The Autophagy-Lysosomal Pathways and Their Emerging Roles in Modulating Proteostasis in Tumors


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Proteostasis in Cancer
2.1. Proteostasis Needs to Be Balanced by Modulation of PNs
2.2. Enhanced Regulation of PNs Is a New Hallmark of Cancer
3. Classical Proteostasis Networks in Cancer
3.1. Protein Synthesis
3.2. Molecular Chaperones
3.3. Trafficking Modules
3.4. Unfolded Protein Response (UPR)
3.5. Stress Responsive Pathways (SRPs)
3.6. Ubiquitin Proteasome System (UPS)
3.7. Secretions
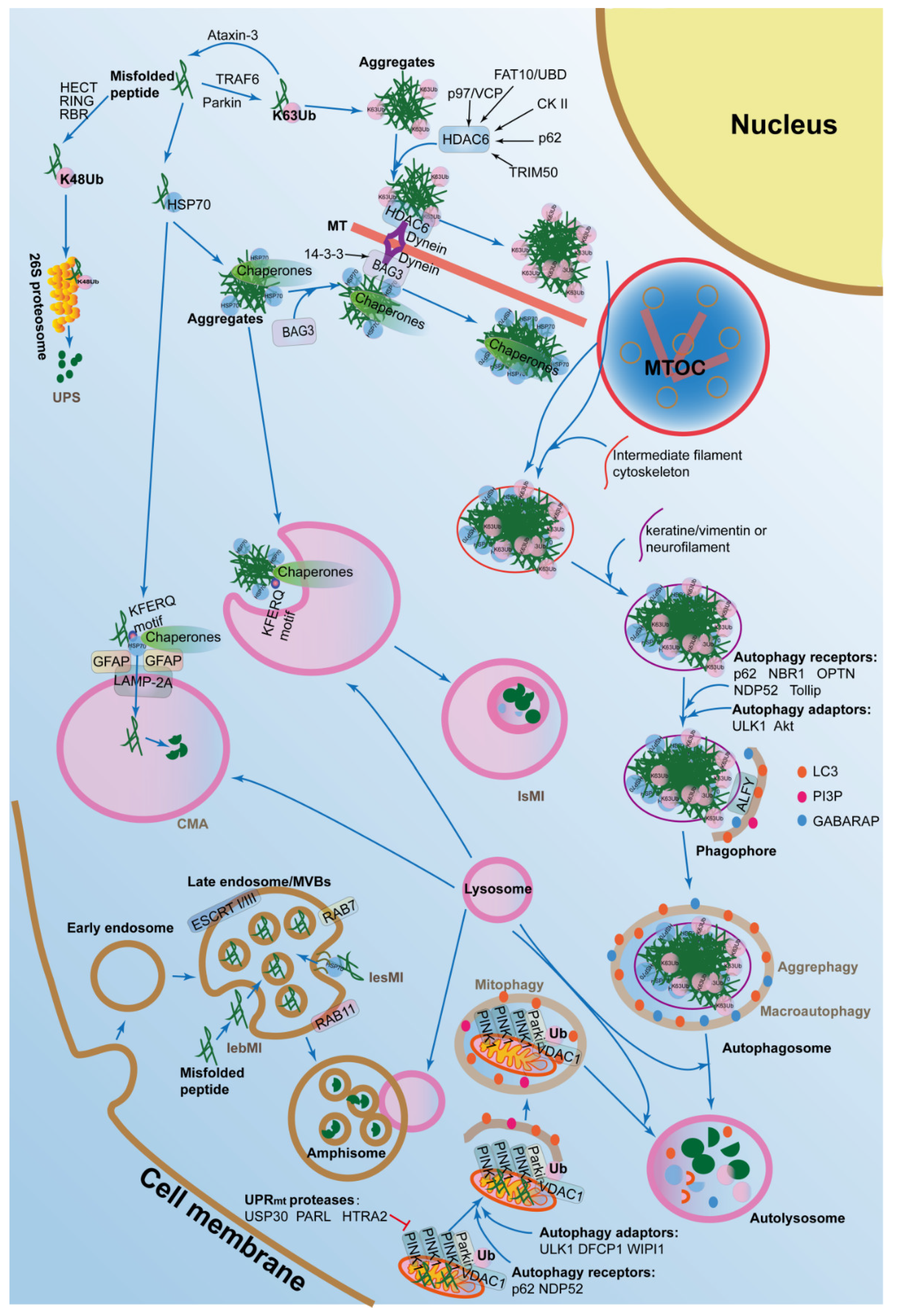
4. The Autophagy-Lysosomal Pathways in Proteostasis
4.1. Macroautophagy and Aggrephagy in Proteostasis
4.2. CMA and Aggrephagy in Proteostasis
4.3. Microautophagy in Proteostasis
4.4. Mitophagy in Proteostasis
5. Proteostasis Regulated by the ALPs Are Important for Tumor Malignancy
6. The ALPs Have Crosstalk with Other PNs in the Regulation of Proteostasis in Cancers
7. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Liu, Y.; Ye, Y. Proteostasis regulation at the endoplasmic reticulum: A new perturbation site for targeted cancer therapy. Cell Res. 2011, 21, 867–883. [Google Scholar] [CrossRef]
- Carvalho, A.S.; Rodriguez, M.S.; Matthiesen, R. Review and Literature Mining on Proteostasis Factors and Cancer. Methods Mol. Biol. (Clifton NJ) 2016, 1449, 71–84. [Google Scholar] [CrossRef]
- Press, M.; Jung, T.; Konig, J.; Grune, T.; Hohn, A. Protein aggregates and proteostasis in aging: Amylin and beta-cell function. Mech. Ageing Dev. 2018. [Google Scholar] [CrossRef]
- Morimoto, R.I.; Cuervo, A.M. Proteostasis and the aging proteome in health and disease. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. 1), S33–S38. [Google Scholar] [CrossRef]
- Amanullah, A.; Upadhyay, A.; Joshi, V.; Mishra, R.; Jana, N.R.; Mishra, A. Progressing neurobiological strategies against proteostasis failure: Challenges in neurodegeneration. Prog. Neurobiol. 2017, 159, 1–38. [Google Scholar] [CrossRef]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science (N. Y.) 2008, 319, 916–919. [Google Scholar] [CrossRef]
- Dufey, E.; Urra, H.; Hetz, C. ER proteostasis addiction in cancer biology: Novel concepts. Semin. Cancer Biol. 2015, 33, 40–47. [Google Scholar] [CrossRef]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. JCB 2018, 217, 51–63. [Google Scholar] [CrossRef]
- Deshaies, R.J. Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Biol. 2014, 12, 94. [Google Scholar] [CrossRef]
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Hartl, F.U. Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. [Google Scholar] [CrossRef]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science (N. Y.) 1973, 181, 223–230. [Google Scholar] [CrossRef]
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef]
- Natalello, A.; Mattoo, R.U.; Priya, S.; Sharma, S.K.; Goloubinoff, P.; Doglia, S.M. Biophysical characterization of two different stable misfolded monomeric polypeptides that are chaperone-amenable substrates. J. Mol. Biol. 2013, 425, 1158–1171. [Google Scholar] [CrossRef]
- Mattoo, R.U.H.; Goloubinoff, P. Molecular chaperones are nanomachines that catalytically unfold misfolded and alternatively folded proteins. Cell. Mol. Life Sci. CMLS 2014, 71, 3311–3325. [Google Scholar] [CrossRef] [Green Version]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science (N. Y.) 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Weaver, B.A.; Cleveland, D.W. Does aneuploidy cause cancer? Curr. Opin. Cell Biol. 2006, 18, 658–667. [Google Scholar] [CrossRef]
- Williams, B.R.; Prabhu, V.R.; Hunter, K.E.; Glazier, C.M.; Whittaker, C.A.; Housman, D.E.; Amon, A. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science (N. Y.) 2008, 322, 703–709. [Google Scholar] [CrossRef]
- Torres, E.M.; Dephoure, N.; Panneerselvam, A.; Tucker, C.M.; Whittaker, C.A.; Gygi, S.P.; Dunham, M.J.; Amon, A. Identification of aneuploidy-tolerating mutations. Cell 2010, 143, 71–83. [Google Scholar] [CrossRef]
- Demirsoy, S.; Martin, S.; Maes, H.; Agostinis, P. Adapt, Recycle, and Move on: Proteostasis and Trafficking Mechanisms in Melanoma. Front. Oncol. 2016, 6, 240. [Google Scholar] [CrossRef]
- Vilchez, D.; Saez, I.; Dillin, A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 2014, 5, 5659. [Google Scholar] [CrossRef] [Green Version]
- Sklirou, A.; Papanagnou, E.D.; Fokialakis, N.; Trougakos, I.P. Cancer chemoprevention via activation of proteostatic modules. Cancer Lett. 2018, 413, 110–121. [Google Scholar] [CrossRef]
- Miller, B.F.; Drake, J.C.; Naylor, B.; Price, J.C.; Hamilton, K.L. The measurement of protein synthesis for assessing proteostasis in studies of slowed aging. Ageing Res. Rev. 2014, 18, 106–111. [Google Scholar] [CrossRef] [Green Version]
- Arnsburg, K.; Kirstein-Miles, J. Interrelation between protein synthesis, proteostasis and life span. Curr. Genom. 2014, 15, 66–75. [Google Scholar] [CrossRef]
- Bustamante, C.J.; Kaiser, C.M.; Maillard, R.A.; Goldman, D.H.; Wilson, C.A.M. Mechanisms of cellular proteostasis: Insights from single-molecule approaches. Annu. Rev. Biophys. 2014, 43, 119–140. [Google Scholar] [CrossRef]
- Fedyukina, D.V.; Cavagnero, S. Protein Folding at the Exit Tunnel. Annu. Rev. Biophys. 2011, 40, 337–359. [Google Scholar] [CrossRef]
- Guerra-Moreno, A.; Isasa, M.; Bhanu, M.K.; Waterman, D.P.; Eapen, V.V.; Gygi, S.P.; Hanna, J. Proteomic Analysis Identifies Ribosome Reduction as an Effective Proteotoxic Stress Response. J. Biol. Chem. 2015, 290, 29695–29706. [Google Scholar] [CrossRef] [Green Version]
- Charmpilas, N.; Daskalaki, I.; Papandreou, M.E.; Tavernarakis, N. Protein synthesis as an integral quality control mechanism during ageing. Ageing Res. Rev. 2015, 23, 75–89. [Google Scholar] [CrossRef]
- Pelletier, J.; Thomas, G.; Volarevic, S. Ribosome biogenesis in cancer: New players and therapeutic avenues. Nat. Rev. Cancer 2018, 18, 51–63. [Google Scholar] [CrossRef]
- Bublik, D.R.; Bursac, S.; Sheffer, M.; Orsolic, I.; Shalit, T.; Tarcic, O.; Kotler, E.; Mouhadeb, O.; Hoffman, Y.; Fuchs, G.; et al. Regulatory module involving FGF13, miR-504, and p53 regulates ribosomal biogenesis and supports cancer cell survival. Proc. Natl. Acad. Sci. USA 2017, 114, E496–E505. [Google Scholar] [CrossRef]
- Sumera, A.; Radhakrishnan, A.; Baba, A.A.; George, E. Review: Beta-thalassemia and molecular chaperones. Blood Cells Mol. Dis. 2015, 54, 348–352. [Google Scholar] [CrossRef]
- Lindquist, S.; Craig, E.A. The heat-shock proteins. Ann. Rev. Genet. 1988, 22, 631–677. [Google Scholar] [CrossRef]
- Requena, J.M.; Montalvo, A.M.; Fraga, J. Molecular Chaperones of Leishmania: Central Players in Many Stress-Related and -Unrelated Physiological Processes. BioMed Res. Int. 2015, 2015, 301326. [Google Scholar] [CrossRef]
- Rappa, F.; Farina, F.; Zummo, G.; David, S.; Campanella, C.; Carini, F.; Tomasello, G.; Damiani, P.; Cappello, F.; de Macario, E.C.; et al. HSP-molecular chaperones in cancer biogenesis and tumor therapy: An overview. Anticancer Res. 2012, 32, 5139–5150. [Google Scholar]
- Nahleh, Z.; Tfayli, A.; Najm, A.; El Sayed, A.; Nahle, Z. Heat shock proteins in cancer: Targeting the ‘chaperones’. Future Med. Chem. 2012, 4, 927–935. [Google Scholar] [CrossRef]
- Macario, A.J.; Cappello, F.; Zummo, G.; Conway de Macario, E. Chaperonopathies of senescence and the scrambling of interactions between the chaperoning and the immune systems. Ann. N. Y. Acad. Sci. 2010, 1197, 85–93. [Google Scholar] [CrossRef]
- Jego, G.; Hazoume, A.; Seigneuric, R.; Garrido, C. Targeting heat shock proteins in cancer. Cancer Lett. 2013, 332, 275–285. [Google Scholar] [CrossRef]
- Calderwood, S.K.; Gong, J. Molecular chaperones in mammary cancer growth and breast tumor therapy. J. Cell. Biochem. 2012, 113, 1096–1103. [Google Scholar] [CrossRef] [Green Version]
- Kloog, Y.; Elad-Sfadia, G.; Haklai, R.; Mor, A. Ras chaperones: New targets for cancer and immunotherapy. Enzymes 2013, 33, 267–289. [Google Scholar] [CrossRef]
- Boridy, S.; Le, P.U.; Petrecca, K.; Maysinger, D. Celastrol targets proteostasis and acts synergistically with a heat-shock protein 90 inhibitor to kill human glioblastoma cells. Cell Death Dis. 2014, 5, e1216. [Google Scholar] [CrossRef]
- Oh, M.; Lee, J.H.; Wang, W.; Lee, H.S.; Lee, W.S.; Burlak, C.; Im, W.; Hoang, Q.Q.; Lim, H.S. Potential pharmacological chaperones targeting cancer-associated MCL-1 and Parkinson disease-associated alpha-synuclein. Proc. Natl. Acad. Sci. USA 2014, 111, 11007–11012. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huang, T.; Bu, G.; Xu, H. Dysregulation of protein trafficking in neurodegeneration. Mol. Neurodegener. 2014, 9, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baloyannis, S.J. Golgi apparatus and protein trafficking in Alzheimer’s disease. J. Alzheimer’s Dis. JAD 2014, 42 (Suppl. 3), S153–S162. [Google Scholar] [CrossRef]
- Tsimberidou, A.M. Targeted therapy in cancer. Cancer Chemother. Pharmacol. 2015, 76, 1113–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gargalionis, A.N.; Karamouzis, M.V.; Adamopoulos, C.; Papavassiliou, A.G. Protein trafficking in colorectal carcinogenesis-targeting and bypassing resistance to currently applied treatments. Carcinogenesis 2015, 36, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.C.; Link, W. Protein localization in disease and therapy. J. Cell Sci. 2011, 124, 3381–3392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niederst, M.J.; Engelman, J.A. Bypass mechanisms of resistance to receptor tyrosine kinase inhibition in lung cancer. Sci. Signal. 2013, 6. [Google Scholar] [CrossRef]
- Goldenring, J.R. A central role for vesicle trafficking in epithelial neoplasia: Intracellular highways to carcinogenesis. Nat. Rev. Cancer 2013, 13, 813–820. [Google Scholar] [CrossRef]
- Smith, M.; Wilkinson, S. ER homeostasis and autophagy. Essays Biochem. 2017, 61, 625–635. [Google Scholar] [CrossRef] [Green Version]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, A.; Ordonez, R.; Reiter, R.J.; Gonzalez-Gallego, J.; Mauriz, J.L. Melatonin and endoplasmic reticulum stress: Relation to autophagy and apoptosis. J. Pineal Res. 2015, 59, 292–307. [Google Scholar] [CrossRef] [PubMed]
- McConkey, D.J. The integrated stress response and proteotoxicity in cancer therapy. Biochem. Biophys. Res. Commun. 2017, 482, 450–453. [Google Scholar] [CrossRef] [Green Version]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Garg, A.D.; Maes, H.; van Vliet, A.R.; Agostinis, P. Targeting the hallmarks of cancer with therapy-induced endoplasmic reticulum (ER) stress. Mol. Cell. Oncol. 2015, 2, e975089. [Google Scholar] [CrossRef]
- Liu, Y.H.; Weng, Y.P.; Tsai, H.Y.; Chen, C.J.; Lee, D.Y.; Hsieh, C.L.; Wu, Y.C.; Lin, J.Y. Aqueous extracts of Paeonia suffruticosa modulates mitochondrial proteostasis by reactive oxygen species-induced endoplasmic reticulum stress in pancreatic cancer cells. Phytomed. Int. J. Phytother. Phytopharmacol. 2018, 46, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Fourcade, S.; Ferrer, I.; Pujol, A. Oxidative stress, mitochondrial and proteostasis malfunction in adrenoleukodystrophy: A paradigm for axonal degeneration. Free Radic. Biol. Med. 2015, 88, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R.I. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008, 22, 1427–1438. [Google Scholar] [CrossRef] [Green Version]
- Bettigole, S.E.; Glimcher, L.H. Endoplasmic reticulum stress in immunity. Annu. Rev. Immunol. 2015, 33, 107–138. [Google Scholar] [CrossRef]
- Haigis, M.C.; Yankner, B.A. The aging stress response. Mol. Cell 2010, 40, 333–344. [Google Scholar] [CrossRef]
- Fawcett, E.M.; Hoyt, J.M.; Johnson, J.K.; Miller, D.L. Hypoxia disrupts proteostasis in Caenorhabditis elegans. Aging Cell 2015, 14, 92–101. [Google Scholar] [CrossRef]
- Tamás, M.; Sharma, S.; Ibstedt, S.; Jacobson, T.; Christen, P. Heavy Metals and Metalloids As a Cause for Protein Misfolding and Aggregation. Biomolecules 2014, 4, 252. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R.I. The heat shock response: Systems biology of proteotoxic stress in aging and disease. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Herr, I.; Debatin, K.-M. Cellular stress response and apoptosis in cancer therapy. Blood 2001, 98, 2603–2614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chircop, M.; Speidel, D. Cellular stress responses in cancer and cancer therapy. Front. Oncol. 2014, 4, 304. [Google Scholar] [CrossRef] [PubMed]
- Zelenka, J.; Koncosova, M.; Ruml, T. Targeting of stress response pathways in the prevention and treatment of cancer. Biotechnol. Adv. 2018, 36, 583–602. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.; Ye, Y. In Search of a Cure for Proteostasis-Addicted Cancer: A AAA Target Revealed. Cancer Cell 2015, 28, 550–552. [Google Scholar] [CrossRef]
- Goldberg, A.L. Development of proteasome inhibitors as research tools and cancer drugs. J. Cell Biol. 2012, 199, 583–588. [Google Scholar] [CrossRef] [Green Version]
- Goy, A.; Younes, A.; McLaughlin, P.; Pro, B.; Romaguera, J.E.; Hagemeister, F.; Fayad, L.; Dang, N.H.; Samaniego, F.; Wang, M.; et al. Phase II study of proteasome inhibitor bortezomib in relapsed or refractory B-cell non-Hodgkin’s lymphoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 667–675. [Google Scholar] [CrossRef]
- Hideshima, T.; Bradner, J.E.; Wong, J.; Chauhan, D.; Richardson, P.; Schreiber, S.L.; Anderson, K.C. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc. Natl. Acad. Sci. USA 2005, 102, 8567–8572. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, O.A.; Wright, J.; Moskowitz, C.; Muzzy, J.; MacGregor-Cortelli, B.; Stubblefield, M.; Straus, D.; Portlock, C.; Hamlin, P.; Choi, E.; et al. Phase II clinical experience with the novel proteasome inhibitor bortezomib in patients with indolent non-Hodgkin’s lymphoma and mantle cell lymphoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Sonneveld, P.; Schuster, M.W.; Irwin, D.; Stadtmauer, E.A.; Facon, T.; Harousseau, J.L.; Ben-Yehuda, D.; Lonial, S.; Goldschmidt, H.; et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 2005, 352, 2487–2498. [Google Scholar] [CrossRef] [PubMed]
- Demo, S.D.; Kirk, C.J.; Aujay, M.A.; Buchholz, T.J.; Dajee, M.; Ho, M.N.; Jiang, J.; Laidig, G.J.; Lewis, E.R.; Parlati, F.; et al. Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res. 2007, 67, 6383–6391. [Google Scholar] [CrossRef] [PubMed]
- Weathington, N.M.; Mallampalli, R.K. Emerging therapies targeting the ubiquitin proteasome system in cancer. J. Clin. Investig. 2014, 124, 6–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, M.; Schmitt, S.; Buac, D.; Dou, Q.P. Targeting the ubiquitin-proteasome system for cancer therapy. Expert Opin. Ther. Targets 2013, 17, 1091–1108. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Deng, Q.; Zhou, J.; Zou, J.; Zhang, Y.; Tan, P.; Zhang, W.; Cui, H. CSN6 controls the proliferation and metastasis of glioblastoma by CHIP-mediated degradation of EGFR. Oncogene 2017, 36, 1134–1144. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, N. The ubiquitin-proteasome system meets angiogenesis. Mol. Cancer Ther. 2012, 11, 538–548. [Google Scholar] [CrossRef]
- Simón, D.; García-García, E.; Royo, F.; Falcón-Pérez, J.M.; Avila, J. Proteostasis of tau. Tau overexpression results in its secretion via membrane vesicles. FEBS Lett. 2012, 586, 47–54. [Google Scholar] [CrossRef]
- Desdin-Mico, G.; Mittelbrunn, M. Role of exosomes in the protection of cellular homeostasis. Cell Adhes. Migr. 2017, 11, 127–134. [Google Scholar] [CrossRef]
- Genereux, J.C.; Qu, S.; Zhou, M.; Ryno, L.M.; Wang, S.; Shoulders, M.D.; Kaufman, R.J.; Lasmezas, C.I.; Kelly, J.W.; Wiseman, R.L. Unfolded protein response-induced ERdj3 secretion links ER stress to extracellular proteostasis. EMBO J. 2015, 34, 4–19. [Google Scholar] [CrossRef]
- De Duve, C. The lysosome turns fifty. Nat. Cell Biol. 2005, 7, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Martini-Stoica, H.; Xu, Y.; Ballabio, A.; Zheng, H. The Autophagy-Lysosomal Pathway in Neurodegeneration: A TFEB Perspective. Trends Neurosci. 2016, 39, 221–234. [Google Scholar] [CrossRef]
- Nedic, O.; Rattan, S.I.; Grune, T.; Trougakos, I.P. Molecular effects of advanced glycation end products on cell signalling pathways, ageing and pathophysiology. Free Radic. Res. 2013, 47 (Suppl. 1), 28–38. [Google Scholar] [CrossRef] [Green Version]
- Alers, S.; Löffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell. Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 Antiapoptotic Proteins Inhibit Beclin 1-Dependent Autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Dong, Z.; Lei, Q.; Yang, J.; Hu, H.; Li, Q.; Ji, Y.; Guo, L.; Zhang, Y.; Liu, Y.; et al. Inactivation/deficiency of DHODH induces cell cycle arrest and programed cell death in melanoma. Oncotarget 2017, 8, 112354–112370. [Google Scholar] [CrossRef] [PubMed]
- Obara, K.; Ohsumi, Y. Dynamics and function of PtdIns(3)P in autophagy. Autophagy 2008, 4, 952–954. [Google Scholar] [CrossRef]
- Metlagel, Z.; Otomo, C.; Ohashi, K.; Takaesu, G.; Otomo, T. Structural insights into E2–E3 interaction for LC3 lipidation. Autophagy 2014, 10, 522–523. [Google Scholar] [CrossRef]
- Shao, Y.; Gao, Z.; Feldman, T.; Jiang, X. Stimulation of ATG12-ATG5 Conjugation by Ribonucleic Acid. Autophagy 2007, 3, 10–16. [Google Scholar] [CrossRef]
- Satoo, K.; Noda, N.N.; Kumeta, H.; Fujioka, Y.; Mizushima, N.; Ohsumi, Y.; Inagaki, F. The structure of Atg4B–LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. EMBO J. 2009, 28, 1341–1350. [Google Scholar] [CrossRef] [Green Version]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER–mitochondria contact sites. Nature 2013, 495, 389. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Bell Biol. 2010, 12, 747. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Klionsky, D.J. The Golgi as a potential membrane source for autophagy. Autophagy 2010, 6, 950–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, N. Kinesin and Dynein Superfamily Proteins and the Mechanism of Organelle Transport. Science (N. Y.) 1998, 279, 519–526. [Google Scholar] [CrossRef]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The Hairpin-type Tail-Anchored SNARE Syntaxin 17 Targets to Autophagosomes for Fusion with Endosomes/Lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Prat, L.; Martinez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodriguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef] [Green Version]
- Carroll, B.; Otten, E.G.; Manni, D.; Stefanatos, R.; Menzies, F.M.; Smith, G.R. Oxidation of SQSTM1/p62 mediates the link between redox state and protein homeostasis. Nat. Commun. 2018, 9, 256. [Google Scholar] [CrossRef] [Green Version]
- Feldmann, A.; Bekbulat, F.; Huesmann, H.; Ulbrich, S.; Tatzelt, J.; Behl, C.; Kern, A. The RAB GTPase RAB18 modulates macroautophagy and proteostasis. Biochem. Biophys. Res. Commun. 2017, 486, 738–743. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Gestwicki, J.E.; Murphy, L.O.; Klionsky, D.J. Potential therapeutic applications of autophagy. Nat. Rev. Drug Discov. 2007, 6, 304. [Google Scholar] [CrossRef]
- Sarkar, S.; Rubinsztein, D.C. Small molecule enhancers of autophagy for neurodegenerative diseases. Mol. BioSyst. 2008, 4, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Wong, E. Chapter Fifteen—Kinetics of Protein Aggregates Disposal by Aggrephagy. In Methods in Enzymology; Galluzzi, L., Bravo-San Pedro, J.M., Kroemer, G., Eds.; Academic Press: New York, NY, USA, 2017; Volume 588, pp. 245–281. [Google Scholar]
- Lim, K.-L.; Dawson, V.L.; Dawson, T.M. Parkin-mediated lysine 63-linked polyubiquitination: A link to protein inclusions formation in Parkinson’s and other conformational diseases? Neurobiol. Aging 2006, 27, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Zucchelli, S.; Marcuzzi, F.; Codrich, M.; Agostoni, E.; Vilotti, S.; Biagioli, M.; Pinto, M.; Carnemolla, A.; Santoro, C.; Gustincich, S.; et al. Tumor necrosis factor receptor-associated factor 6 (TRAF6) associates with huntingtin protein and promotes its atypical ubiquitination to enhance aggregate formation. J. Biol. Chem. 2011, 286, 25108–25117. [Google Scholar] [CrossRef] [PubMed]
- Kopito, R.R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000, 10, 524–530. [Google Scholar] [CrossRef]
- Lamark, T.; Johansen, T. Aggrephagy: Selective disposal of protein aggregates by macroautophagy. Int. J. Cell Biol. 2012, 2012, 736905. [Google Scholar] [CrossRef] [PubMed]
- Burnett, B.G.; Pittman, R.N. The polyglutamine neurodegenerative protein ataxin 3 regulates aggresome formation. Proc. Natl. Acad. Sci. USA 2005, 102, 4330–4335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.-P. The Deacetylase HDAC6 Regulates Aggresome Formation and Cell Viability in Response to Misfolded Protein Stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, H.; Ali, Y.O.; Ravichandran, M.; Dong, A.; Qiu, W.; MacKenzie, F.; Dhe-Paganon, S.; Arrowsmith, C.H.; Zhai, R.G. Protein Aggregates Are Recruited to Aggresome by Histone Deacetylase 6 via Unanchored Ubiquitin C Termini. J. Biol. Chem. 2012, 287, 2317–2327. [Google Scholar] [CrossRef] [PubMed]
- Watabe, M.; Nakaki, T. Protein kinase CK2 regulates the formation and clearance of aggresomes in response to stress. J. Cell Sci. 2011, 124, 1519–1532. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Seibenhener, M.L.; Calderilla-Barbosa, L.; Diaz-Meco, M.T.; Moscat, J.; Jiang, J.; Wooten, M.W.; Wooten, M.C. SQSTM1/p62 interacts with HDAC6 and regulates deacetylase activity. PLoS ONE 2013, 8, e76016. [Google Scholar] [CrossRef]
- Fusco, C.; Micale, L.; Egorov, M.; Monti, M.; D’Addetta, E.V.; Augello, B.; Cozzolino, F.; Calcagni, A.; Fontana, A.; Polishchuk, R.S.; et al. The E3-ubiquitin ligase TRIM50 interacts with HDAC6 and p62, and promotes the sequestration and clearance of ubiquitinated proteins into the aggresome. PLoS ONE 2012, 7, e40440. [Google Scholar] [CrossRef] [PubMed]
- Kalveram, B.; Schmidtke, G.; Groettrup, M. The ubiquitin-like modifier FAT10 interacts with HDAC6 and localizes to aggresomes under proteasome inhibition. J. Cell Sci. 2008, 121, 4079–4088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Arias, E. Methods to Study Chaperone-Mediated Autophagy. Methods Enzymol. 2017, 588, 283–305. [Google Scholar] [CrossRef]
- Gamerdinger, M.; Kaya, A.M.; Wolfrum, U.; Clement, A.M.; Behl, C. BAG3 mediates chaperone-based aggresome-targeting and selective autophagy of misfolded proteins. EMBO Rep. 2011, 12, 149–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Graham, K.; Foote, M.; Liang, F.; Rizkallah, R.; Hurt, M.; Wang, Y.; Wu, Y.; Zhou, Y. 14-3-3 protein targets misfolded chaperone-associated proteins to aggresomes. J. Cell Sci. 2013, 126, 4173–4186. [Google Scholar] [CrossRef] [Green Version]
- Sahu, R.; Kaushik, S.; Clement, C.C.; Cannizzo, E.S.; Scharf, B.; Follenzi, A.; Potolicchio, I.; Nieves, E.; Cuervo, A.M.; Santambrogio, L. Microautophagy of cytosolic proteins by late endosomes. Dev. Cell 2011, 20, 131–139. [Google Scholar] [CrossRef]
- Morozova, K.; Clement, C.C.; Kaushik, S.; Stiller, B.; Arias, E.; Ahmad, A.; Rauch, J.N.; Chatterjee, V.; Melis, C.; Scharf, B.; et al. Structural and Biological Interaction of hsc-70 Protein with Phosphatidylserine in Endosomal Microautophagy. J. Biol. Chem. 2016, 291, 18096–18106. [Google Scholar] [CrossRef]
- Kawamura, N.; Sun-Wada, G.-H.; Aoyama, M.; Harada, A.; Takasuga, S.; Sasaki, T.; Wada, Y. Delivery of endosomes to lysosomes via microautophagy in the visceral endoderm of mouse embryos. Nat. Commun. 2012, 3, 1071. [Google Scholar] [CrossRef] [Green Version]
- Chanoca, A.; Kovinich, N. Anthocyanin Vacuolar Inclusions Form by a Microautophagy Mechanism. Plant Cell 2015, 27, 2545–2559. [Google Scholar] [CrossRef] [Green Version]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Bell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309. [Google Scholar] [CrossRef]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science (N. Y.) 2011, 331, 456–461. [Google Scholar] [CrossRef]
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016, 44, D1251–D1257. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.B.; Jasper, H. Mitochondrial Proteostasis in the Control of Aging and Longevity. Cell Metab. 2014, 20, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Moehle, E.A.; Shen, K.; Dillin, A. Mitochondrial Proteostasis in the Context of Cellular and Organismal Health and Aging. J. Biol. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, V.; Romani, M.; Mouchiroud, L.; Beck, J.S.; Zhang, H.; D’Amico, D.; Moullan, N.; Potenza, F.; Schmid, A.W.; Rietsch, S.; et al. Enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity. Nature 2017, 552, 187–193. [Google Scholar] [CrossRef]
- Drake, J.C.; Yan, Z. Mitophagy in maintaining skeletal muscle mitochondrial proteostasis and metabolic health with ageing. J. Physiol. 2017, 595, 6391–6399. [Google Scholar] [CrossRef]
- Laker, R.C.; Drake, J.C.; Wilson, R.J.; Lira, V.A.; Lewellen, B.M.; Ryall, K.A.; Fisher, C.C.; Zhang, M.; Saucerman, J.J.; Goodyear, L.J.; et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 2017, 8, 548. [Google Scholar] [CrossRef]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [Green Version]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H. Targeting autophagy in lymphomas: A double-edged sword? Int. J. Hematol. 2018, 107, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Noonan, J.; Zarrer, J.; Murphy, B.M. Targeting Autophagy in Glioblastoma. Crit. Rev. Oncog. 2016, 21, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Yuan, M.; Yu, Q.; Zhou, X.; Min, W.; Gao, D. Autophagy regulation and its role in gastric cancer and colorectal cancer. Cancer biomark. Sect. A Dis. Mark. 2016, 17, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Burada, F.; Nicoli, E.R.; Ciurea, M.E.; Uscatu, D.C.; Ioana, M.; Gheonea, D.I. Autophagy in colorectal cancer: An important switch from physiology to pathology. World J. Gastrointest. Oncol. 2015, 7, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Jain, K.; Paranandi, K.S.; Sridharan, S.; Basu, A. Autophagy in breast cancer and its implications for therapy. Am. J. Cancer Res. 2013, 3, 251–265. [Google Scholar]
- Miettinen, T.P.; Bjorklund, M. The mevalonate pathway as a metabolic requirement for autophagy-implications for growth control, proteostasis, and disease. Mol. Cell. Oncol. 2016, 3, e1143546. [Google Scholar] [CrossRef]
- Zismanov, V.; Attar-Schneider, O.; Lishner, M.; Heffez Aizenfeld, R.; Tartakover Matalon, S.; Drucker, L. Multiple myeloma proteostasis can be targeted via translation initiation factor eIF4E. Int. J. Oncol. 2015, 46, 860–870. [Google Scholar] [CrossRef]
- Milan, E.; Perini, T.; Resnati, M.; Orfanelli, U.; Oliva, L.; Raimondi, A.; Cascio, P.; Bachi, A.; Marcatti, M.; Ciceri, F.; et al. A plastic SQSTM1/p62-dependent autophagic reserve maintains proteostasis and determines proteasome inhibitor susceptibility in multiple myeloma cells. Autophagy 2015, 11, 1161–1178. [Google Scholar] [CrossRef]
- Li, X.; Zhu, F.; Jiang, J.; Sun, C.; Zhong, Q.; Shen, M.; Wang, X.; Tian, R.; Shi, C.; Xu, M.; et al. Simultaneous inhibition of the ubiquitin-proteasome system and autophagy enhances apoptosis induced by ER stress aggravators in human pancreatic cancer cells. Autophagy 2016, 12, 1521–1537. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.Y.; Yi, H.S.; Kim, H.W.; Shong, M. Dysregulation of mitophagy in carcinogenesis and tumor progression. Biochim. Biophys. Acta Bioenerget. 2017, 1858, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Papa, L.; Germain, D. SirT3 Regulates the Mitochondrial Unfolded Protein Response. Mol. Cell. Biol. 2014, 34, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Manning, B.D. mTORC1 signaling activates NRF1 to increase cellular proteasome levels. Cell Cycle (Georget. TX) 2015, 14, 2011–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, K.H.; Dai, C. mTORC1 senses stresses: Coupling stress to proteostasis. BioEssays News Rev. Mol. Cell. Dev. Biol. 2017, 39. [Google Scholar] [CrossRef]
- Smith, M.D.; Harley, M.E.; Kemp, A.J.; Wills, J.; Lee, M.; Arends, M.; von Kriegsheim, A.; Behrends, C.; Wilkinson, S. CCPG1 Is a Non-canonical Autophagy Cargo Receptor Essential for ER-Phagy and Pancreatic ER Proteostasis. Dev. Cell 2018, 44, 217–232.e211. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, K.; De, S.; Mukherjee, S.; Das, S.; Ghosh, A.N.; Sengupta, S.B. Withaferin A induced impaired autophagy and unfolded protein response in human breast cancer cell-lines MCF-7 and MDA-MB-231. Toxicol. In Vitro 2017, 44, 330–338. [Google Scholar] [CrossRef]
- Choutka, C.; DeVorkin, L.; Go, N.E.; Hou, Y.C.; Moradian, A.; Morin, G.B.; Gorski, S.M. Hsp83 loss suppresses proteasomal activity resulting in an upregulation of caspase-dependent compensatory autophagy. Autophagy 2017, 13, 1573–1589. [Google Scholar] [CrossRef] [Green Version]
- Klimek, C.; Kathage, B.; Wordehoff, J.; Hohfeld, J. BAG3-mediated proteostasis at a glance. J. Cell Sci. 2017, 130, 2781–2788. [Google Scholar] [CrossRef]
- Powers, E.T.; Morimoto, R.I.; Dillin, A.; Kelly, J.W.; Balch, W.E. Biological and Chemical Approaches to Diseases of Proteostasis Deficiency. Ann. Rev. Biochem. 2009, 78, 959–991. [Google Scholar] [CrossRef]
- Adams, J. Proteasome inhibition in cancer: Development of PS-341. Semin. Oncol. 2001, 28, 613–619. [Google Scholar] [CrossRef]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The Role of Autophagy in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2011, 10, 1533. [Google Scholar] [CrossRef] [PubMed]
- Moschovi, M.; Critselis, E.; Cen, O.; Adamaki, M.; Lambrou, G.I.; Chrousos, G.P.; Vlahopoulos, S. Drugs acting on homeostasis: Challenging cancer cell adaptation. Expert Rev. Anticancer Ther. 2015, 15, 1405–1417. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, Z.; Cui, H. The Autophagy-Lysosomal Pathways and Their Emerging Roles in Modulating Proteostasis in Tumors. Cells 2019, 8, 4. https://doi.org/10.3390/cells8010004
Dong Z, Cui H. The Autophagy-Lysosomal Pathways and Their Emerging Roles in Modulating Proteostasis in Tumors. Cells. 2019; 8(1):4. https://doi.org/10.3390/cells8010004
Chicago/Turabian StyleDong, Zhen, and Hongjuan Cui. 2019. "The Autophagy-Lysosomal Pathways and Their Emerging Roles in Modulating Proteostasis in Tumors" Cells 8, no. 1: 4. https://doi.org/10.3390/cells8010004
APA StyleDong, Z., & Cui, H. (2019). The Autophagy-Lysosomal Pathways and Their Emerging Roles in Modulating Proteostasis in Tumors. Cells, 8(1), 4. https://doi.org/10.3390/cells8010004