Genetic Renal Diseases: The Emerging Role of Zebrafish Models

 ,
,  ,
,
Abstract
:1. Introduction
2. Methods for Genetic Modeling
2.1. Morpholino Antisense Oligonucleotides
2.2. CRISPR-Cas9
3. Assessment of the Renal Phenotype
3.1. Evaluation of Zebrafish Survival, Development, and Morphology
3.2. Evaluation of Glomerular Function
3.3. Evaluation of Tubular Function
3.3.1. Tubular Endocytosis
3.3.2. Ion and Small Solute Transport
3.4. Evaluation of Renal Cysts
4. Characterized Zebrafish Models of Genetic Renal Diseases
5. Drug Discovery and Validation
6. Limitations of Zebrafish Models
7. Future Perspectives
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lieschke, G.J.; Currie, P.D. Animal models of human disease: Zebrafish swim into view. Nat. Rev. Genet. 2007, 8, 353–367. [Google Scholar] [CrossRef] [PubMed]
- MacRae, C.A.; Peterson, R.T. Zebrafish as tools for drug discovery. Nat. Rev. Drug Discov. 2015, 14, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Martin, P. Imaging innate immune responses at tumour initiation: New insights from fish and flies. Nat. Rev. Cancer 2015, 15, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlegel, A.; Gut, P. Metabolic insights from zebrafish genetics, physiology, and chemical biology. Cell. Mol. Life Sci. 2015, 72, 2249–2260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Zhao, L.; Page-McCaw, P.S.; Chen, W. Zebrafish genome engineering using the CRISPR–Cas9 system. Trends Genet. 2016, 32, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Diep, C.Q.; Peng, Z.; Ukah, T.K.; Kelly, P.M.; Daigle, R.V.; Davidson, A.J. Development of the zebrafish mesonephros. Genesis 2015, 53, 257–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habuka, M.; Fagerberg, L.; Odeberg, J. The Kidney Transcriptome and Proteome Defined by Transcriptomics and Antibody-Based Profiling. PLoS ONE 2014, 9, e116125. [Google Scholar] [CrossRef] [PubMed]
- Wingert, R.A.; Selleck, R.; Yu, J.; Song, H.D.; Chen, Z.; Song, A.; Zhou, Y.; Thisse, B.; Thisse, C.; McMahon, A.P.; et al. The cdx genes and retinoic acid control the positioning and segmentation of the zebrafish pronephros. PLoS Genet. 2007, 3, 1922–1938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wingert, R.A.; Davidson, A.J. The zebrafish pronephros: A model to study nephron segmentation. Kidney Int. 2008, 73, 1120–1127. [Google Scholar] [CrossRef] [PubMed]
- Desgrange, A.; Cereghini, S. Nephron patterning: Lessons from xenopus, zebrafish, and mouse studies. Cells 2015, 4, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Drummond, I.A.; Majumdar, A.; Hentschel, H.; Elger, M.; Solnica-Krezel, L.; Schier, A.F.; Neuhauss, S.C.; Stemple, D.L.; Zwartkruis, F.; Rangini, Z.; et al. Early development of the zebrafish pronephros and analysis of mutations affecting pronephric function. Development 1998, 125, 4655–4667. [Google Scholar] [PubMed]
- Krause, M.; Rak-Raszewska, A.; Pietilä, I.; Quaggin, S.E.; Vainio, S. Signaling during kidney development. Cells 2015, 4, 112–132. [Google Scholar] [CrossRef] [PubMed]
- McCampbell, K.K.; Springer, K.N.; Wingert, R.A. Analysis of nephron composition and function in the adult zebrafish kidney. J. Vis. Exp. 2014, 90, e51644. [Google Scholar] [CrossRef] [PubMed]
- Rider, S.A.; Christian, H.C.; Mullins, L.J.; Howarth, A.R.; MacRae, C.A.; Mullins, J.J. Zebrafish mesonephric renin cells are functionally conserved and comprise two distinct morphological populations. Am. J. Physiol.-Ren. Physiol. 2017, 312, F778–F790. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Mckee, M.; Huang, H.D.; Xiang, A.; Davidson, A.J.; Lu, H.A.J. A zebrafish model of conditional targeted podocyte ablation and regeneration. Kidney Int. 2013, 83, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Diep, C.Q.; Ma, D.; Deo, R.C.; Holm, T.M.; Naylor, R.W.; Arora, N.; Wingert, R.A.; Bollig, F.; Djordjevic, G.; Lichman, B.; et al. Identification of adult nephron progenitors capable of kidney regeneration in zebrafish. Nature 2011, 470, 95–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felsenfeld, A.L. Defining the boundaries of zebrafish developmental genetics. Nat. Genet. 1996, 14, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; Nüsslein-Volhard, C. Fishing for genes controlling development. Curr. Opin. Genet. Dev. 1996, 6, 461–468. [Google Scholar] [CrossRef]
- Walker, C.; Streisinger, G. Induction of mutations by Y-rays in pregonial germ cells of zebrafish embryos. Genetics 1983, 103, 125–136. [Google Scholar] [PubMed]
- Grunwald, D.; Streisinger, G. Induction of recessive lethal and specific locus mutations in the zebrafish with ethyl nitrosourea. Genet. Res. 1992, 59, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Lawson, N.D.; Wolfe, S.A. Forward and reverse genetic approaches for the analysis of vertebrate development in the zebrafish. Dev. Cell 2011, 21, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Gaiano, N.; Culp, P.; Burns, J.C.; Friedmann, T.; Yee, J.K.; Hopkins, N. Integration and germ-line transmission of a pseudotyped retroviral vector in zebrafish. Science 1994, 265, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Amsterdam, A.; Burgess, S.; Golling, G.; Chen, W.; Sun, Z.; Townsend, K.; Farrington, S.; Haldi, M.; Hopkins, N. A large-scale insertional mutagenesis screen in zebrafish. Genes Dev. 1999, 13, 2713–2724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amsterdam, A.; Varshney, G.K.; Burgess, S.M. Retroviral-mediated insertional mutagenesis in zebrafish. Methods Cell Biol. 2011, 104, 59–82. [Google Scholar] [CrossRef] [PubMed]
- Nasevicius, A.; Ekker, S.C. Effective targeted gene “knockdown” in zebrafish. Nat. Genet. 2000, 26, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Tsai, S.Q.; Sander, J.D.; Peterson, R.T.; Yeh, J.-R.J.; Joung, J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 227–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, P.; Zhu, Z.; Lin, S.; Zhang, B. Reverse genetic approaches in zebrafish. J. Genet. Genom. 2012, 39, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Eisen, J.S.; Smith, J.C. Controlling morpholino experiments: Don’t stop making antisense. Development 2008, 135, 1735–1743. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Kontarakis, Z.; Gerri, C.; Nolte, H.; Hölper, S.; Krüger, M.; Stainier, D.Y.R. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 2015, 524, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Robu, M.E.; Larson, J.D.; Nasevicius, A.; Beiraghi, S.; Brenner, C.; Farber, S.A.; Ekker, S.C. p53 activation by knockdown technologies. PLoS Genet. 2007, 3, 787–801. [Google Scholar] [CrossRef] [PubMed]
- Stainier, D.Y.R.; Raz, E.; Lawson, N.D.; Ekker, S.C.; Burdine, R.D.; Eisen, J.S.; Ingham, P.W.; Schulte-Merker, S.; Yelon, D.; Weinstein, B.M.; et al. Guidelines for morpholino use in zebrafish. PLoS Genet. 2017, 13, e1007000. [Google Scholar] [CrossRef] [PubMed]
- Blum, M.; De Robertis, E.M.; Wallingford, J.B.; Niehrs, C. Morpholinos: Antisense and sensibility. Dev. Cell 2015, 35, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Mekler, V.; Minakhin, L.; Severinov, K. Mechanism of duplex DNA destabilization by RNA-guided Cas9 nuclease during target interrogation. Proc. Natl. Acad. Sci. USA 2017, 114, 5443–5448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, A.V.; Nuñez, J.K.; Doudna, J.A. Biology and applications of CRISPR systems: Harnessing nature’s toolbox for genome engineering. Cell 2016, 164, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Marraffini, L.A. CRISPR-Cas immunity in prokaryotes. Nature 2015, 526, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Bassett, A.R.; Tibbit, C.; Ponting, C.P.; Liu, J.L. Highly efficient targeted mutagenesis of Drosophila with the CRISPR/Cas9 system. Cell Rep. 2013, 4, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Xie, K.; Yang, Y. RNA-Guided genome editing in plants using a CRISPR-Cas system. Mol. Plant 2013, 6, 1975–1983. [Google Scholar] [CrossRef] [PubMed]
- Friedland, A.E.; Tzur, Y.B.; Esvelt, K.M.; Colaiácovo, M.P.; Church, G.M.; Calarco, J.A. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat. Methods 2013, 10, 741–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Wang, H.; Shivalila, C.S.; Cheng, A.W.; Shi, L.; Jaenisch, R. XOne-step generation of mice carrying reporter and conditional alleles by CRISPR/cas-mediated genome engineering. Cell 2013, 154, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Cyranoski, D. CRISPR gene-editing tested in a person for the first time. Nature 2016, 539, 479. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.R.; Heckler, K.; Stoll, S.J.; Hillebrands, J.L.; Kynast, K.; Herpel, E.; Porubsky, S.; Elger, M.; Hadaschik, B.; Bieback, K.; et al. ELMO1 protects renal structure and ultrafiltration in kidney development and under diabetic conditions. Sci. Rep. 2016, 6, 37172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, C.; Niewoehner, O.; Duerst, A.; Jinek, M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 2014, 513, 569–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Zhou, Y.; Qi, X.; Chen, J.; Chen, W.; Qiu, G.; Wu, Z.; Wu, N. CRISPR/Cas9 in zebrafish: An efficient combination for human genetic diseases modeling. Hum. Genet. 2017, 136, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.B.; Komor, A.C.; Levy, J.M.; Packer, M.S.; Zhao, K.T.; Liu, D.R. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 2017, 35, 371–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmonem, M.A.; Khalil, R.; Khodaparast, L.; Khodaparast, L.; Arcolino, F.O.; Morgan, J.; Pastore, A.; Tylzanowski, P.; Ny, A.; Lowe, M.; et al. Cystinosis (ctns) zebrafish mutant shows pronephric glomerular and tubular dysfunction. Sci. Rep. 2017, 7, 42583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oltrabella, F.; Pietka, G.; Ramirez, I.B.R.; Mironov, A.; Starborg, T.; Drummond, I.A.; Hinchliffe, K.A.; Lowe, M. The Lowe syndrome protein OCRL1 Is required for endocytosis in the zebrafish pronephric tubule. PLoS Genet. 2015, 11, e1005058. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z. A genetic screen in zebrafish identifies cilia genes as a principal cause of cystic kidney. Development 2004, 131, 4085–4093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Galeano, M.C.R.; Ott, E.; Kaeslin, G.; Kausalya, P.J.; Kramer, C.; Ortiz-Brüchle, N.; Hilger, N.; Metzis, V.; Hiersche, M.; et al. Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease. Nat. Genet. 2017, 49, 1025–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.S.; Hwang, K.S.; Oh, H.W.; Ji-Ae, K.; Kim, H.T.; Cho, H.S.; Lee, J.J.; Yeong Ko, J.; Choi, J.H.; Jeong, Y.M.; et al. IFT46 plays an essential role in cilia development. Dev. Biol. 2015, 400, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Bizet, A.A.; Becker-Heck, A.; Ryan, R.; Weber, K.; Filhol, E.; Krug, P.; Halbritter, J.; Delous, M.; Lasbennes, M.C.; Linghu, B.; et al. Mutations in TRAF3IP1/IFT54 reveal a new role for IFT proteins in microtubule stabilization. Nat. Commun. 2015, 6, 8666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, N.; Lu, J.; Sun, Y. Evidence of a role of inositol polyphosphate 5-phosphatase INPP5E in cilia formation in zebrafish. Vis. Res. 2012, 75, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Kramer-Zucker, A.G.; Wiessner, S.; Jensen, A.M.; Drummond, I.A. Organization of the pronephric filtration apparatus in zebrafish requires nephrin, podocin and the FERM domain protein mosaic eyes. Dev. Biol. 2005, 285, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Hanke, N.; Staggs, L.; Schroder, P.; Litteral, J.; Fleig, S.; Kaufeld, J.; Pauli, C.; Haller, H.; Schiffer, M. “Zebrafishing” for novel genes relevant to the glomerular filtration barrier. Biomed Res. Int. 2013, 2013, 658270. [Google Scholar] [CrossRef] [PubMed]
- Christou-Savina, S.; Beales, P.L.; Osborn, D.P.S. Evaluation of zebrafish kidney function using a fluorescent clearance assay. J. Vis. Exp. 2015, e52540. [Google Scholar] [CrossRef] [PubMed]
- Hanke, N.; King, B.L.; Vaske, B.; Haller, H.; Schiffer, M. A fluorescence-based assay for proteinuria screening in larval zebrafish (Danio rerio). Zebrafish 2015, 12, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Eneman, B.; Elmonem, M.A.; Van Den Heuvel, L.P.; Khodaparast, L.; Khodaparast, L.; Van Geet, C.; Freson, K.; Levtchenko, E. Pituitary adenylate cyclase-activating polypeptide (PACAP) in zebrafish models of nephrotic syndrome. PLoS ONE 2017, 12, e0182100. [Google Scholar] [CrossRef] [PubMed]
- Kotb, A.M.; Simon, O.; Blumenthal, A.; Vogelgesang, S.; Dombrowski, F.; Amann, K.; Zimmermann, U.; Endlich, K.; Endlich, N. Knockdown of ApoL1 in zebrafish larvae affects the glomerular filtration barrier and the expression of nephrin. PLoS ONE 2016, 11, e0153768. [Google Scholar] [CrossRef] [PubMed]
- Kotb, A.M.; Müller, T.; Xie, J.; Anand-Apte, B.; Endlich, K.; Endlich, N. Simultaneous assessment of glomerular filtration and barrier function in live zebrafish. Am. J. Physiol. Ren. Physiol. 2014, 307, F1427–F1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hentschel, D.M.; Park, K.M.; Cilenti, L.; Zervos, A.S.; Drummond, I.; Bonventre, J.V. Acute renal failure in zebrafish: A novel system to study a complex disease. Am. J. Physiol. Ren. Physiol. 2005, 288, F923–F929. [Google Scholar] [CrossRef] [PubMed]
- Rider, S.A.; Tucker, C.S.; Del-Pozo, J.; Rose, K.N.; MacRae, C.A.; Bailey, M.A.; Mullins, J.J. Techniques for the in vivo assessment of cardio-renal function in zebrafish (Danio rerio) larvae. J. Physiol. 2012, 590, 1803–1809. [Google Scholar] [CrossRef] [PubMed]
- Gorgulho, R.; Jacinto, R.; Lopes, S.S.; Pereira, S.A.; Tranfield, E.M.; Martins, G.G.; Gualda, E.J.; Derks, R.J.E.; Correia, A.C.; Steenvoorden, E.; et al. Usefulness of zebrafish larvae to evaluate drug-induced functional and morphological renal tubular alterations. Arch. Toxicol. 2017, 92, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Pottel, H. Measuring and estimating glomerular filtration rate in children. Pediatr. Nephrol. 2017, 32, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Farage, E.; Sugimoto, M.; Anand-Apte, B. A novel transgenic zebrafish model for blood-brain and blood-retinal barrier development. BMC Dev. Biol. 2010, 10, 76. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Boucher, R.C.; Bollig, F.; Englert, C.; Hildebrandt, F. Characterization of mesonephric development and regeneration using transgenic zebrafish. Am. J. Physiol. Ren. Physiol. 2010, 299, F1040–F1047. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Hildebrandt, F. Inducible podocyte injury and proteinuria in transgenic zebrafish. J. Am. Soc. Nephrol. 2012, 23, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Tan, X.F.; Lim, T.K.; Lin, Q.; Gong, Z. Comprehensive and quantitative proteomic analyses of zebrafish plasma reveals conserved protein profiles between genders and between zebrafish and human. Sci. Rep. 2016, 6, 24329. [Google Scholar] [CrossRef] [PubMed]
- Noël, E.S.; Dos Reis, M.; Arain, Z.; Ober, E.A. Analysis of the albumin/α-fetoprotein/afamin/group specific component gene family in the context of zebrafish liver differentiation. Gene Expr. Patterns 2010, 10, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Siegerist, F.; Zhou, W.; Endlich, K.; Endlich, N. 4D in vivo imaging of glomerular barrier function in a zebrafish podocyte injury model. Acta Physiol. 2017, 220, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.; Christensen, E.I.; Birn, H. Megalin and cubilin in proximal tubule protein reabsorption: From experimental models to human disease. Kidney Int. 2016, 89, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Donnai, D.; Barrow, M. Diaphragmatic hernia, exomphalos, absent corpus callosum, hypertelorism, myopia, and sensorineural deafness: A newly recognized autosomal recessive disorder? Am. J. Med. Genet. 1993, 47, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Hultenby, K.; Axelsson, J.; Nordström, J.; He, B.; Wernerson, A.; Lindström, K. Proximal tubular expression patterns of megalin and cubilin in proteinuric nephropathies. Kidney Int. Rep. 2017, 47, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Anzenberger, U.; Bit-avragim, N.; Rohr, S.; Rudolph, F.; Dehmel, B.; Willnow, T.E.; Abdelilah-seyfried, S. Elucidation of megalin/LRP2-dependent endocytic transport processes in the larval zebrafish pronephros. J. Cell Sci. 2006, 2, 2127–2137. [Google Scholar] [CrossRef] [PubMed]
- Kur, E.; Christa, A.; Veth, K.N.; Gajera, C.R.; Andrade-Navarro, M.A.; Zhang, J.; Willer, J.R.; Gregg, R.G.; Abdelilah-Seyfried, S.; Bachmann, S.; et al. Loss of Lrp2 in zebrafish disrupts pronephric tubular clearance but not forebrain development. Dev. Dyn. 2011, 240, 1567–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leheste, J.R.; Rolinski, B.; Vorum, H.; Hilpert, J.; Nykjaer, A.; Jacobsen, C.; Aucouturier, P.; Moskaug, J.O.; Otto, A.; Christensen, E.I.; et al. Megalin knockout mice as an animal model of low molecular weight proteinuria. Am. J. Pathol. 1999, 155, 1361–1370. [Google Scholar] [CrossRef]
- Birn, H.; Vorum, H.; Verroust, P.J.; Moestrup, S.K.; Christensen, E.I. Receptor-associated protein is important for normal processing of megalin in kidney proximal tubules. J. Am. Soc. Nephrol. 2000, 11, 191–202. [Google Scholar] [PubMed]
- Nykjaer, A.; Dragun, D.; Walther, D.; Vorum, H.; Jacobsen, C.; Herz, J.; Melsen, F.; Christensen, E.I.; Willnow, T.E. An endocytic pathway essential for renal uptake and activation of the steroid 25-(OH) vitamin D3. Cell 1999, 96, 507–515. [Google Scholar] [CrossRef]
- Sander, V.; Patke, S.; Sahu, S.; Teoh, C.L.; Peng, Z.; Chang, Y.-T.; Davidson, A.J. The small molecule probe PT-Yellow labels the renal proximal tubules in zebrafish. Chem. Commun. 2015, 51, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, Z.H.; Zhou, L.; Li, Z.; Gui, J.F. Grouper tshβ promoter-driven transgenic zebrafish marks proximal kidney tubule development. PLoS ONE 2014, 9, e97806. [Google Scholar] [CrossRef] [PubMed]
- Cianciolo Cosentino, C.; Skrypnyk, N.I.; Brilli, L.L.; Chiba, T.; Novitskaya, T.; Woods, C.; West, J.; Korotchenko, V.N.; McDermott, L.; Day, B.W.; et al. Histone deacetylase inhibitor enhances recovery after AKI. J. Am. Soc. Nephrol. 2013, 24, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Seiler, C.; Pack, M. Transgenic labeling of the zebrafish pronephric duct and tubules using a promoter from the enpep gene. Gene Expr. Patterns 2011, 11, 118–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kersten, S.; Arjona, F.J. Ion transport in the zebrafish kidney from a human disease angle: Possibilities, considerations, and future perspectives. Am. J. Physiol. Ren. Physiol. 2017, 312, 172–189. [Google Scholar] [CrossRef] [PubMed]
- Guh, Y.; Lin, C.; Hwang, P. Osmoregulation in zebrafish: Ion transport mechanisms and functional regulation. EXCLI J. 2015, 14, 627–659. [Google Scholar] [PubMed]
- Castillo, J.; Crespo, D.; Capilla, E.; Díaz, M.; Chauvigné, F.; Cerdà, J.; Planas, J.V. Evolutionary structural and functional conservation of an ortholog of the GLUT2 glucose transporter gene (SLC2A2) in zebrafish. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1570–R1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.; Su, C.; Hwang, P. Calcium-sensing receptor mediates Ca2+ homeostasis by modulating expression of PTH and stanniocalcin. Endocrinology 2017, 155, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Corre, T.; Arjona, F.J.; Hayward, C.; Youhanna, S.; de Baaij, J.H.F.; Belge, H.; Nägele, N.; Debaix, H.; Lamparter, D.; Macé, A.; et al. Genome-wide meta-analysis unravels interactions between magnesium homeostasis and metabolic phenotypes. J. Am. Soc. Nephrol. 2018, 29, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.; Willer, J.; Marjoram, L.; Bagwell, J.; Mankiewicz, J.; Leshchiner, I.; Goessling, W.; Bagnat, M.; Katsanis, N. Rapid identification of kidney cyst mutations by whole exome sequencing in zebrafish. Development 2013, 140, 4445–4451. [Google Scholar] [CrossRef] [PubMed]
- Bollig, F.; Mehringer, R.; Perner, B.; Hartung, C.; Schäfer, M.; Schartl, M.; Volff, J.N.; Winkler, C.; Englert, C. Identification and comparative expression analysis of a second wt1 gene in zebrafish. Dev. Dyn. 2006, 235, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Xiao, A.; Wecker, A.; McBride, D.A.; Choi, S.Y.; Zhou, W.; Lipschutz, J.H. A possible zebrafish model of polycystic kidney disease: Knockdown of wnt5a causes cysts in zebrafish kidneys. J. Vis. Exp. 2014. [Google Scholar] [CrossRef] [PubMed]
- Gee, H.Y.; Sadowski, C.E.; Aggarwal, P.K.; Porath, J.D.; Yakulov, T.A.; Schueler, M.; Lovric, S.; Ashraf, S.; Braun, D.A.; Halbritter, J.; et al. FAT1 mutations cause a glomerulotubular nephropathy. Nat. Commun. 2016, 7, 10822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, M.Y.; Ma, T.L.; Hung, C.C.; Tian, Y.C.; Chen, Y.C.; Yang, C.W.; Cheng, Y.C. Metformin inhibits cyst formation in a zebrafish model of polycystin-2 deficiency. Sci. Rep. 2017, 7, 7161. [Google Scholar] [CrossRef] [PubMed]
- Borovina, A.; Superina, S.; Voskas, D.; Ciruna, B. Vangl2 directs the posterior tilting and asymmetric localization of motile primary cilia. Nat. Cell Biol. 2010, 12, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Baier, H. Lamina-specific axonal projections in the zebrafish tectum require the type IV collagen Dragnet. Nat. Neurosci. 2007, 10, 1529–1537. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, D.J.; Whitfield, T.T.; Hukriede, N.A.; Lam, W.K.; Weinberg, E.S. The zebrafish dog-eared mutation disrupts eya1, a gene required for cell survival and differentiation in the inner ear and lateral line. Dev. Biol. 2005, 277, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Shmukler, B.E.; Reimold, F.R.; Heneghan, J.F.; Chen, C.Y.; Zhao, T.X.; Paw, B.H.; Alper, S.L. Molecular cloning and functional characterization of zebrafish Slc4a3/Ae3 anion exchanger. Pflugers Arch. J. Physiol. 2014, 466, 1605–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Yan, J.; Tseng, Y.; Chen, R.; Hwang, P. Molecular physiology of an extra-renal Cl− uptake mechanism for body fluid Cl− homeostasis. Int. J. Biol. Sci. 2015, 11, 1190–1203. [Google Scholar] [CrossRef] [PubMed]
- Elizondo, M.R.; Arduini, B.L.; Paulsen, J.; MacDonald, E.L.; Sabel, J.L.; Henion, P.D.; Cornell, R.A.; Parichy, D.M. Defective skeletogenesis with kidney stone formation in dwarf zebrafish mutant for trpm7. Curr. Biol. 2005, 15, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Bolar, N.A.; Golzio, C.; Živná, M.; Hayot, G.; Van Hemelrijk, C.; Schepers, D.; Vandeweyer, G.; Hoischen, A.; Huyghe, J.R.; Raes, A.; et al. Heterozygous loss-of-function SEC61A1 mutations cause autosomal-dominant tubulo-interstitial and glomerulocystic kidney disease with anemia. Am. J. Hum. Genet. 2016, 99, 174–187. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Wei, J.; Li, Y.; Wang, J.; Li, W.; Wang, K.; Hong, X.; Zhao, L.; Chen, C.; Min, J.; et al. Zebrafish slc30a10 deficiency revealed a novel compensatory mechanism of Atp2c1 in maintaining manganese homeostasis. PLoS Genet. 2017, 13, e1006892. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, F.; Mozere, M.; Zdebik, A.A.; Stanescu, H.C.; Tobin, J.; Beales, P.L.; Kleta, R.; Bockenhauer, D.; Russell, C. Generation and validation of a zebrafish model of EAST (epilepsy, ataxia, sensorineural deafness and tubulopathy) syndrome. Dis. Model. Mech. 2013, 6, 652–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sussman, C.R.; Zhao, J.H.; Plata, C.; Lu, J.; Daly, C.; Angle, N.; DiPiero, J.; Drummond, I.A.; Liang, J.O.; Boron, W.F.; et al. Cloning, localization, and functional expression of the electrogenic Na+ bicarbonate cotransporter (NBCe1) from zebrafish. Am. J. Physiol. Physiol. 2009, 297, C865–C875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinkes, B.; Wiggins, R.C.; Gbadegesin, R.; Vlangos, C.N.; Seelow, D.; Nürnberg, G.; Garg, P.; Verma, R.; Chaib, H.; Hoskins, B.E.; et al. Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat. Genet. 2006, 38, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Hentschel, D.M.; Mengel, M.; Boehme, L.; Liebsch, F.; Albertin, C.; Bonventre, J.V.; Haller, H.; Schiffer, M. Rapid screening of glomerular slit diaphragm integrity in larval zebrafish. Am. J. Physiol. Ren. Physiol. 2007, 293, F1746–F1750. [Google Scholar] [CrossRef] [PubMed]
- Perner, B.; Englert, C.; Bollig, F. The Wilms tumor genes wt1a and wt1b control different steps during formation of the zebrafish pronephros. Dev. Biol. 2007, 309, 87–96. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Ebarasi, L.; Zhao, Z.; Guo, J.; Ojala, J.R.M.; Hultenby, K.; De Val, S.; Betsholtz, C.; Tryggvason, K. Lmx1b and FoxC combinatorially regulate podocin expression in podocytes. J. Am. Soc. Nephrol. 2014, 25, 2764–2777. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Gu, S.; Yu, P.; Yu, F.; Feng, C.; Gao, N.; Du, J. Deficiency of smarcal1 causes cell cycle arrest and developmental abnormalities in zebrafish. Dev. Biol. 2010, 339, 89–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, B.R.; Howell, D.N.; Soldano, K.; Garrett, M.E.; Katsanis, N.; Telen, M.J.; Davis, E.E.; Ashley-Koch, A.E. In vivo modeling implicates APOL1 in nephropathy: Evidence for dominant negative effects and epistasis under anemic stress. PLoS Genet. 2015, 11, e1005349. [Google Scholar] [CrossRef]
- Sun, H.; Al-Romaih, K.I.; MacRae, C.A.; Pollak, M.R. Human kidney disease-causing INF2 mutations perturb Rho/Dia signaling in the glomerulus. EBioMedicine 2014, 1, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Wang, D.; Mataleena, P.; He, B.; Niu, D.; Katayama, K.; Xu, X.; Ojala, J.R.M.; Wang, W.; Shu, Q.; et al. Myo1e impairment results in actin reorganization, podocyte dysfunction, and proteinuria in zebrafish and cultured podocytes. PLoS ONE 2013, 8, e72750. [Google Scholar] [CrossRef] [PubMed]
- Gbadegesin, R.A.; Hall, G.; Adeyemo, A.; Hanke, N.; Tossidou, I.; Burchette, J.; Wu, G.; Homstad, A.; Sparks, M.A.; Gomez, J.; et al. Mutations in the gene that encodes the F-actin binding protein anillin cause FSGS. J. Am. Soc. Nephrol. 2014, 25, 1991–2002. [Google Scholar] [CrossRef] [PubMed]
- Ebarasi, L.; He, L.; Hultenby, K.; Takemoto, M.; Betsholtz, C.; Tryggvason, K.; Majumdar, A. A reverse genetic screen in the zebrafish identifies crb2b as a regulator of the glomerular filtration barrier. Dev. Biol. 2009, 334, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.H.; Chang, C.F.; Lai, Y.Y.; Sun, C.Y.; Ding, Y.J.; Tsai, J.N. von Hippel-Lindau gene plays a role during zebrafish pronephros development. In Vitro Cell. Dev. Biol.-Anim. 2015, 51, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Van Rooijen, E.; van de Hoek, G.; Logister, I.; Ajzenberg, H.; Knoers, N.V.A.M.; van Eeden, F.; Voest, E.E.; Schulte-Merker, S.; Giles, R.H. The von Hippel-Lindau gene is required to maintain renal proximal tubule and glomerulus integrity in zebrafish larvae. Nephron 2018, 138, 310–323. [Google Scholar] [CrossRef] [PubMed]
- Yeo, N.C.; O’Meara, C.C.; Bonomo, J.A.; Veth, K.N.; Tomar, R.; Flister, M.J.; Drummond, I.A.; Bowden, D.W.; Freedman, B.I.; Lazar, J.; et al. Shroom3 contributes to the maintenance of the glomerular filtration barrier integrity. Genome Res. 2015, 25, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Mangos, S.; Lam, P.Y.; Zhao, A.; Liu, Y.; Mudumana, S.; Vasilyev, A.; Liu, A.; Drummond, I.A. The ADPKD genes pkd1a/b and pkd2 regulate extracellular matrix formation. Dis. Model. Mech. 2010, 3, 354–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, P.; Sieben, C.J.; Xu, X.; Harris, P.C.; Lin, X. Autophagy activators suppress cystogenesis in an autosomal dominant polycystic kidney disease model. Hum. Mol. Genet. 2017, 26, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Schottenfeld, J.; Sullivan-Brown, J.; Burdine, R.D. Zebrafish curly up encodes a Pkd2 ortholog that restricts left-side-specific expression of southpaw. Development 2007, 134, 1605–1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slanchev, K.; Pütz, M.; Schmitt, A.; Kramer-Zucker, A.; Walz, G. Nephrocystin-4 is required for pronephric duct-dependent cloaca formation in zebrafish. Hum. Mol. Genet. 2011, 20, 3119–3128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otto, E.A.; Schermer, B.; Obara, T.; O’Toole, J.F.; Hiller, K.S.; Mueller, A.M.; Ruf, R.G.; Hoefele, J.; Beekmann, F.; Landau, D.; et al. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat. Genet. 2003, 34, 413–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Dai, J.; Attanasio, M.; Hildebrandt, F. Nephrocystin-3 is required for ciliary function in zebrafish embryos. AJP Ren. Physiol. 2010, 299, F55–F62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schäfer, T.; Pütz, M.; Lienkamp, S.; Ganner, A.; Bergbreiter, A.; Ramachandran, H.; Gieloff, V.; Gerner, M.; Mattonet, C.; Czarnecki, P.G.; et al. Genetic and physical interaction between the NPHP5 and NPHP6 gene products. Hum. Mol. Genet. 2008, 17, 3655–3662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayer, J.A.; Otto, E.A.; O’Toole, J.F.; Nurnberg, G.; Kennedy, M.A.; Becker, C.; Hennies, H.C.; Helou, J.; Attanasio, M.; Fausett, B.V.; et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat. Genet. 2006, 38, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Epting, D.; Slanchev, K.; Engel, C.; Walz, G.; Kramer-Zucker, A. A complex of BBS1 and NPHP7 is required for cilia motility in zebrafish. PLoS ONE 2013, 8, e72549. [Google Scholar] [CrossRef] [PubMed]
- Fukui, H.; Shiba, D.; Asakawa, K.; Kawakami, K.; Yokoyama, T. The ciliary protein Nek8/Nphp9 acts downstream of Inv/Nphp2 during pronephros morphogenesis and left-right establishment in zebrafish. FEBS Lett. 2012, 586, 2273–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otto, E.A.; Hurd, T.W.; Airik, R.; Chaki, M.; Zhou, W.; Stoetzel, C.; Patil, S.B.; Levy, S.; Ghosh, A.K.; Murga-Zamalloa, C.A.; et al. Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat. Genet. 2010, 42, 840–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, R.; Failler, M.; Reilly, M.L.; Garfa-traore, M.; Filhol, E.; Reboul, T.; Bole-feysot, C.; Nitschké, P.; Saunier, S. Functional characterization of tektin-1 in motile cilia and evidence for TEKT1 as a new candidate gene for motile ciliopathies. Hum. Mol. Genet. 2018, 27, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Chaki, M.; Airik, R.; Ghosh, A.K.; Giles, R.H.; Chen, R.; Slaats, G.G.; Wang, H.; Hurd, T.W.; Zhou, W.; Cluckey, A.; et al. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell 2012, 150, 533–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Jin, M.; Hu, R.; Wang, H.; Zhang, F.; Yuan, S.; Cao, Y. The Joubert syndrome protein Inpp5e controls ciliogenesis by regulating phosphoinositides at the apical membrane. J. Am. Soc. Nephrol. 2017, 28, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Logan, C.V.; Mougou-Zerelli, S.; Lee, J.H.; Silhavy, J.L.; Brancati, F.; Iannicelli, M.; Travaglini, L.; Romani, S.; Illi, B.; et al. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat. Genet. 2010, 42, 619–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simms, R.J.; Hynes, A.M.; Eley, L.; Inglis, D.; Chaudhry, B.; Dawe, H.R.; Sayer, J.A. Modelling a ciliopathy: Ahi1 knockdown in model systems reveals an essential role in brain, retinal, and renal development. Cell. Mol. Life Sci. 2012, 69, 993–1009. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.; Simms, R.J.; Abdelhamed, Z.; Dawe, H.R.; Szymanska, K.; Logan, C.V.; Wheway, G.; Pitt, E.; Gull, K.; Knowles, M.A.; et al. A meckelin-filamin a interaction mediates ciliogenesis. Hum. Mol. Genet. 2012, 21, 1272–1286. [Google Scholar] [CrossRef] [PubMed]
- Khanna, H.; Davis, E.E.; Murga-Zamalloa, C.A.; Estrada-Cuzcano, A.; Lopez, I.; Den Hollander, A.I.; Zonneveld, M.N.; Othman, M.I.; Waseem, N.; Chakarova, C.F.; et al. A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies. Nat. Genet. 2009, 41, 739–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantagrel, V.; Silhavy, J.L.; Bielas, S.L.; Swistun, D.; Marsh, S.E.; Bertrand, J.Y.; Audollent, S.; Attié-Bitach, T.; Holden, K.R.; Dobyns, W.B.; et al. Mutations in the cilia gene ARL13B Lead to the classical form of Joubert syndrome. Am. J. Hum. Genet. 2008, 83, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Gorden, N.T.; Arts, H.H.; Parisi, M.A.; Coene, K.L.M.; Letteboer, S.J.F.; van Beersum, S.E.C.; Mans, D.A.; Hikida, A.; Eckert, M.; Knutzen, D.; et al. CC2D2A is mutated in Joubert syndrome and interacts with the ciliopathy-associated basal body protein CEP290. Am. J. Hum. Genet. 2008, 83, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, M.I.; Romio, L.; Castro, S.; Collins, J.E.; Goulding, D.A.; Stemple, D.L.; Woolf, A.S.; Wilson, S.W. Convergent extension movements and ciliary function are mediated by ofd1, a zebrafish orthologue of the human oral-facial-digital type 1 syndrome gene. Hum. Mol. Genet. 2009, 18, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.E.; Zhang, Q.; Liu, Q.; Diplas, B.H.; Davey, L.M.; Hartley, J.; Stoetzel, C.; Szymanska, K.; Ramaswami, G.; Logan, C.V.; et al. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet. 2011, 43, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dibella, L.M.; Park, A.; Sun, Z. Zebrafish Tsc1 reveals functional interactions between the cilium and the TOR pathway. Hum. Mol. Genet. 2009, 18, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Speirs, C.K.; Solnica-Krezel, L.; Ess, K.C. Zebrafish model of tuberous sclerosis complex reveals cell-autonomous and non-cell-autonomous functions of mutant tuberin. Dis. Model. Mech. 2011, 4, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Halbritter, J.; Bizet, A.A.; Schmidts, M.; Porath, J.D.; Braun, D.A.; Gee, H.Y.; McInerney-Leo, A.M.; Krug, P.; Filhol, E.; Davis, E.E.; et al. Defects in the IFT-B component IFT172 cause jeune and mainzer-saldino syndromes in humans. Am. J. Hum. Genet. 2013, 93, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Lunt, S.C.; Haynes, T.; Perkins, B.D. Zebrafish ift57, ift88, and ift172 intraflagellar transport mutants disrupt cilia but do not affect hedgehog signaling. Dev. Dyn. 2009, 238, 1744–1759. [Google Scholar] [CrossRef] [PubMed]
- Schueler, M.; Braun, D.A.; Chandrasekar, G.; Gee, H.Y.; Klasson, T.D.; Halbritter, J.; Bieder, A.; Porath, J.D.; Airik, R.; Zhou, W.; et al. DCDC2 mutations cause a renal-hepatic ciliopathy by disrupting Wnt signaling. Am. J. Hum. Genet. 2015, 96, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, R.; Schmidts, M.; Faqeih, E.; Hashem, A.; Lausch, E.; Holder, I.; Superti-Furga, A.; Mitchison, H.M.; Almoisheer, A.; Alamro, R.; et al. A founder CEP120 mutation in Jeune asphyxiating thoracic dystrophy expands the role of centriolar proteins in skeletal ciliopathies. Hum. Mol. Genet. 2015, 24, 1410–1419. [Google Scholar] [CrossRef] [PubMed]
- Stoetzel, C.; Bär, S.; De Craene, J.O.; Scheidecker, S.; Etard, C.; Chicher, J.; Reck, J.R.; Perrault, I.; Geoffroy, V.; Chennen, K.; et al. A mutation in VPS15 (PIK3R4) causes a ciliopathy and affects IFT20 release from the cis-Golgi. Nat. Commun. 2016, 7, 13586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majumdar, A.; Lun, K.; Brand, M.; Drummond, I.A. Zebrafish no isthmus reveals a role for pax2.1 in tubule differentiation and patterning events in the pronephric primordia. Development 2000, 127, 2089–2098. [Google Scholar]
- Lopez-Rivera, E.; Liu, Y.P.; Verbitsky, M.; Anderson, B.R.; Capone, V.P.; Otto, E.A.; Yan, Z.; Mitrotti, A.; Martino, J.; Steers, N.J.; et al. Genetic drivers of kidney defects in the DiGeorge syndrome. N. Engl. J. Med. 2017, 376, 742–754. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, L.L.; Grimaldi, M.; Kostun, Z.; Wingert, R.A.; Selleck, R.; Davidson, A.J. Wt1a, Foxc1a, and the Notch mediator Rbpj physically interact and regulate the formation of podocytes in zebrafish. Dev. Biol. 2011, 358, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Naylor, R.W.; Przepiorski, A.; Ren, Q.; Yu, J.; Davidson, A.J. HNF1β is essential for nephron segmentation during nephrogenesis. J. Am. Soc. Nephrol. 2013, 24, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.; Taylor, J.C.; Winyard, P.; Baker, K.F.; Sullivan-Brown, J.; Schild, R.; Knuppel, T.; Zurowska, A.M.; Caldas-Alfonso, A.; Litwin, M.; et al. SIX2 and BMP4 mutations associate with anomalous kidney development. J. Am. Soc. Nephrol. 2008, 19, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Brophy, P.D.; Rasmussen, M.; Parida, M.; Bonde, G.; Darbro, B.W.; Hong, X.; Clarke, J.C.; Peterson, K.A.; Denegre, J.; Schneider, M.; et al. A gene implicated in activation of retinoic acid receptor targets is a novel renal agenesis gene in humans. Genetics 2017, 207, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Knapp, M.; Suzuki, K.; Kajioka, D.; Schmidt, J.M.; Winkler, J.; Yilmaz, Ö.; Pleschka, M.; Cao, J.; Kockum, C.C.; et al. ISL1 is a major susceptibility gene for classic bladder exstrophy and a regulator of urinary tract development. Sci. Rep. 2017, 7, 42170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanna-Cherchi, S.; Sampogna, R. V.; Papeta, N.; Burgess, K.E.; Nees, S.N.; Perry, B.J.; Choi, M.; Bodria, M.; Liu, Y.; Weng, P.L.; et al. Mutations in DSTYK and dominant urinary tract malformations. N. Engl. J. Med. 2013, 369, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Strähle, U.; Scholz, S.; Geisler, R.; Greiner, P.; Hollert, H.; Rastegar, S.; Schumacher, A.; Selderslaghs, I.; Weiss, C.; Witters, H.; et al. Zebrafish embryos as an alternative to animal experiments-A commentary on the definition of the onset of protected life stages in animal welfare regulations. Reprod. Toxicol. 2012, 33, 128–132. [Google Scholar] [CrossRef] [PubMed]
- North, T.E.; Goessling, W.; Walkley, C.R.; Lengerke, C.; Kopani, K.R.; Lord, A.M.; Weber, G.J.; Bowman, T.V.; Jang, I.H.; Grosser, T.; et al. Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature 2007, 447, 1007–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagannathan-Bogdan, M.; Zon, L.I. Hematopoiesis. Development 2013, 140, 2463–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gut, P.; Baeza-Raja, B.; Andersson, O.; Hasenkamp, L.; Hsiao, J.; Hesselson, D.; Akassoglou, K.; Verdin, E.; Hirschey, M.D.; Stainier, D.Y.R. Whole-organism screening for gluconeogenesis identifies activators of fasting metabolism. Nat. Chem. Biol. 2013, 9, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Semanchik, N.; Lee, S.H.; Somlo, S.; Barbano, P.E.; Coifman, R.; Sun, Z. Chemical modifier screen identifies HDAC inhibitors as suppressors of PKD models. Proc. Natl. Acad. Sci. USA 2009, 106, 21819–21824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grampa, V.; Delous, M.; Zaidan, M.; Odye, G.; Thomas, S.; Elkhartoufi, N.; Filhol, E.; Niel, O.; Silbermann, F.; Lebreton, C.; et al. Novel NEK8 mutations cause severe syndromic renal cystic dysplasia through YAP dysregulation. PLoS Genet. 2016, 12, e1005894. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; Tsai, I.C.; Morleo, M.; Oh, E.C.; Leitch, C.C.; Massa, F.; Lee, B.H.; Parker, D.S.; Finley, D.; Zaghloul, N.A.; et al. Ciliopathy proteins regulate paracrine signaling by modulating proteasomal degradation of mediators. J. Clin. Investig. 2014, 124, 2059–2070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstone, J.V.; Mcarthur, A.G.; Kubota, A.; Zanette, J.; Parente, T.; Jönsson, M.E.; Nelson, D.R.; Stegeman, J.J. Identification and developmental expression of the full complement of Cytochrome P450 genes in zebrafish. BMC Genom. 2010, 11, 643. [Google Scholar] [CrossRef] [PubMed]
- Kithcart, A.; MacRae, C.A. Using zebrafish for high-throughput screening of novel cardiovascular drugs. JACC Basic Transl. Sci. 2017, 2, 1–12. [Google Scholar] [CrossRef]
- Glasauer, S.M.K.; Neuhauss, S.C.F. Whole-genome duplication in teleost fishes and its evolutionary consequences. Mol. Genet. Genom. 2014, 289, 1045–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Higgins, P.J.; Zhang, W. Development and diseases of the collecting duct system. Results Probl. Cell Differ. 2017, 60, 165–203. [Google Scholar] [PubMed]
- Tingaud-Sequeira, A.; Calusinska, M.; Finn, R.N.; Chauvigné, F.; Lozano, J.; Cerdà, J. The zebrafish genome encodes the largest vertebrate repertoire of functional aquaporins with dual paralogy and substrate specificities similar to mammals. BMC Evol. Biol. 2010, 10, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanukoglu, I.; Hanukoglu, A. Epithelial sodium channel (ENaC) family: Phylogeny, structure-function, tissue distribution, and associated inherited diseases. Gene 2016, 579, 95–132. [Google Scholar] [CrossRef] [PubMed]
- Vliegenthart, A.D.B.; Tucker, C.S.; Del Pozo, J.; Dear, J.W. Zebrafish as model organisms for studying drug-induced liver injury. Br. J. Clin. Pharmacol. 2014, 78, 1217–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoriello, C.; Zon, L.I. Hooked! modeling human disease in zebrafish. J. Clin. Investig. 2012, 122, 2337–2343. [Google Scholar] [CrossRef] [PubMed]
- McCampbell, K.K.; Springer, K.N.; Wingert, R.A. Atlas of cellular dynamics during zebrafish adult kidney regeneration. Stem Cells Int. 2015, 2015, 547636. [Google Scholar] [CrossRef] [PubMed]
- Warejko, J.K.; Tan, W.; Daga, A.; Schapiro, D.; Lawson, J.A.; Shril, S.; Lovric, S.; Ashraf, S.; Rao, J.; Hermle, T.; et al. Whole exome sequencing of patients with steroid-resistant nephrotic syndrome. Clin. J. Am. Soc. Nephrol. 2018, 13, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Daga, A.; Majmundar, A.J.; Braun, D.A.; Gee, H.Y.; Lawson, J.A.; Shril, S.; Jobst-Schwan, T.; Vivante, A.; Schapiro, D.; Tan, W.; et al. Whole exome sequencing frequently detects a monogenic cause in early onset nephrolithiasis and nephrocalcinosis. Kidney Int. 2018, 93, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Heidet, L.; Morinière, V.; Henry, C.; De Tomasi, L.; Reilly, M.L.; Humbert, C.; Alibeu, O.; Fourrage, C.; Bole-Feysot, C.; Nitschké, P.; et al. Targeted exome sequencing identifies PBX1 as involved in monogenic congenital anomalies of the kidney and urinary tract. J. Am. Soc. Nephrol. 2017, 28, 2901–2914. [Google Scholar] [CrossRef] [PubMed]
- Panizzi, J.R.; Becker-Heck, A.; Castleman, V.H.; Al-Mutairi, D.A.; Liu, Y.; Loges, N.T.; Pathak, N.; Austin-Tse, C.; Sheridan, E.; Schmidts, M.; et al. CCDC103 mutations cause primary ciliary dyskinesia by disrupting assembly of ciliary dynein arms. Nat. Genet. 2012, 44, 714–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austin-Tse, C.; Halbritter, J.; Zariwala, M.A.; Gilberti, R.M.; Gee, H.Y.; Hellman, N.; Pathak, N.; Liu, Y.; Panizzi, J.R.; Patel-King, R.S.; et al. Zebrafish ciliopathy screen plus human mutational analysis identifies C21orf59 and CCDC65 defects as causing primary ciliary dyskinesia. Am. J. Hum. Genet. 2013, 93, 672–686. [Google Scholar] [CrossRef] [PubMed]
- Bedell, V.M.; Westcot, S.E.; Ekker, S.C. Lessons from morpholino-based screening in zebrafish. Brief. Funct. Genom. 2011, 10, 181–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baxendale, S.; van Eeden, F.; Wilkinson, R. The Power of Zebrafish in personalised medicine. Adv. Exp. Med. Biol. 2017, 1007, 179–197. [Google Scholar] [PubMed]
- Roy, B.; Zhao, J.; Yang, C.; Luo, W.; Xiong, T.; Li, Y.; Fang, X.; Gao, G.; Singh, C.O.; Madsen, L.; et al. CRISPR/cascade 9-mediated genome editing-challenges and opportunities. Front. Genet. 2018, 9, 240. [Google Scholar] [CrossRef] [PubMed]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Prew, M.S.; Tsai, S.Q.; Topkar, V.V.; Nguyen, N.T.; Zheng, Z.; Gonzales, A.P.W.; Li, Z.; Peterson, R.T.; Yeh, J.R.J.; et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 2015, 523, 481–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canzar, S.; Salzberg, S.L. Short Read Mapping: An Algorithmic Tour. Proc. IEEE 2017, 105, 436–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abadi, S.; Yan, W.X.; Amar, D.; Mayrose, I. A machine learning approach for predicting CRISPR-Cas9 cleavage efficiencies and patterns underlying its mechanism of action. PLoS Comput. Biol. 2017, 13, e1005807. [Google Scholar] [CrossRef] [PubMed]
- Listgarten, J.; Weinstein, M.; Kleinstiver, B.P.; Sousa, A.A.; Joung, J.K.; Crawford, J.; Gao, K.; Hoang, L.; Elibol, M.; Doench, J.G.; et al. Prediction of off-target activities for the end-to-end design of CRISPR guide RNAs. Nat. Biomed. Eng. 2018, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Frock, R.L.; Hu, J.; Meyers, R.M.; Ho, Y.J.; Kii, E.; Alt, F.W. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat. Biotechnol. 2015, 33, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015, 33, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Bae, S.; Park, J.; Kim, E.; Kim, S.; Yu, H.R.; Hwang, J.; Kim, J.I.; Kim, J.S. Digenome-seq: Genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat. Methods 2015, 12, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Cameron, P.; Fuller, C.K.; Donohoue, P.D.; Jones, B.N.; Thompson, M.S.; Carter, M.M.; Gradia, S.; Vidal, B.; Garner, E.; Slorach, E.M.; et al. Mapping the genomic landscape of CRISPR-Cas9 cleavage. Nat. Methods 2017, 14, 600–606. [Google Scholar] [CrossRef] [PubMed]
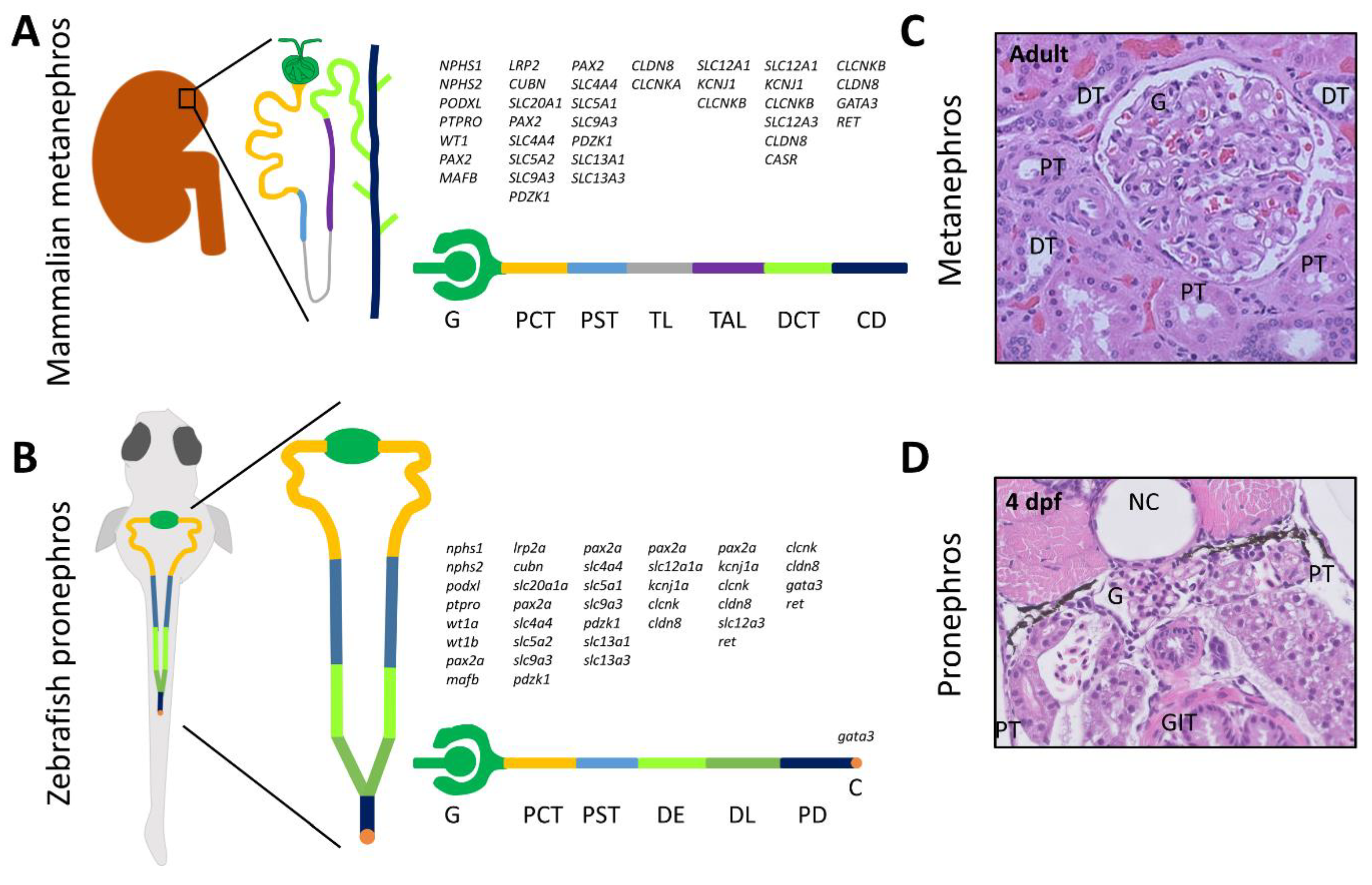




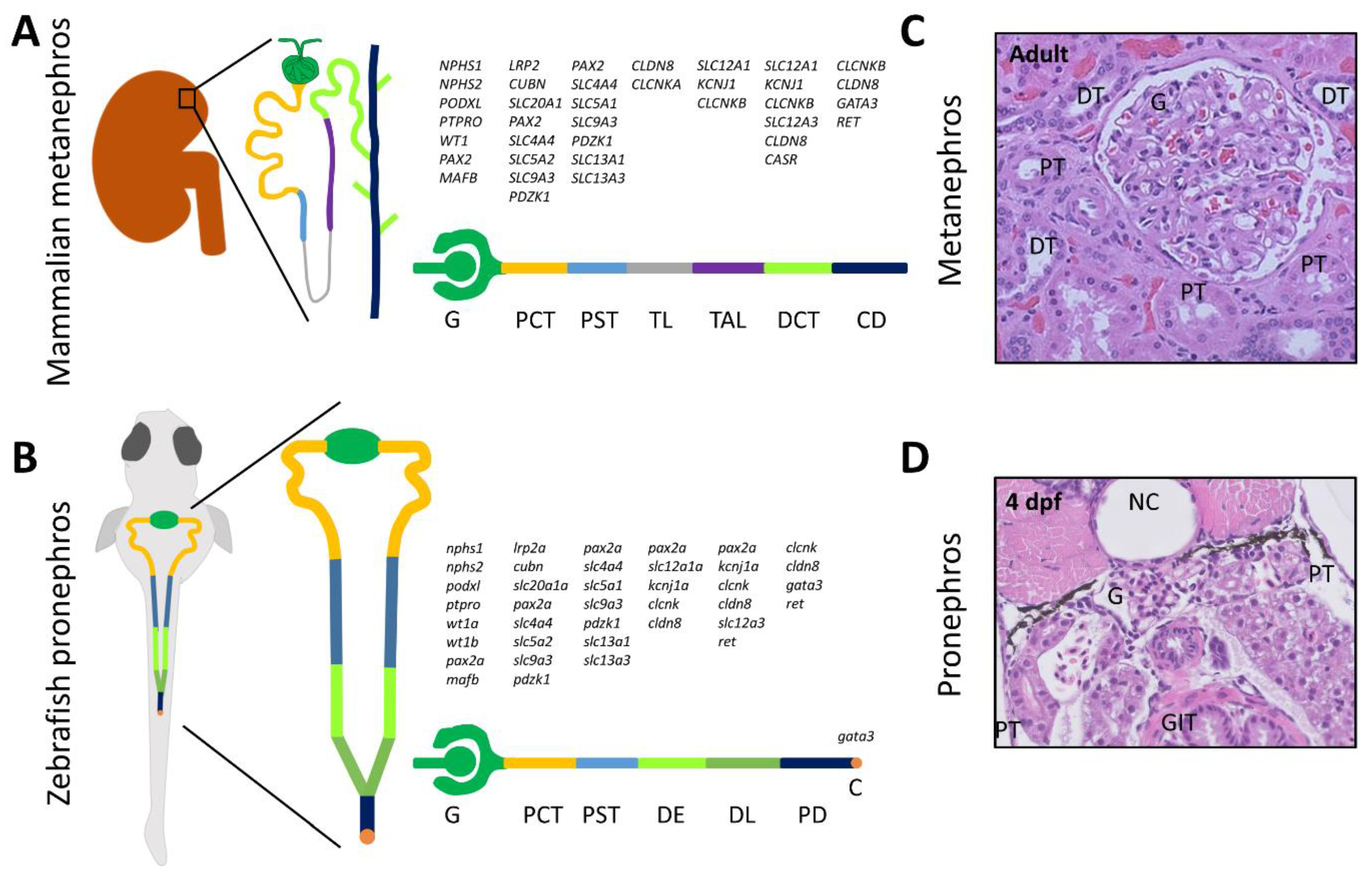{kind=link}
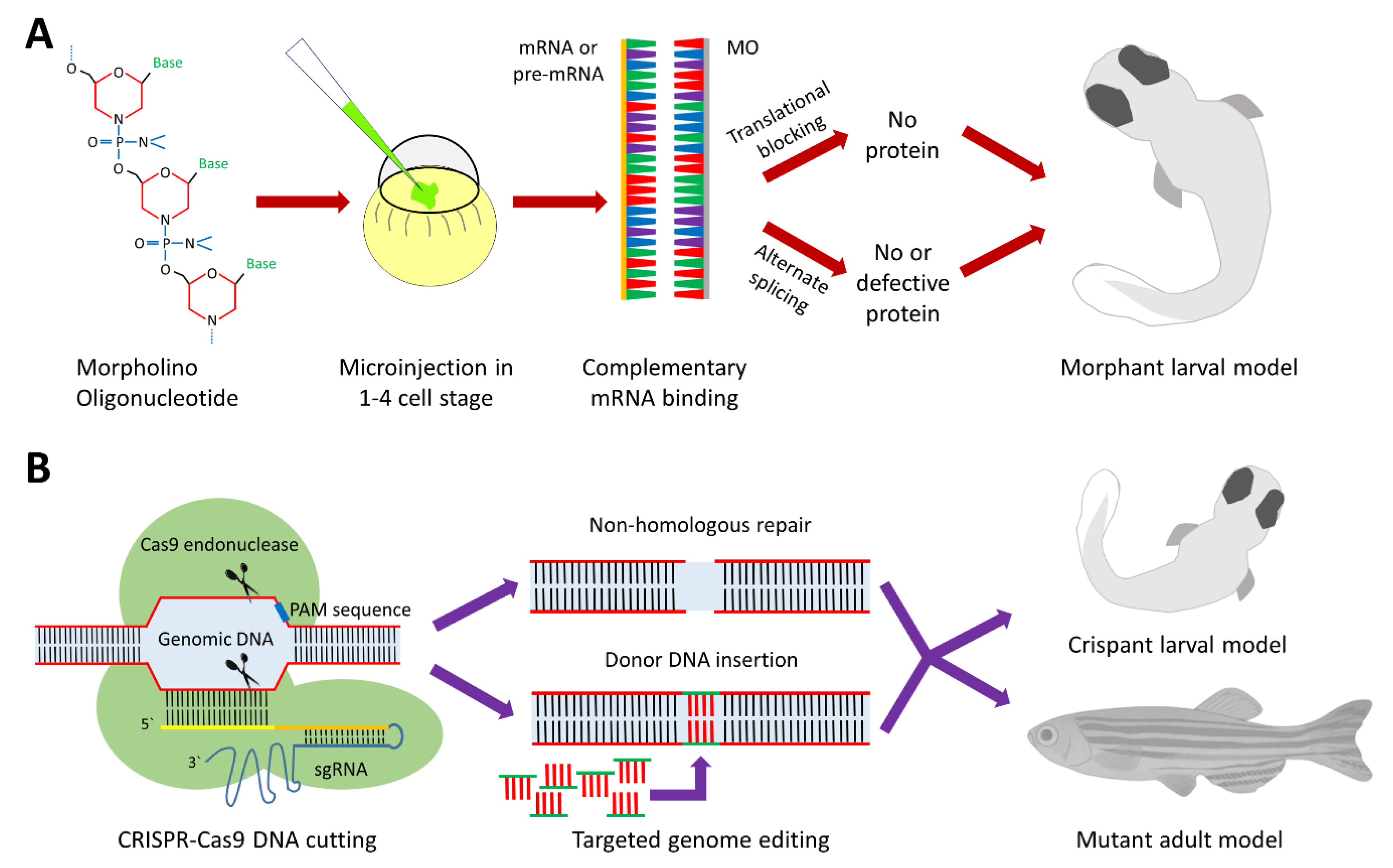{kind=link}
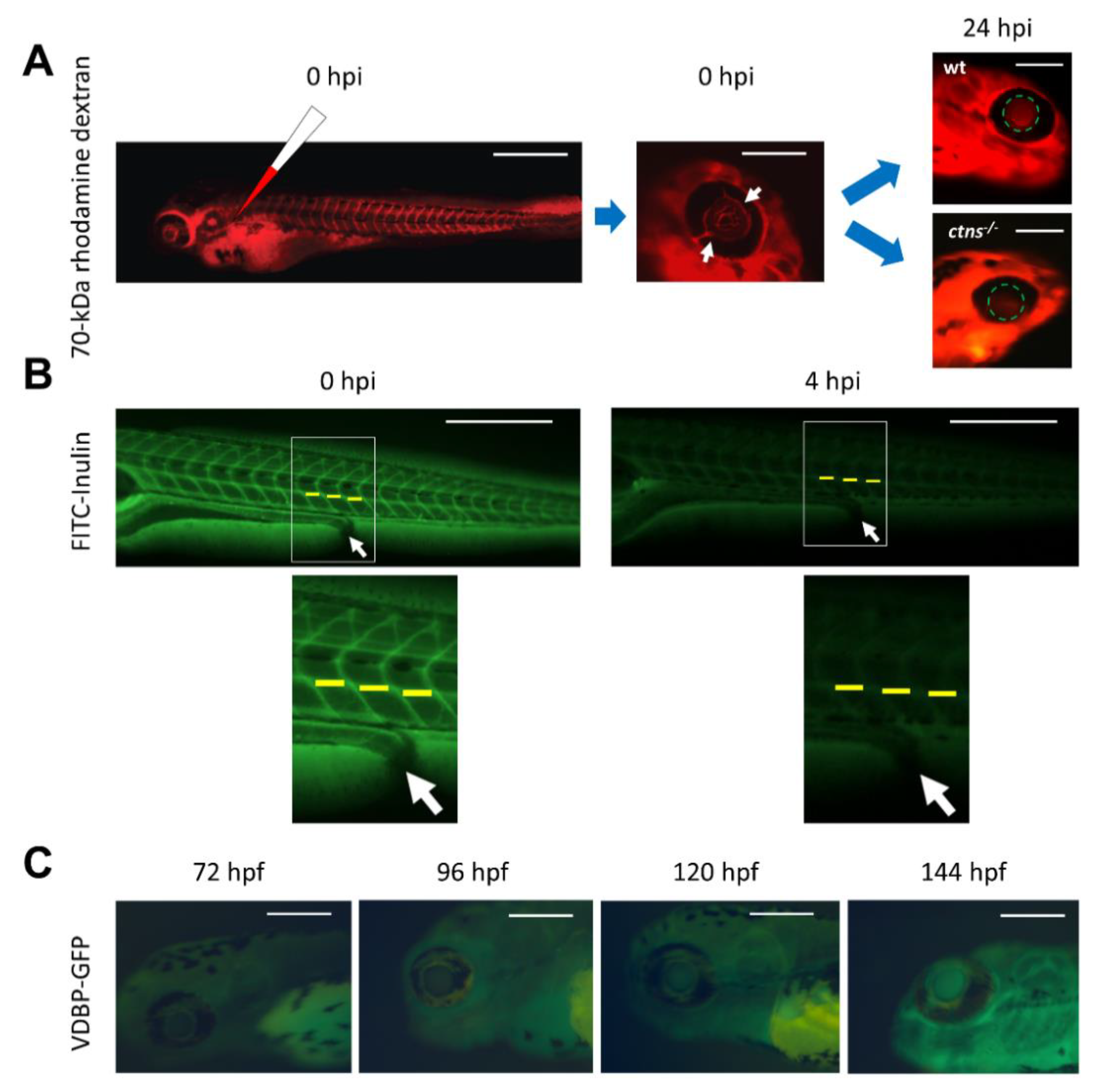{kind=link}
{kind=link}
| Forward Genetics | Reverse Genetics | |||
|---|---|---|---|---|
| ENU Mutagenesis | Retroviral Insertion | MO | CRISPR-Cas9 | |
| Technique first described in zebrafish | Grunwald and Streisinger (1992) [21] | Lin et al. (1994) [23] | Nasevicius and Ekker (2000) [26] | Hwang et al. (2013) [27] |
| Genetic target | Genomic DNA | Genomic DNA | mRNA | Genomic DNA |
| Stage of inducing mutagenesis | Adult males | 512–2048 cell stage (blastulae) | 1–4 cell stage | 1 cell stage |
| Mutation site | Random | Random | No DNA mutations | specific DNA sequence |
| Mutational effect | Mainly deficiency | Mainly deficiency | Deficiency | Deficiency/Gain |
| Difficulty of confirming the mutant genotype | Difficult | Less difficult | Easy | Easy |
| Efficiency of mutagenesis | Medium | Low | High | High |
| Mutant model | Permanent | Permanent | Transient | Permanent |
| Time, effort and resources | +++ | ++++ | + | ++ |
| Off-target effects | + | + | +++ | + |
| Disease | OMIM | Heredity | Gene | Methodology | Phenotype | Ref. |
|---|---|---|---|---|---|---|
| Tubular disorders | ||||||
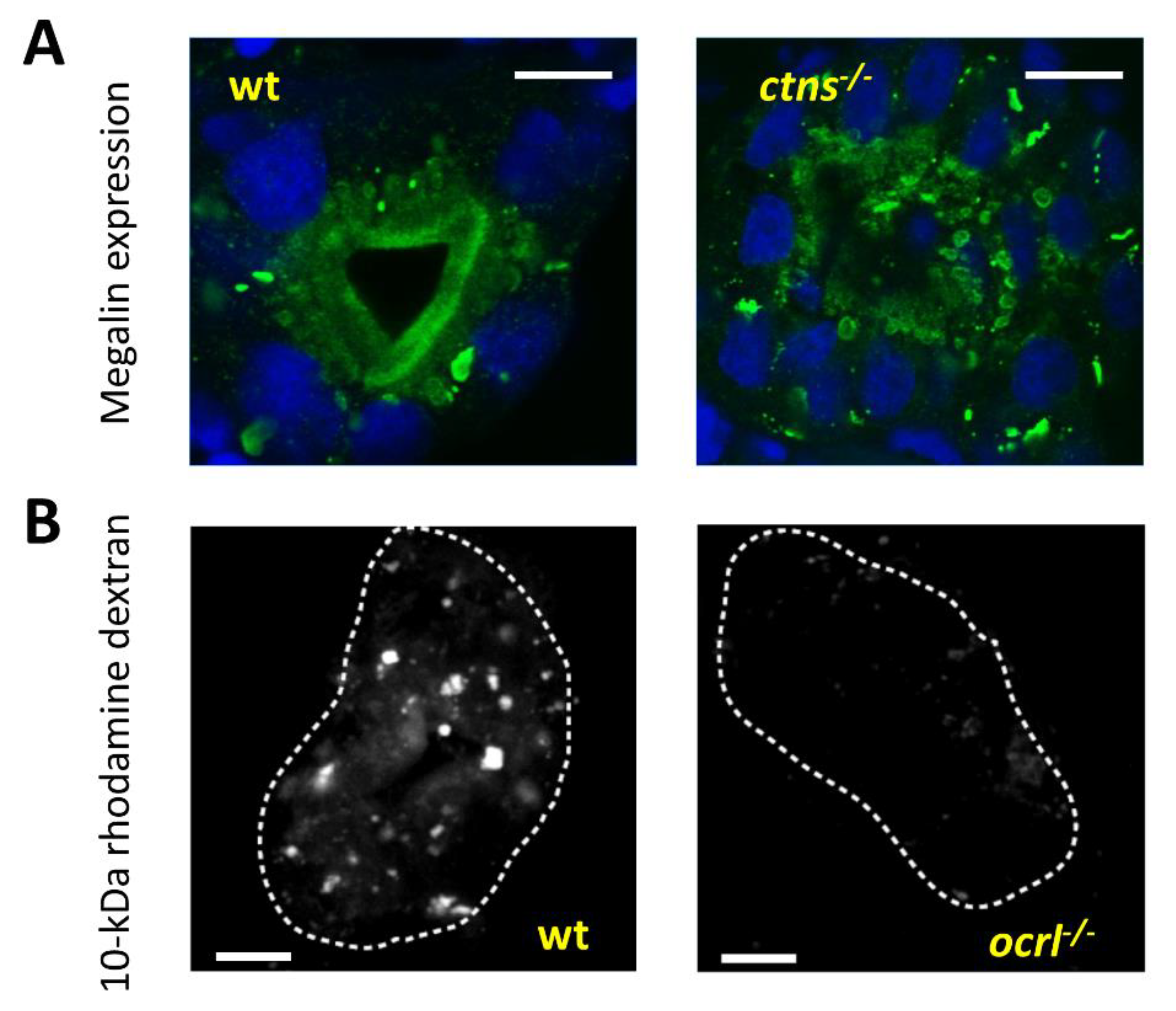
| Cystinosis | 219800 | AR | ctns | MO, ENU | Cystine accumulation, increased embryonic mortality, delayed development, apoptosis, defective glomerular permeability, altered tubular reabsorption, and megalin expression | [48] |
| Donnai–Barrow syndrome | 222448 | AR | lrp2a,b | MO, ENU | Defective endocytosis in larvae and bug eyes in adults | [75,76] |
| Lowe syndrome | 300555 | AR | ocrl | MO, Retroviral insertion | Increased embryonic mortality, delayed development, impaired pronephric endocytosis, altered megalin subcellular localization in proximal tubules | [49] |
| ADTKD | 617056 | AD | sec61a1 | MO, CRISPR | Convolution defects of the pronephric tubules, pronephric tubular atrophy | [100] |
| Hypermanganesemia with dystonia type 1 | 613280 | AR | slc30a10 | CRISPR | Hypermanganesemia and fatty liver in larvae and dystonia, cirrhosis, and neurological deficits in adults | [101] |
| SeSAME syndrome | 612780 | AR | kcnj10a | MO | Dilated pronephric duct, pericardial edema, neurological manifestation | [102] |
| Proximal RTA with ocular anomalies | 604278 | AR | slc4a4 | MO | Impaired renal electrolyte balance, edema, altered brain and eye development | [103] |
| Familial Hypocalciuric Hypercalcemia type I | 145980 | AD | casr | MO | Increased calcium content, impaired regulation of calcium metabolism | [87] |
| Hypomagnesemia * | ------------ | ------------ | arl15b | MO | Pronephric magnesium wasting, cardiovascular impairments, poorly metabolized yolk | [88] |
| Glomerular disorders | ||||||
| SRNS1 (Finish type) | 256300 | AR | nphs1 | MO | Ultrastructural glomerular damage, proteinuria, edema, increased embryonic mortality | [55] |
| SRNS2 | 600995 | AR | nphs2 | MO | Ultrastructural glomerular damage, proteinuria, edema, increased embryonic mortality | [55] |
| SRNS3 | 610725 | AR | plce1 | MO | Ultrastructural glomerular damage, proteinuria, edema | [104] |
| SRNS4 | 607832 | AR, AD | cd2ap | MO | Ultrastructural glomerular damage, proteinuria, edema | [105] |
| Denys–Drash syndrome | 194080 | AD | wt1a,b | MO | Ultrastructural glomerular damage, proteinuria, edema, deformity, high embryonic mortality | [106] |
| Nail-patella syndrome | 161200 | AD | lmx1b | MO | Ultrastructural glomerular damage, proteinuria, edema | [107] |
| Schimke Immuno-Osseous Dysplasia | 242900 | AR | smarcal1 | MO | Increased embryonic mortality, delayed development, increased apoptosis, edema, deformity | [108] |
| FSGS4 | 612551 | AR | apol1 | MO | Ultrastructural glomerular damage, proteinuria, edema | [109] |
| FSGS5 | 613237 | AD | inf2 | MO | Ultrastructural glomerular damage, proteinuria, edema | [110] |
| FSGS6 | 614131 | AR | myo1e | MO | Pericardial edema, pronephric cysts | [111] |
| FSGS8 | 616032 | AD | anln | MO | Ultrastructural glomerular damage, proteinuria, edema | [112] |
| FSGS9 | 616220 | AR | crb2b | MO | Ultrastructural glomerular damage, proteinuria, edema | [113] |
| Von Hippel–Lindau disease | 193300 | AD | vhl | MO, ENU | Ultrastructural glomerular damage, proteinuria, edema, proximal tubular damage, increased angiogenesis | [114,115] |
| Glomerulopathy * | ------------ | ------------ | shroom3 | MO | Ultrastructural glomerular damage, proteinuria, edema, gastrulation defects | [116] |
| Glomerulopathy * | ------------ | ------------ | fat1 | MO | Impaired podocyte migration, glomerular defects, pronephric cysts | [92] |
| Renal ciliopathies | ||||||
| ADPKD | 173900 | AD | pkd1a,b | MO, TALENs | Dorsal axis curvature in morphants and hydrocephalus, craniofacial defects, and pronephric cysts in both | [117,118] |
| 613095 | AD | pkd2 | MO, ENU | Dorsal axis curvature, hydrocephalus, pronephric cysts in morphants, and organ laterality defects in both | [117,119] | |
| ARPKD | 617610 | AR | dzip1l | MO, CRISPR | Pronephric cysts, curved body, hydrocephalus, otolith defects | [51] |
| NPHP1 | 256100 | AR | nphp1 | MO | Pronephric cysts, duct dilatations, deformity | [120] |
| NPHP2 | 602088 | AR | invs | MO | Pronephric cysts, ventral axis curvature, randomization of heart looping | [121] |
| NPHP3 | 604837 | AR | nphp3 | MO | Pronephric cysts, curved body, hydrocephalus, left right asymmetry | [122] |
| NPHP4 | 606966 | AR | nphp4 | MO | Pronephric cysts, curved body, hydrocephalus, pericardial edema | [120] |
| NPHP5 | 609254 | AR | iqcb1 | MO | Pronephric cysts, curved body, hydrocephalus, pericardial edema | [123] |
| NPHP6 | 610188 | AR | cep290 | MO | Pronephric cysts, curved body, hydrocephalus, retinitis pigmentosa, cerebellar defects | [124] |
| NPHP7 | 611498 | AR | glis2 | MO | Pronephric cysts, convergent extension defects, curved body, hydrocephalus, abnormal cardiac looping | [125] |
| NPHP9 | 613824 | AR | nek8 | MO | Pronephric cysts, developmental delay, curved body, abnormal cardiac looping | [126] |
| NPHP10 | 613615 | AR | sdccag8 | MO | Pronephric cysts, developmental delay, curved body, hydrocephalus | [127] |
| NPHP13 | 614377 | AR | wdr19 | MO | Pronephric cysts, hydrocephalus, microphthalmia, body curvature | [128] |
| NPHP15 | 614845 | AR | cep164 | MO | Ventral body axis curvature, abnormal heart looping, pronephric tubule cysts, hydrocephalus heart looping | [129] |
| SLNS9 | 616629 | AR | traf3ip1 | MO | Pronephric cysts, microphthalmia, retinitis pigmentosa | [53] |
| JBTS 1 | 213300 | AR | inpp5e | MO, CRISPR | Left–right body axis asymmetry, microphthalmia and disruption of apicobasal polarity in morphants and pronephric cysts, pericardial effusion and body curvature in both morphants and mutants | [54,130] |
| JBTS 2 | 608091 | AR | tmem216 | MO | Pronephric cysts, body axis asymmetry, gastrulation defects | [131] |
| JBTS 3 | 608629 | AR | ahi1 | MO | Pronephric cysts, cardiac asymmetry, brain, eye and ear abnormalities | [132] |
| JBTS 6 | 610688 | AR | tmem67 | MO | Pronephric cysts, pronephric duct dilatation, notochord anomalies, abnormal eye formation | [133] |
| JBTS 7 | 611560 | AR | rpgrip1l | MO | Gastrulation defects, shortened body axis, thin somites with broad lateral extensions, minor kinking of the notochord, underdeveloped anterior structures | [134] |
| JBTS 8 | 612291 | AR | arl13b | Retroviral insertion | Pronephric cysts, curved body | [135] |
| JBTS 9 | 612285 | AR | cc2d2a | ENU | Pronephric cysts, pericardial edema, curved body | [136] |
| JBTS 10 | 300804 | XLR | ofd1 | MO | Curved body, hydrocephalus, pericardial edema, randomized laterality of brain and heart | [137] |
| JBTS 11 | 613820 | AD, AR | ttc21b | MO | Gastrulation defects, shortened body axis, kinking of the notochord, broadening of somites | [138] |
| BBS 1 | 209900 | AR, DR | bbs1 | MO | Pronephric cysts, convergent extension defects, curved body, hydrocephalus, abnormal heart looping | [125] |
| TSC 1 | 191100 | AD | tsc1a | MO | Pronephric cysts, asymmetry defects, curved body | [139] |
| TSC 2 | 613254 | AD | tsc2 | ENU | Abnormal brain development, increased embryonic mortality, enlarged liver, abnormal cilia | [140] |
| Short-rib thoracic dysplasia with or without polydactyly | 615630 | AR | ift172 | MO, Retroviral insertion | Ventral body-axis curvature, formation of renal cysts, cartilage defects with hypoplasia | [141,142] |
| 611263 | AR | ift80 | MO | Abnormal brain development, increased embryonic mortality, enlarged liver, abnormal cilia | [141] | |
| ------------ | AR | tekt1 | MO | Ventral body-axis curvature, formation of renal cysts, cartilage defects with hypoplasia | [128] | |
| Renal-hepatic ciliopathy | 616217 | AR | dcdc2 | MO | Pronephric cysts, hydrocephalus, ventralized body axis, pericardial edema | [143] |
| Jeune thoracic dystrophy | 616300 | AR | cep120 | MO | Abnormal body curvature, hydrocephalus, otolith defects, abnormal renal and craniofacial development | [144] |
| Ciliopathy * | ------------ | ------------ | pik3r4 | MO | Pronephric cysts, hydrocephalus, curved body | [145] |
| CAKUT | ||||||
| Papillorenal syndrome | 616002 | AD | pax2a | ENU | Abnormal pronephros development, defective tubular differentiation and patterning | [146] |
| DiGeorge syndrome | 188400 | AD | crkl, aifm3, snap29 | MO, CRISPR | Major convolution defects, reduced length of pronephric tubules | [147] |
| Denys–Drash syndrome | 194080 | AD | wt1a | MO | Disruption of glomerular morphogenesis and differentiation | [148] |
| Renal cysts and diabetes syndrome | 137920 | AD | hnf1ba,b | MO, Retroviral insertion | Abnormal nephron segmentation, tubular dysfunction | [149] |
| Renal hypodysplasia | 604994 | AD | six2 | MO | Altered renal morphology, dorsalization of the embryo | [150] |
| Renal hypodysplasia Bilateral renal agenesis * | 112262 | AD | bmp4 | MO | Altered renal morphology, ventralization of the embryo | [150] |
| ------------ | AD | greb1l | ENU, MO, CRISPR | Dilated tubules, deformed junction between proximal convoluted tubules and the neck, pronephric cysts, pericardial edema, early mortality | [151] | |
| Classic bladder exstrophy | 600057 | XLR | isl1 | MO | Abnormal urinary tract development | [152] |
| CAKUT1 | 612666 | AD | dstyk | MO | Cloacal deformities, growth retardation, pericardial edema, small fins, abnormal jaw development | [153] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elmonem, M.A.; Berlingerio, S.P.; Van den Heuvel, L.P.; De Witte, P.A.; Lowe, M.; Levtchenko, E.N. Genetic Renal Diseases: The Emerging Role of Zebrafish Models. Cells 2018, 7, 130. https://doi.org/10.3390/cells7090130
Elmonem MA, Berlingerio SP, Van den Heuvel LP, De Witte PA, Lowe M, Levtchenko EN. Genetic Renal Diseases: The Emerging Role of Zebrafish Models. Cells. 2018; 7(9):130. https://doi.org/10.3390/cells7090130
Chicago/Turabian StyleElmonem, Mohamed A., Sante Princiero Berlingerio, Lambertus P. Van den Heuvel, Peter A. De Witte, Martin Lowe, and Elena N. Levtchenko. 2018. "Genetic Renal Diseases: The Emerging Role of Zebrafish Models" Cells 7, no. 9: 130. https://doi.org/10.3390/cells7090130
APA StyleElmonem, M. A., Berlingerio, S. P., Van den Heuvel, L. P., De Witte, P. A., Lowe, M., & Levtchenko, E. N. (2018). Genetic Renal Diseases: The Emerging Role of Zebrafish Models. Cells, 7(9), 130. https://doi.org/10.3390/cells7090130