface localization. Growth Factors 1991, 4, 265–275. [Google Scholar] [CrossRef]
- Arnaud, E.; Touriol, C.; Boutonnet, C.; Gensac, M.C.; Vagner, S.; Prats, H.; Prats, A.C. A new 34-kilodalton isoform of human fibroblast growth factor 2 is cap dependently synthesized by using a non-AUG start codon and behaves as a survival factor. Mol. Cell Biol. 1999, 19, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Bastaki, M.; Nelli, E.E.; Dell’Era, P.; Rusnati, M.; Molinari-Tosatti, M.P.; Parolini, S.; Auerbach, R.; Ruco, L.P.; Possati, L.; Presta, M. Basic fibroblast growth factor-induced angiogenic phenotype in mouse endothelium. A study of aortic and microvascular endothelial cell lines. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 454–464. [Google Scholar] [CrossRef]
- Johnson, D.E.; Williams, L.T. Structural and functional diversity in the FGF receptor multigene family. Adv. Cancer Res. 1993, 60, 1–41. [Google Scholar]
- Bikfalvi, A.; Klein, S.; Pintucci, G.; Rifkin, D.B. Biological roles of fibroblast growth factor-2. Endocr. Rev. 1997, 18, 26–45. [Google Scholar] [CrossRef]
- Chamorro-Jorganes, A.; Araldi, E.; Penalva, L.O.; Sandhu, D.; Fernandez-Hernando, C.; Suarez, Y. MicroRNA-16 and microRNA-424 regulate cell-autonomous angiogenic functions in endothelial cells via targeting vascular endothelial growth factor receptor-2 and fibroblast growth factor receptor-1. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2595–2606. [Google Scholar] [CrossRef]
- Santulli, G. MicroRNAs and Endothelial (Dys) Function. J. Cell Physiol. 2016, 231, 1638–1644. [Google Scholar] [CrossRef] [PubMed]
- Finnerty, J.R.; Wang, W.X.; Hebert, S.S.; Wilfred, B.R.; Mao, G.; Nelson, P.T. The miR-15/107 group of microRNA genes: Evolutionary biology, cellular functions, and roles in human diseases. J. Mol. Biol. 2010, 402, 491–509. [Google Scholar] [CrossRef] [PubMed]
- Forrest, A.R.; Kanamori-Katayama, M.; Tomaru, Y.; Lassmann, T.; Ninomiya, N.; Takahashi, Y.; de Hoon, M.J.; Kubosaki, A.; Kaiho, A.; Suzuki, M.; et al. Induction of microRNAs, mir-155, mir-222, mir-424 and mir-503, promotes monocytic differentiation through combinatorial regulation. Leukemia 2010, 24, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, M.; Coppola, V.; Addario, A.; Patrizii, M.; Maugeri-Sacca, M.; Memeo, L.; Colarossi, C.; Francescangeli, F.; Biffoni, M.; Collura, D.; et al. Control of tumor and microenvironment cross-talk by miR-15a and miR-16 in prostate cancer. Oncogene 2011, 30, 4231–4242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osterloh, A. Immune response against rickettsiae: Lessons from murine infection models. Med. Microbiol. Immunol. 2017, 206, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Alhassan, A.; Liu, H.; McGill, J.; Cerezo, A.; Jakkula, L.; Nair, A.D.S.; Winkley, E.; Olson, S.; Marlow, D.; Sahni, A.; et al. Rickettsia rickettsii whole cell antigens offer protection against Rocky Mountain spotted fever in the canine host. Infect. Immun. 2018. [Google Scholar] [CrossRef] [PubMed]
- Rogelj, S.; Klagsbrun, M.; Atzmon, R.; Kurokawa, M.; Haimovitz, A.; Fuks, Z.; Vlodavsky, I. Basic fibroblast growth factor is an extracellular matrix component required for supporting the proliferation of vascular endothelial cells and the differentiation of PC12 cells. J. Cell Biol. 1989, 109, 823–831. [Google Scholar] [CrossRef] [Green Version]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef]
- Tomlin, H.; Piccinini, A.M. A complex interplay between the extracellular matrix and the innate immune response to microbial pathogens. Immunology 2018, 155, 186–201. [Google Scholar] [CrossRef]
- Hanneken, A.; Ying, W.; Ling, N.; Baird, A. Identification of soluble forms of the fibroblast growth factor receptor in blood. Proc. Natl. Acad. Sci. USA 1994, 91, 9170–9174. [Google Scholar] [CrossRef]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahni, A.; Narra, H.P.; Patel, J.; Sahni, S.K. MicroRNA-Regulated Rickettsial Invasion into Host Endothelium via Fibroblast Growth Factor 2 and Its Receptor FGFR1. Cells 2018, 7, 240. https://doi.org/10.3390/cells7120240
Sahni A, Narra HP, Patel J, Sahni SK. MicroRNA-Regulated Rickettsial Invasion into Host Endothelium via Fibroblast Growth Factor 2 and Its Receptor FGFR1. Cells. 2018; 7(12):240. https://doi.org/10.3390/cells7120240
Chicago/Turabian StyleSahni, Abha, Hema P. Narra, Jignesh Patel, and Sanjeev K. Sahni. 2018. "MicroRNA-Regulated Rickettsial Invasion into Host Endothelium via Fibroblast Growth Factor 2 and Its Receptor FGFR1" Cells 7, no. 12: 240. https://doi.org/10.3390/cells7120240
APA StyleSahni, A., Narra, H. P., Patel, J., & Sahni, S. K. (2018). MicroRNA-Regulated Rickettsial Invasion into Host Endothelium via Fibroblast Growth Factor 2 and Its Receptor FGFR1. Cells, 7(12), 240. https://doi.org/10.3390/cells7120240