CEP55 Promotes Cell Motility via JAK2–STAT3–MMPs Cascade in Hepatocellular Carcinoma



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Gene Expression and Survival Analysis
2.3. Quantitative Real-Time PCR
2.4. Lentivirus Construction and Infection
- CEP55 shRNA1: 5′-ACCTCACGAATTGCTGAAC-3′; CEP55 shRNA2: 5′-GACTGAGAGACCAACTGAA-3′;
- CEP55 shRNA3: 5′-GCTCCAAACTGCTTCAACT-3′; CEP55 shRNA4: 5′-CTGAAAGATGCTCTGGAGA-3′; and
- JAK2 shRNA: 5′-GCAGAATTAGCAAACCTTATA-3′; STAT3 shRNA: 5′-TGTTCTCTATCAGCACAAT-3′.
2.5. Western Blot
2.6. Co-Immunoprecipitation (Co-IP) Assay
2.7. Cell Wound Scratch Assay
2.8. Trans-Well Assay
2.9. Statistical Analysis
3. Results
3.1. CEP55 Expression Is Upregulated in HCC Tissues and Cell Lines
3.2. Overexpression of CEP55 Is a Poor Prognostic Factor for HCC Patients
3.3. CEP55 Knockdown Suppresses the Motility Activity of HCC Cells
3.4. CEP55 Promotes Expression of MMPs in HCC Cells
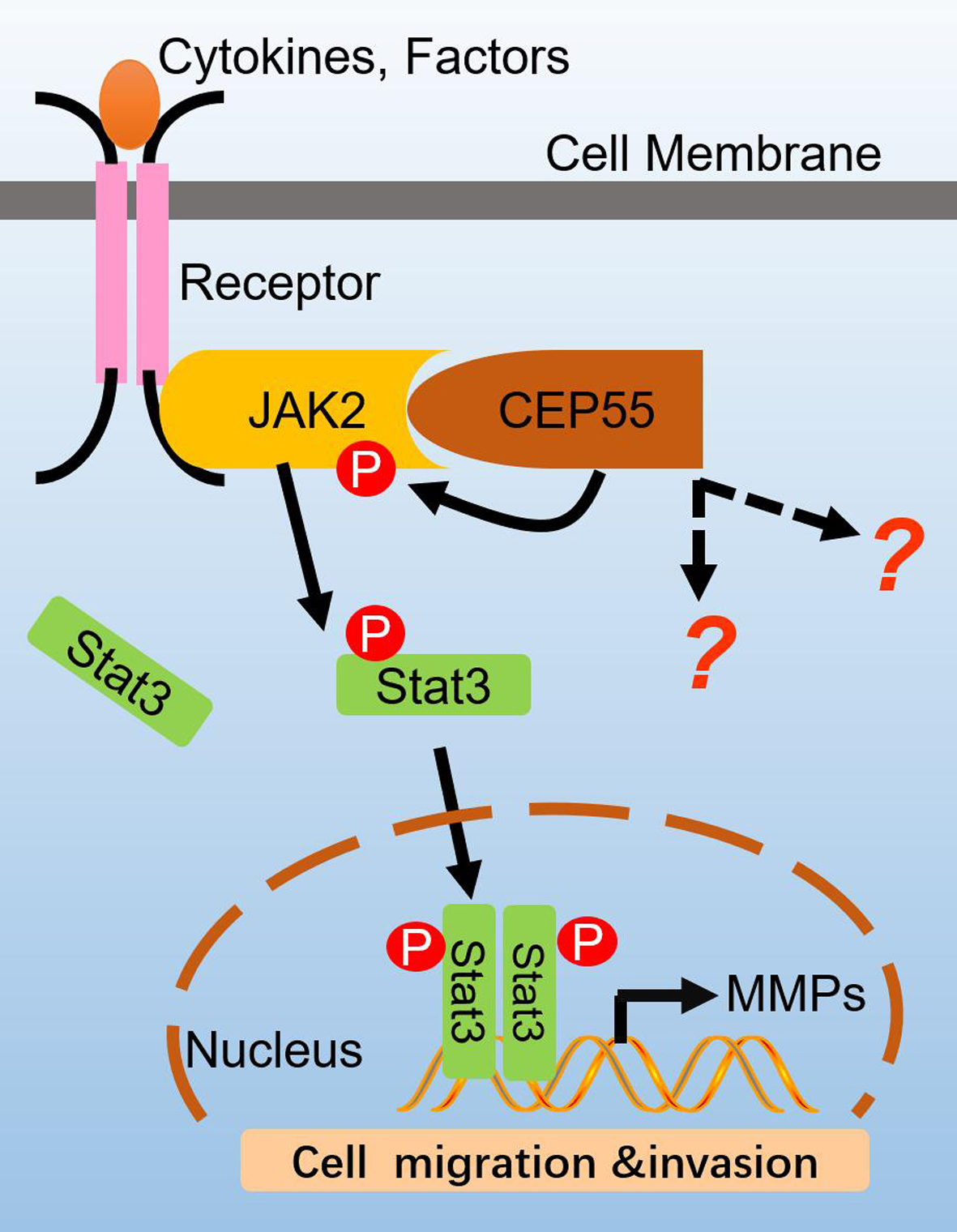
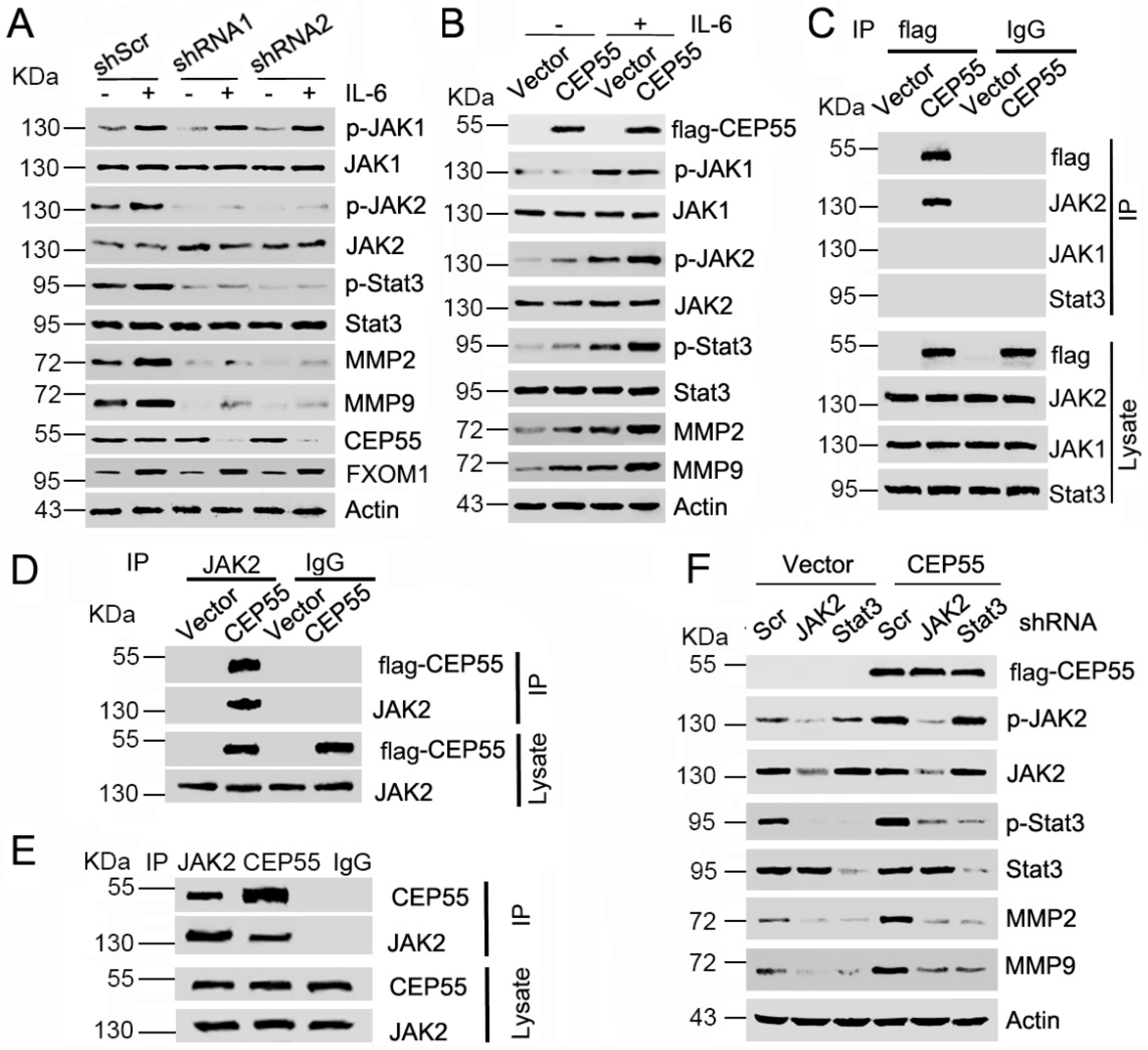
3.5. CEP55 Stimulates the JAK2–STAT3–MMP Axis in HCC Cells
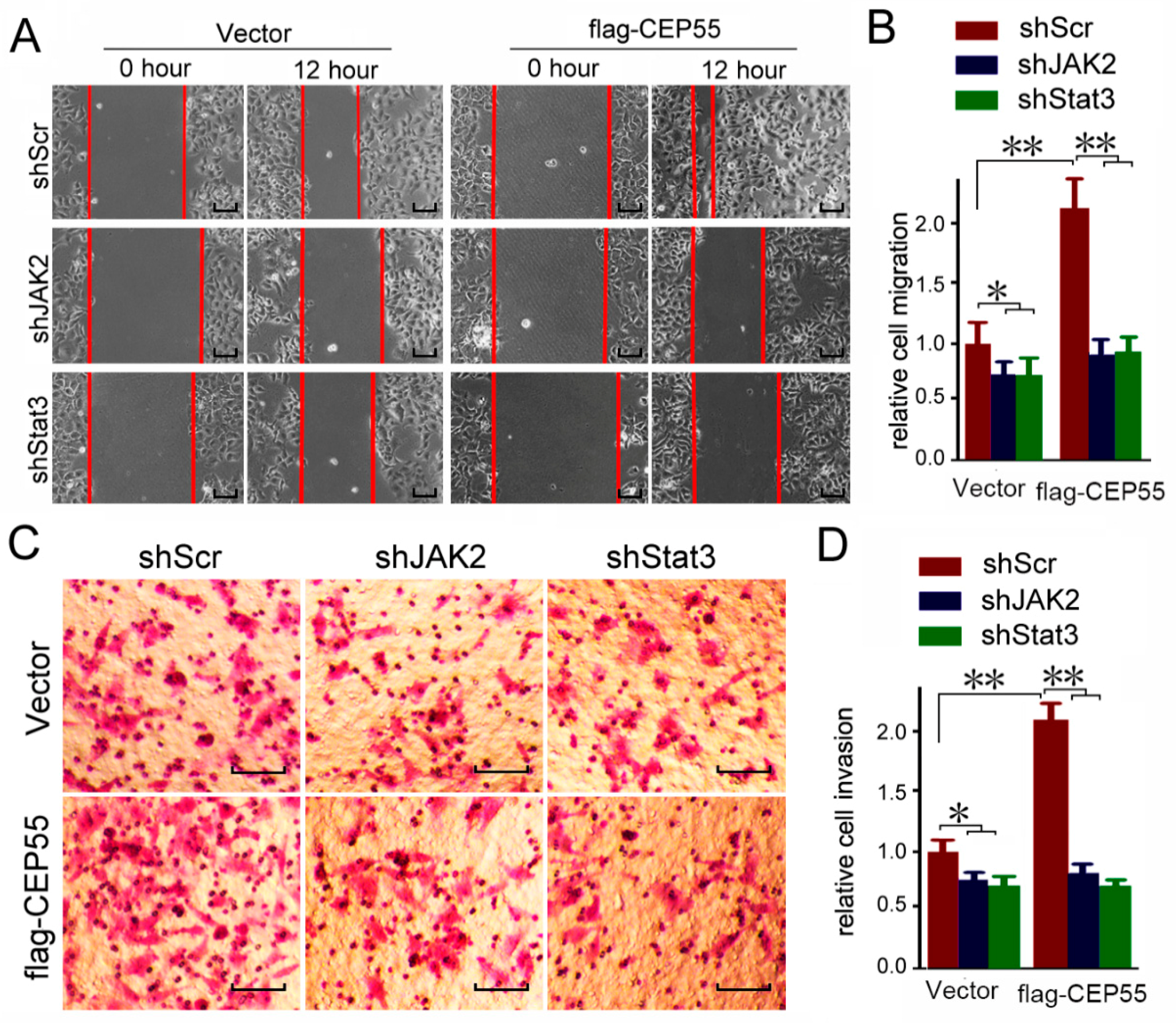
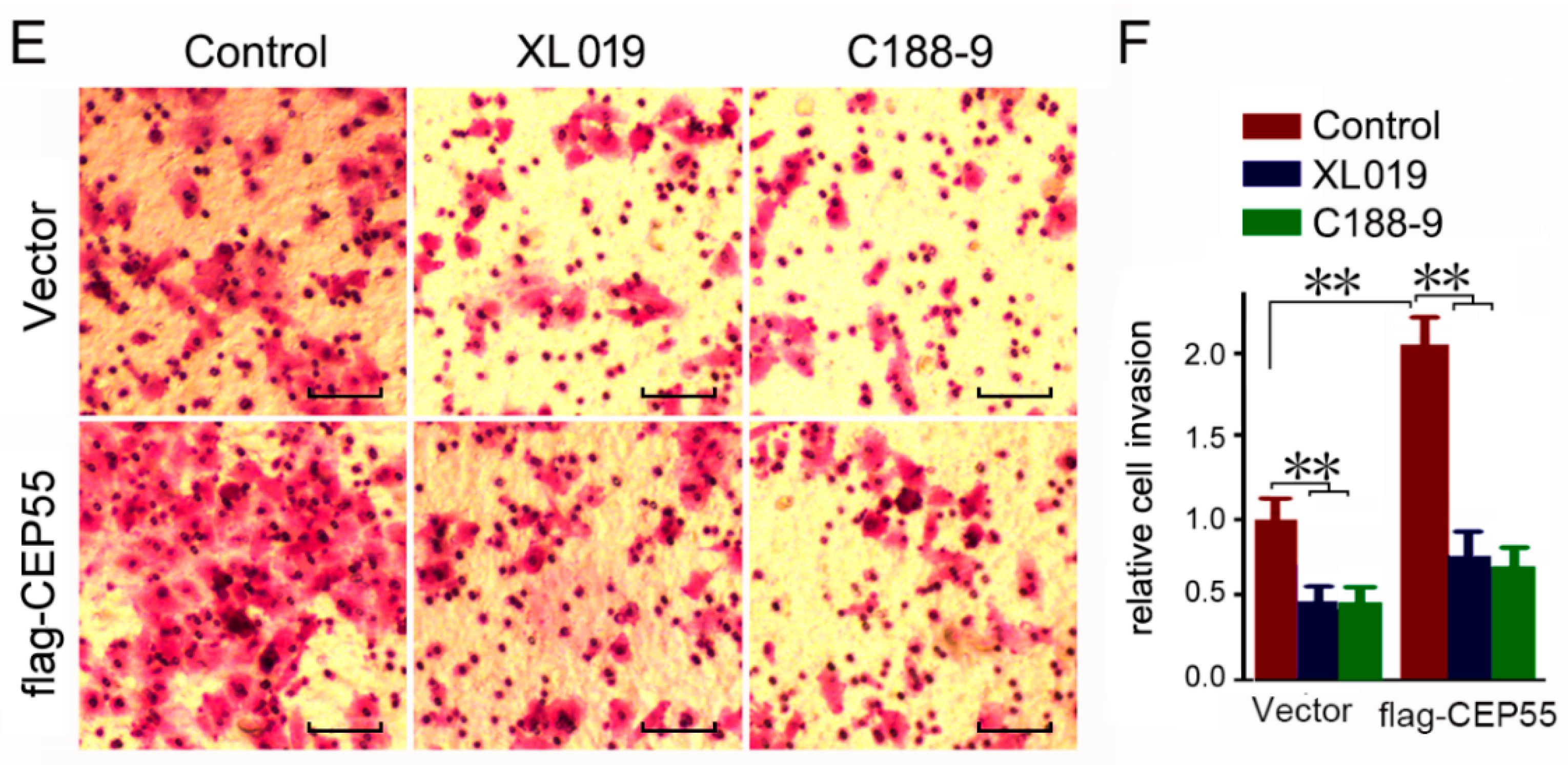
3.6. JAK2 or STAT3 Blocking Abrogates the Pro-Migration and Pro-Invasion Effects of CEP55
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.; Mahato, R.I. Recent advances in hepatocellular carcinoma therapy. Pharmacol. Ther. 2017, 173, 106–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbro, M.; Zhou, B.B.; Takahashi, M.; Sarcevic, B.; Lal, P.; Graham, M.E.; Gabrielli, B.G.; Robinson, P.J.; Nigg, E.A.; Ono, Y.; et al. Cdk1/Erk2- and Plk1-dependent phosphorylation of a centrosome protein, Cep55, is required for its recruitment to midbody and cytokinesis. Dev. Cell 2005, 9, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Garay, I.; Rustom, A.; Gerdes, H.H.; Kutsche, K. The novel centrosomal associated protein CEP55 is present in the spindle midzone and the midbody. Genomics 2006, 87, 243–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.M.; Seki, A.; Fang, G. CEP55, a microtubule-bundling protein, associates with centralspindlin to control the midbody integrity and cell abscission during cytokinesis. Mol. Biol. Cell 2006, 17, 3881–3896. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, J.; Sinha, D.; Srihari, S.; Kalimutho, M.; Khanna, K.K. Beyond cytokinesis: The emerging roles of CEP55 in tumorigenesis. Oncogene 2016, 35, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Lu, P.J.; Chen, Y.C.; Fu, S.L.; Wu, K.J.; Tsou, A.P.; Lee, Y.C.; Lin, T.C.; Hsu, S.L.; Lin, W.J.; et al. FLJ10540-elicited cell transformation is through the activation of PI3-kinase/AKT pathway. Oncogene 2007, 26, 4272–4283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.H.; Chien, C.Y.; Huang, C.C.; Hwang, C.F.; Chuang, H.C.; Fang, F.M.; Huang, H.Y.; Chen, C.M.; Liu, H.L.; Huang, C.Y. Expression of FLJ10540 is correlated with aggressiveness of oral cavity squamous cell carcinoma by stimulating cell migration and invasion through increased FOXM1 and MMP-2 activity. Oncogene 2009, 28, 2723–2737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.H.; Lai, J.M.; Chou, T.Y.; Chen, C.Y.; Su, L.J.; Lee, Y.C.; Cheng, T.S.; Hong, Y.R.; Chou, C.K.; Jacqueline, W.P.; et al. Vegfa upregulates FLJ10540 and modulates migration and invasion of lung cancer via PI3K/AKT pathway. PLoS ONE 2009, 4, e5052. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Chang, A.Y.; Li, S.H.; Tsai, H.T.; Shiu, L.Y.; Su, L.J.; Wang, W.L.; Chiu, T.J.; Luo, S.D.; Huang, T.L.; et al. Suppression of aurora-a-FLJ10540 signaling axis prohibits the malignant state of head and neck cancer. Mol. Cancer 2015, 14. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jin, T.; Dai, X.; Xu, J. Lentivirus-mediated knockdown of CEP55 suppresses cell proliferation of breast cancer cells. Biosci. Trends 2016, 10, 67–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Niu, C.; He, W.; Hou, T.; Sun, X.; Xu, L.; Zhang, Y. Upregulation of centrosomal protein 55 is associated with unfavorable prognosis and tumor invasion in epithelial ovarian carcinoma. Tumour Biol. 2016, 37, 6239–6254. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Wang, Z.; Jia, Y. CEP55 overexpression predicts poor prognosis in patients with locally advanced esophageal squamous cell carcinoma. Oncol. Lett. 2017, 13, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Shin, G.; Kang, T.W.; Yang, S.; Baek, S.J.; Jeong, Y.S.; Kim, S.Y. Gent: Gene expression database of normal and tumor tissues. Cancer Inform. 2011, 10, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cbio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, H.Y.; Fattet, L.; Yang, J. Molecular pathways: Linking tumor microenvironment to epithelial-mesenchymal transition in metastasis. Clin. Cancer Res. 2015, 5, 962–968. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Yang, G.; Jiang, T.; Huang, K.; Cao, J.; Qiu, Z. Effects of IL-6 and AG490 on regulation of STAT3 signaling pathway and invasion of human pancreatic cancer cells in vitro. J. Exp. Clin. Cancer Res. 2010, 29, 51. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Li, G.; Li, R.; Shen, J.; He, Q.; Deng, L.; Zhang, C.; Zhang, J. IL-6 promotion of glioblastoma cell invasion and angiogenesis in U251 and T98G cell lines. J. Neurooncol. 2010, 100, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Shiu, L.Y.; Su, L.J.; Huang, C.Y.; Huang, S.C.; Huang, C.C.; Yin, Y.F.; Wang, W.S.; Tsai, H.T.; Fang, F.M.; et al. FLJ10540 is associated with tumor progression in nasopharyngeal carcinomas and contributes to nasopharyngeal cell proliferation, and metastasis via osteopontin/CD44 pathway. J. Transl. Med. 2012, 10, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, D.; Kalimutho, M.; Bowles, J.; Chan, A.L.; Merriner, D.J.; Bain, A.L.; Simmons, J.L.; Freire, R.; Lopez, J.A.; Hobbs, R.M.; et al. Cep55 overexpression causes male-specific sterility in mice by suppressing Foxo1 nuclear retention through sustained activation of PI3K/Akt signaling. FASEB J. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Liu, M.; Wang, H.; Yu, S.; Jiang, Z.; Sun, J.; Han, K.; Shen, J.; Zhu, M.; Lin, Z.; et al. Centrosomal protein of 55 regulates glucose metabolism, proliferation and apoptosis of glioma cells via the Akt/mTOR signaling pathway. J. Cancer 2016, 7, 1431–1440. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Hara, A.; Kuno, T.; Kitaori, N.; Huilan, Z.; Mori, H.; Toida, M.; Shibata, T. Matrix metalloproteinases 2 and 9 in oral squamous cell carcinomas: Manifestation and localization of their activity. J. Cancer Res. Clin. Oncol. 2005, 131, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Huynh, J.; Etemadi, N.; Hollande, F.; Ernst, M.; Buchert, M. The JAK/STAT3 axis: A comprehensive drug target for solid malignancies. Semin. Cancer Biol. 2017, 45, 13–22. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | CEP55 mRNA Expression # | p-Value (χ2) | |
|---|---|---|---|
| Low | High | ||
| Gender | |||
| Male | 86 | 75 | 0.1540 |
| Female | 30 | 41 | |
| Age (year) | |||
| ≤50 | 18 | 30 | 0.0739 |
| >50 | 98 | 86 | |
| AFP (ng/mL) | |||
| ≤200 | 101 | 69 | <0.0001 * |
| >200 | 15 | 47 | |
| Hepatitis status | |||
| Negative | 55 | 47 | 0.3545 |
| Positive | 61 | 69 | |
| Vascular invasion | |||
| No | 91 | 72 | 0.0095 * |
| Yes | 25 | 44 | |
| Histologic grade | |||
| I | 19 | 5 | <0.0001 * |
| II | 64 | 43 | |
| III–IV | 33 | 68 | |
| TNM stage | |||
| I | 74 | 52 | 0.02 * |
| II | 22 | 30 | |
| III–IV | 20 | 32 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Gao, J.; Li, D.; Yin, Y. CEP55 Promotes Cell Motility via JAK2–STAT3–MMPs Cascade in Hepatocellular Carcinoma. Cells 2018, 7, 99. https://doi.org/10.3390/cells7080099
Li M, Gao J, Li D, Yin Y. CEP55 Promotes Cell Motility via JAK2–STAT3–MMPs Cascade in Hepatocellular Carcinoma. Cells. 2018; 7(8):99. https://doi.org/10.3390/cells7080099
Chicago/Turabian StyleLi, Minjing, Ju Gao, Defang Li, and Yancun Yin. 2018. "CEP55 Promotes Cell Motility via JAK2–STAT3–MMPs Cascade in Hepatocellular Carcinoma" Cells 7, no. 8: 99. https://doi.org/10.3390/cells7080099
APA StyleLi, M., Gao, J., Li, D., & Yin, Y. (2018). CEP55 Promotes Cell Motility via JAK2–STAT3–MMPs Cascade in Hepatocellular Carcinoma. Cells, 7(8), 99. https://doi.org/10.3390/cells7080099