Prohibitins: A Critical Role in Mitochondrial Functions and Implication in Diseases


{kind=link}
Abstract
:1. Introduction
2. Role of Prohibitins in the Oxidative Phosphorylation System
3. Role of Prohibitins in Mitochondrial Biogenesis
4. Role of Prohibitins in Unfolded Protein Response (UPRmt)
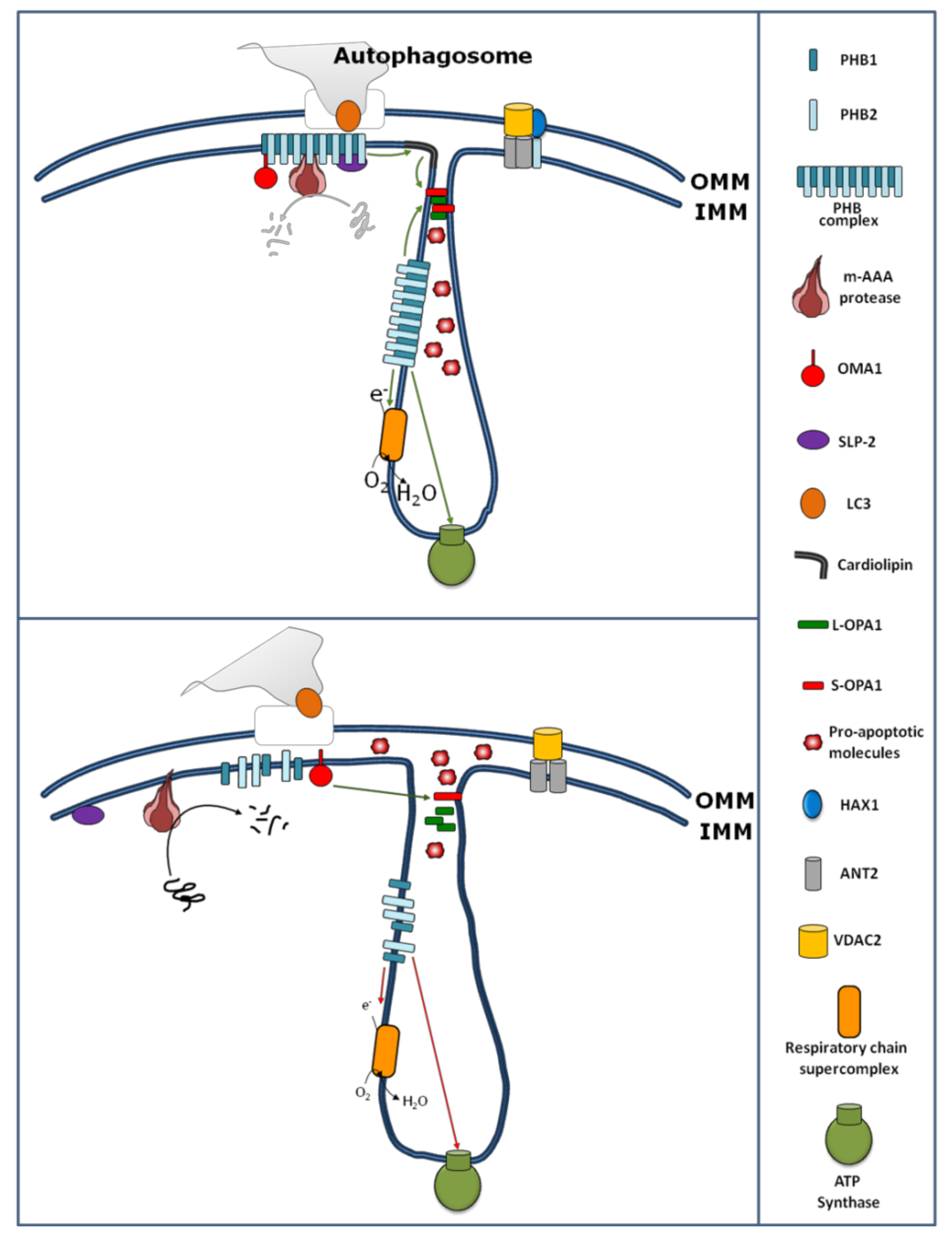
5. Role of Prohibitins in Mitochondrial Dynamics and Ultrastructure
6. Role of Prohibitins in Apoptosis
7. Prohibitins in Mitophagy
8. Pathological Role of Prohibitins
8.1. Prohibitin and Aging
8.2. Prohibitins in Parkinson’s and Alzheimer’s Diseases
8.3. Prohibitins in Kidney Diseases
8.4. Prohibitins in Cardiac Diseases
8.5. Prohibitins and Cancer
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Papa, S.; Martino, P.L.; Capitanio, G.; Gaballo, A.; De Rasmo, D.; Signorile, A.; Petruzzella, V. The oxidative phosphorylation system in mammalian mitochondria. Adv. Exp. Med. Biol. 2012, 942, 3–37. [Google Scholar] [PubMed]
- Estaquier, J.; Vallette, F.; Vayssiere, J.L.; Mignotte, B. The mitochondrial pathways of apoptosis. Adv. Exp. Med. Biol. 2012, 942, 157–183. [Google Scholar] [PubMed]
- Holloszy, J.O. Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J. Biol. Chem. 1967, 242, 2278–2282. [Google Scholar] [PubMed]
- Huttemann, M.; Lee, I.; Samavati, L.; Yu, H.; Doan, J.W. Regulation of mitochondrial oxidative phosphorylation through cell signalling. Biochim. Biophys. Acta 2007, 1773, 1701–1720. [Google Scholar] [CrossRef] [PubMed]
- De Rasmo, D.; Signorile, A.; Papa, F.; Roca, E.; Papa, S. cAMP/Ca2+ response element-binding protein plays a central role in the biogenesis of respiratory chain proteins in mammalian cells. IUBMB Life 2010, 62, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Benard, G.; Massa, F.; Puente, N.; Lourenço, J.; Bellocchio, L.; Soria-Gómez, E.; Matias, I.; Delamarre, A.; Metna-Laurent, M.; Cannich, A.; et al. Mitochondrial CB(1) receptors regulate neuronal energy metabolism. Nat. Neurosci. 2012, 15, 558–564. [Google Scholar] [CrossRef] [PubMed]
- De Rasmo, D.; Micelli, L.; Santeramo, A.; Signorile, A.; Lattanzio, P.; Papa, S. cAMP regulates the functional activity, coupling efficiency and structural organization of mammalian FOF1 ATP synthase. Biochim. Biophys. Acta 2016, 1857, 350–358. [Google Scholar] [CrossRef]
- McClung, J.K.; Danner, D.B.; Stewart, D.A.; Smith, J.R.; Schneider, E.L.; Lumpkin, C.K.; Dell’Orco, R.T.; Nuell, M.J. Isolation of a cDNA that hybrid selects antiproliferative mRNA from rat liver. Biochem. Biophys. Res. Commun. 1989, 164, 1316–1322. [Google Scholar] [CrossRef]
- Terashima, M.; Kim, K.M.; Adachi, T.; Nielsen, P.J.; Reth, M.; Köhler, G.; Lamers, M.C. The IgM antigen receptor of B lymphocytes is associated with prohibitin and a prohibitin-related protein. EMBO J. 1994, 13, 3782–3792. [Google Scholar] [CrossRef]
- Back, J.W.; Sanz, M.A.; De Jong, L.; De Koning, L.J.; Nijtmans, L.G.; De Koster, C.G.; Grivell, L.A.; Van Der Spek, H.; Muijsers, A.O. A structure for the yeast prohibitin complex: Structure prediction and evidence from chemical crosslinking and mass spectrometry. Protein Sci. 2002, 11, 2471–2478. [Google Scholar] [CrossRef]
- Steglich, G.; Neupert, W.; Langer, T. Prohibitins regulate membrane protein degradation by the m-AAA protease in mitochondria. Mol. Cell. Biol. 1999, 19, 3435–3442. [Google Scholar] [CrossRef] [PubMed]
- Tatsuta, T.; Model, K.; Langer, T. Formation of membrane-bound ring complexes by prohibitins in mitochondria. Mol. Biol. Cell 2005, 16, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Artal-Sanz, M.; Tsang, W.Y.; Willems, E.M.; Grivell, L.A.; Lemire, B.D.; van der Spek, H.; Nijtmans, L.G. The mitochondrial prohibitin complex is essential for embryonic viability and germline function in Caenorhabditis elegans. J. Biol. Chem. 2003, 278, 32091–32099. [Google Scholar] [CrossRef] [PubMed]
- Nijtmans, L.G.; de Jong, L.; Artal Sanz, M.; Coates, P.J.; Berden, J.A.; Back, J.W.; Muijsers, A.O.; van der Spek, H.; Grivell, L.A. Prohibitins act as a membrane-bound chaperone for the stabilization of mitochondrial proteins. EMBO J. 2000, 19, 2444–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artal-Sanz, M.; Tavernarakis, N. Prohibitin couples diapause signalling to mitochondrial metabolism during ageing in C. elegans. Nature 2009, 461, 793–797. [Google Scholar] [CrossRef]
- Koushyar, S.; Jiang, W.G.; Dart, D.A. Unveiling the potential of prohibitin in cancer. Cancer Lett. 2015, 369, 316–322. [Google Scholar] [CrossRef] [Green Version]
- Ferrer, I.; Perez, E.; Dalfó, E.; Barrachina, M. Abnormal levels of prohibitin and ATP synthase in the substantia nigra and frontal cortex in Parkinson’s disease. Neurosci. Lett. 2007, 415, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Ali, N.; Banerjee, E.; Singh, R.; Naskar, A.; Paidi, R.K.; Mohanakumar, K.P. Low Levels of Prohibitin in Substantia Nigra Makes Dopaminergic Neurons Vulnerable in Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 804–821. [Google Scholar] [CrossRef]
- Supale, S.; Thorel, F.; Merkwirth, C.; Gjinovci, A.; Herrera, P.L.; Scorrano, L.; Meda, P.; Langer, T.; Maechler, P. Loss of prohibitin induces mitochondrial damages altering β-cell function and survival and is responsible for gradual diabetes development. Diabetes 2013, 62, 3488–3499. [Google Scholar] [CrossRef]
- Ising, C.; Koehler, S.; Brähler, S.; Merkwirth, C.; Höhne, M.; Baris, O.R.; Hagmann, H.; Kann, M.; Fabretti, F.; Dafinger, C.; et al. Inhibition of insulin/IGF-1 receptor signaling protects from mitochondria-mediated kidney failure. EMBO. Mol. Med. 2015, 7, 275–287. [Google Scholar] [CrossRef] [Green Version]
- Merkwirth, C.; Martinelli, P.; Korwitz, A.; Morbin, M.; Brönneke, H.S.; Jordan, S.D.; Rugarli, E.I.; Langer, T. Loss of prohibitin membrane scaffolds impairs mitochondrial architecture and leads to tau hyperphosphorylation and neurodegeneration. PLoS Genet. 2012, 8, e1003021. [Google Scholar] [CrossRef] [PubMed]
- Fusaro, G.; Wang, S.; Chellappan, S. Differential regulation of Rb family proteins and prohibitin during camptothecin-induced apoptosis. Oncogene 2002, 21, 4539–4548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasashima, K.; Ohta, E.; Kagawa, Y.; Endo, H. Mitochondrial functions and estrogen receptor-dependent nuclear translocation of pleiotropic human prohibitin 2. J. Biol. Chem. 2006, 281, 36401–36410. [Google Scholar] [CrossRef] [PubMed]
- Merkwirth, C.; Dargazanli, S.; Tatsuta, T.; Geimer, S.; Löwer, B.; Wunderlich, F.T.; von Kleist-Retzow, J.C.; Waisman, A.; Westermann, B.; Langer, T. Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev. 2008, 22, 476–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thuaud, F.; Ribeiro, N.; Nebigil, C.G.; Désaubry, L. Prohibitin ligands in cell death and survival: Mode of action and therapeutic potential. Chem. Biol. 2013, 20, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Bavelloni, A.; Piazzi, M.; Raffini, M.; Faenza, I.; Blalock, W.L. Prohibitin 2: At a communications crossroads. IUBMB Life 2015, 67, 239–254. [Google Scholar] [CrossRef] [Green Version]
- Merkwirth, C.; Langer, T. Prohibitin function within mitochondria: Essential roles for cell proliferation and cristae morphogenesis. Biochim. Biophys. Acta 2009, 1793, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Dong, P.; Zhang, Z.; Li, C.; Li, Y.; Liao, Y.; Li, X.; Wu, Z.; Guo, S.; Mai, S.; et al. Akt phosphorylates Prohibitin 1 to mediate its mitochondrial localization and promote proliferation of bladder cancer cells. Cell Death Dis. 2015, 6, e1660. [Google Scholar] [CrossRef] [Green Version]
- Bayot, A.; Gareil, M.; Rogowska-Wrzesinska, A.; Roepstorff, P.; Friguet, B.; Bulteau, A.L. Identification of novel oxidized protein substrates and physiological partners of the mitochondrial ATP-dependent Lon-like protease Pim1. J. Biol. Chem. 2010, 285, 11445–11457. [Google Scholar] [CrossRef]
- McBride, H.; Soubannier, V. Mitochondrial Function: OMA1 and OPA1, the Grandmasters of Mitochondrial Health. Curr. Biol. 2010, 20, R274–R276. [Google Scholar] [CrossRef] [Green Version]
- Da Cruz, S.; Parone, P.A.; Gonzalo, P.; Bienvenut, W.V.; Tondera, D.; Jourdain, A.; Quadroni, M.; Martinou, J.C. SLP-2 interacts with prohibitins in the mitochondrial inner membrane and contributes to their stability. Biochim. Biophys. Acta 2008, 1783, 904–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter-Dennerlein, R.; Korwitz, A.; Haag, M.; Tatsuta, T.; Dargazanli, S.; Baker, M.; Decker, T.; Lamkemeyer, T.; Rugarli, E.I.; Langer, T. DNAJC19, a mitochondrial cochaperone associated with cardiomyopathy, forms a complex with prohibitins to regulate cardiolipin remodeling. Cell Metab. 2014, 20, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Ban, T.; Ishihara, T.; Kohno, H.; Saita, S.; Ichimura, A.; Maenaka, K.; Oka, T.; Mihara, K.; Ishihara, N. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat. Cell Biol. 2017, 19, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, K.; Gohil, V.; Stuart, R.A.; Hunte, C.; Brandt, U.; Greenberg, M.L.; Schägger, H. Cardiolipin stabilizes respiratory chain supercomplexes. J. Biol. Chem. 2003, 278, 52873–52880. [Google Scholar] [CrossRef] [PubMed]
- Jian, C.; Xu, F.; Hou, T.; Sun, T.; Li, J.; Cheng, H.; Wang, X. Deficiency of PHB complex impairs respiratory supercomplex formation and activates mitochondrial flashes. J. Cell Sci. 2017, 130, 2620–2630. [Google Scholar] [CrossRef] [Green Version]
- Osman, C.; Wilmes, C.; Tatsuta, T.; Langer, T. Prohibitins interact genetically with Atp23, novel processing peptidase and chaperone for the F1Fo-ATP synthase. Mol. Biol. Cell 2007, 18, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Chiang, W.C.; Sumpter, R., Jr.; Mishra, P.; Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 2017, 168, 224–238. [Google Scholar] [CrossRef]
- Frezza, C.; Cipolat, S.; Martins de Brito, O.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef]
- Tsutsumi, T.; Matsuda, M.; Aizaki, H.; Moriya, K.; Miyoshi, H.; Fujie, H.; Shintani, Y.; Yotsuyanagi, H.; Miyamura, T.; Suzuki, T.; et al. Proteomics analysis of mitochondrial proteins reveals overexpression of a mitochondrial protein chaperon, prohibitin, in cells expressing hepatitis C virus core protein. Hepatology 2009, 50, 378–386. [Google Scholar] [CrossRef] [Green Version]
- Schleicher, M.; Shepherd, B.R.; Suarez, Y.; Fernandez-Hernando, C.; Yu, J.; Pan, Y.; Acevedo, L.M.; Shadel, G.S.; Sessa, W.C. Prohibitin-1 maintains the angiogenic capacity of endothelial cells by regulating mitochondrial function and senescence. J. Cell Biol. 2008, 180, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Lu, G.M. Reduction of prohibitin expression contributes to left ventricular hypertrophy via enhancement of mitochondrial reactive oxygen species formation in spontaneous hypertensive rats. Free Radic. Res. 2014, 49, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.J.; Kahl, A.; Qian, L.; Stepanova, A.; Starkov, A.; Manfredi, G.; Iadecola, C.; Zhou, P. Prohibitin is a positive modulator of mitochondrial function in PC12 cells under oxidative stress. J. Neurochem. 2018, 146, 235–250. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Guo, J.; Wang, H.; Shi, Y.; Yuan, Y.; Hou, S. Prohibitin 1 gene delivery promotes functional recovery in rats with spinal cord injury. Neuroscience 2014, 286C, 27–36. [Google Scholar] [CrossRef]
- Liu, X.; Ren, Z.; Zhan, R.; Wang, X.; Wang, X.; Zhang, Z.; Leng, X.; Yang, Z.; Qian, L. Prohibitin protects against oxidative stress-induced cell injury in cultured neonatal cardiomyocyte. Cell Stress Chaperones 2009, 14, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Bourges, I.; Ramus, C.; Mousson de Camaret, B.; Beugnot, R.; Remacle, C.; Cardol, P.; Hofhaus, G.; Issartel, J.P. Structural organization of mitochondrial human complex I: Role of the ND4 and ND5 mitochondria-encoded subunits and interaction with prohibitin. Biochem. J. 2004, 383, 491–499. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Cooper, H.M.; Reyes, A.; Di Re, M.; Sembongi, H.; Litwin, T.R.; Gao, J.; Neuman, K.C.; Fearnley, I.M.; Spinazzola, A.; et al. Mitochondrial nucleoid interacting proteins support mitochondrial protein synthesis. Nucl. Acids Res. 2012, 40, 6109–6121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amunts, A.; Brown, A.; Toots, J.; Scheres, S.H.W.; Ramakrishnan, V. Ribosome. The structure of the human mitochondrial ribosome. Science 2015, 348, 95–98. [Google Scholar] [CrossRef]
- Mitsopoulos, P.; Lapohos, O.; Weraarpachai, W.; Antonicka, H.; Chang, Y.H.; Madrenas, J. Stomatin-like protein 2 deficiency results in impaired mitochondrial translation. PLoS ONE 2017, 12, e0179967. [Google Scholar] [CrossRef] [PubMed]
- Strub, G.M.; Paillard, M.; Liang, J.; Gomez, L.; Allegood, J.C.; Hait, N.C.; Maceyka, M.; Price, M.M.; Chen, Q.; Simpson, D.C.; et al. Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 2011, 25, 600–612. [Google Scholar] [CrossRef] [Green Version]
- Wegrzyn, J.; Potla, R.; Chwae, Y.J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef]
- Szczepanek, K.; Chen, Q.; Larner, A.C.; Lesnefsky, E.J. Cytoprotection by the modulation of mitochondrial electron transport chain: The emerging role of mitochondrial STAT3. Mitochondrion 2012, 12, 180–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Yu, C.; Souza, R.F.; Theiss, A.L. Prohibitin 1 modulates mitochondrial function of Stat3. Cell. Signal. 2014, 26, 2086–2095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Liu, D.; Lin, Y.; Kang, T.; Huang, B.; Xu, W.; Garcia-Barrio, M.; Olatinwo, M.; Matthews, R.; Chen, Y.E.; Thompson, W.E. Mitochondrial dysfunction and adipogenic reduction by prohibitin silencing in 3T3-L1 cells. PLoS ONE 2012, 7, e34315. [Google Scholar]
- Ande, S.R.; Nguyen, K.H.; Padilla-Meier, G.P.; Wahida, W.; Nyomba, B.L.; Mishra, S. Prohibitin overexpression in adipocytes induces mitochondrial biogenesis, leads to obesity development, and affects glucose homeostasis in a sex-specific manner. Diabetes 2014, 63, 3734–3741. [Google Scholar] [CrossRef] [PubMed]
- Fusaro, G.; Dasgupta, P.; Rastogi, S.; Joshi, B.; Chellappan, S. Prohibitin induces the transcriptional activity of p53 and is exported from the nucleus upon apoptotic signaling. J. Biol. Chem. 2003, 278, 47853–47861. [Google Scholar] [CrossRef] [PubMed]
- Kasashima, K.; Sumitani, M.; Satoh, M.; Endo, H. Human prohibitin 1 maintains the organization and stability of the mitochondrial nucleoids. Exp. Cell. Res. 2008, 314, 988–996. [Google Scholar] [CrossRef]
- Patron, M.; Sprenger, H.G.; Langer, T. m-AAA proteases, mitochondrial calcium homeostasis and neurodegeneration. Cell Res. 2018, 28, 296–306. [Google Scholar] [CrossRef] [Green Version]
- Munch, C. The different axes of the mammalian mitochondrial unfolded protein response. BMC Biol. 2018, 16, 81. [Google Scholar] [CrossRef]
- Coates, P.J.; Nenutil, R.; McGregor, A.; Picksley, S.M.; Crouch, D.H.; Hall, P.A.; Wright, E.G. Mammalian prohibitin proteins respond to mitochondrial stress and decrease during cellular senescence. Exp. Cell Res. 2001, 265, 262–273. [Google Scholar] [CrossRef]
- Nijtmans, L.G.; Artal Sanz, M.; Bucko, M.; Farhoud, M.H.; Feenstra, M.; Hakkaart, G.A.; Zeviani, M.; Grivell, L.A. Shy1p occurs in a high molecular weight complex and is required for efficient assembly of cytochrome c oxidase in yeast. FEBS Lett. 2001, 498, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Hernando-Rodríguez, B.; Artal-Sanz, M. Mitochondrial Quality Control Mechanisms and the PHB (Prohibitin) Complex. Cells 2018, 7, 238. [Google Scholar] [CrossRef] [PubMed]
- Pernas, L.; Scorrano, L. Mito-Morphosis: Mitochondrial Fusion, Fission, and Cristae Remodeling as Key Mediators of Cellular Function. Annu. Rev. Physiol. 2016, 78, 505–531. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liu, C.Y.; Zhang, X.J.; Feng, C.; Zhou, L.Y.; Zhao, Y.; Li, P.F. miR-361-regulated prohibitin inhibits mitochondrial fission and apoptosis and protects heart from ischemia injury. Cell Death Differ. 2015, 22, 1058–1068. [Google Scholar] [CrossRef] [PubMed]
- Signorile, A.; Santeramo, A.; Tamma, G.; Pellegrino, T.; D’Oria, S.; Lattanzio, P.; De Rasmo, D. Mitochondrial cAMP prevents apoptosis modulating Sirt3 protein level and OPA1 processing in cardiac myoblast cells. Biochim. Biophys. Acta 2017, 1864, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Wai, T.; Baker, M.J.; Kladt, N.; Schauss, A.C.; Rugarli, E.; Langer, T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 2014, 204, 919–929. [Google Scholar] [CrossRef] [Green Version]
- Rainbolt, T.K.; Lebeau, J.; Puchades, C.; Wiseman, R.L. reciprocal degradation of YME1L and OMA1 adapts mitochondrial proteolytic activity during stress. Cell Rep. 2016, 14, 2041–2049. [Google Scholar] [CrossRef]
- Sato, S.; Murata, A.; Orihara, T.; Shirakawa, T.; Suenaga, K.; Kigoshi, H.; Uesugi, M. Marine natural product aurilide activates the OPA1-mediated apoptosis by binding to prohibitin. Chem. Biol. 2011, 18, 131–139. [Google Scholar] [CrossRef]
- Zhang, K.; Li, H.; Song, Z. Membrane depolarization activates the mitochondrial protease OMA1 by stimulating self-cleavage. EMBO Rep. 2014, 15, 576–585. [Google Scholar] [CrossRef] [Green Version]
- Klecker, T.; Wemmer, M.; Haag, M.; Weig, A.; Böckler, S.; Langer, T.; Nunnari, J.; Westermann, B. Interaction of MDM33 with mitochondrial inner membrane homeostasis pathways in yeast. Sci. Rep. 2015, 5, 18344. [Google Scholar] [CrossRef] [Green Version]
- Muraguchi, T.; Kawawa, A.; Kubota, S. Prohibitin protects against hypoxia-induced H9c2 cardiomyocyte cell death. Biomed. Res. 2010, 31, 113–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osman, C.; Merkwirth, C.; Langer, T. Prohibitins and the functional compartmentalization of mitochondrial membranes. J. Cell Sci. 2009, 122, 3823–3830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalvez, F.; Schug, Z.T.; Houtkooper, R.H.; MacKenzie, E.D.; Brooks, D.G.; Wanders, R.J.; Petit, P.X.; Vaz, F.M.; Gottlieb, E. Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J. Cell Biol. 2008, 183, 681–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Gestel, R.A.; Rijken, P.J.; Surinova, S.; O’Flaherty, M.; Heck, A.J.; Killian, J.A.; de Kroon, A.I.; Slijper, M. The influence of the acyl chain composition of cardiolipin on the stability of mitochondrial complexes; an unexpected effect of cardiolipin in alpha-ketoglutarate dehydrogenase and prohibitin complexes. J. Proteomics 2010, 73, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Ryan, M.T.; Schlame, M.; Zhao, M.; Gu, Z.; Klingenberg, M.; Pfanner, N.; Greenberg, M.L. Absence of cardiolipin in the crd1 null mutant results in decreased mitochondrial membrane potential and reduced mitochondrial function. J. Biol. Chem. 2000, 275, 22387–22394. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, R.; Lartigue, L.; Perkins, G.; Scott, R.T.; Dixit, A.; Kushnareva, Y.; Kuwana, T.; Ellisman, M.H.; Newmeyer, D.D. Opa1-mediated cristae opening is Bax/Bak and BH3 dependent, required for apoptosis, and independent of Bak oligomerization. Mol. Cell 2008, 31, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Quintana-Cabrera, R.; Quirin, C.; Glytsou, C.; Corrado, M.; Urbani, A.; Pellattiero, A.; Calvo, E.; Vázquez, J.; Enríquez, J.A.; Gerle, C.; et al. The cristae modulator Optic atrophy 1 requires mitochondrial ATP synthase oligomers to safeguard mitochondrial function. Nat. Commun. 2018, 9, 3399. [Google Scholar] [CrossRef]
- Li, J.; Aung, L.H.; Long, B.; Qin, D.; An, S.; Li, P. miR-23a binds to p53 and enhances its association with miR-128 promoter. Sci. Rep. 2015, 5, 16422. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.; Zhai, J.; Zhu, H.; Kyprianou, N. Prohibitin regulates TGF-beta induced apoptosis as a downstream effector of Smad dependent and -independent signaling. Prostate 2010, 70, 17–26. [Google Scholar] [CrossRef]
- Wong, P.F.; Cheong, W.F.; Shu, M.H.; The, C.H.; Chan, K.L.; AbuBakar, S. Eurycomanone suppresses expression of lung cancer cell tumor markers, prohibitin, annexin 1 and endoplasmic reticulum protein 28. Phytomedicine 2012, 19, 138–144. [Google Scholar] [CrossRef]
- Peng, Y.T.; Chen, P.; Ouyang, R.Y.; Song, L. Multifaceted role of prohibitin in cell survival and apoptosis. Apoptosis 2015, 20, 1135–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tortelli, T.C., Jr.; de Godoy, L.M.F.; de Souza, G.A.; Bonatto, D.; Otake, A.H.; de Freitas Saito, R.; Rosa, J.C.; Greene, L.J.; Chammas, R. Accumulation of prohibitin is a common cellular response to different stressing stimuli and protects melanoma cells from ER stress and chemotherapy-induced cell death. Oncotarget 2017, 8, 43114–43129. [Google Scholar]
- Satheesh Kumar, M.K.; Nair, S.; Mony, U.; Kalingavarman, S.; Venkat, R.; Sivanarayanan, T.B.; Unni, A.K.K.; Rajeshkannan, R.; Anandakuttan, A.; Radhakrishnan, S.; et al. Significance of elevated Prohibitin 1 levels in Multiple Sclerosis patients lymphocytes towards the assessment of subclinical disease activity and its role in the central nervous system pathology of disease. Int. J. Biol. Macromol. 2018, 110, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coates, P.J.; Jamieson, D.J.; Smart, K.; Prescott, A.R.; Hall, P.A. The prohibitin family of mitochondrial proteins regulate replicative lifespan. Curr. Biol. 1997, 7, 607–610. [Google Scholar] [CrossRef] [Green Version]
- Kenyon, C.J. The genetics of ageing. Nature 2010, 464, 504–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.Y.; Wang, D.; Kim, A.K.; Lau, E.; Lin, A.J.; Liem, D.A.; Zhang, J.; Zong, N.C.; Lam, M.P.; Ping, P. Metabolic labeling reveals proteome dynamics of mouse mitochondria. Mol. Cell. Proteomics 2012, 11, 1586–1594. [Google Scholar] [CrossRef] [PubMed]
- Dieteren, C.E.; Koopman, W.J.; Swarts, H.G.; Peters, J.G.; Maczuga, P.; Gemst, J.J.; Masereeuw, R.; Smeitink, J.A.; Nijtmans, L.G.; Willems, P.H. Subunit-specific incorporation efficiency and kinetics in mitochondrial complex I homeostasis. J. Biol. Chem. 2012, 287, 41851–41860. [Google Scholar] [CrossRef]
- De Rasmo, D.; Signorile, A.; Santeramo, A.; Larizza, M.; Lattanzio, P.; Capitanio, G.; Papa, S. Intramitochondrial adenylyl cyclase controls the turnover of nuclear-encoded subunits and activity of mammalian complex I of the respiratory chain. Biochim. Biophys. Acta 2015, 1853, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Lazarou, M.; McKenzie, M.; Ohtake, A.; Thorburn, D.R.; Ryan, M.T. Analysis of the assembly profiles for mitochondrial- and nuclear-DNA-encoded subunits into complex I. Mol. Cell. Biol. 2007, 27, 4228–4237. [Google Scholar] [CrossRef]
- De Rasmo, D.; Signorile, A.; Larizza, M.; Pacelli, C.; Cocco, T.; Papa, S. Activation of the cAMP cascade in human fibroblast cultures rescues the activity of oxidatively damaged complex I. Free Radic. Biol. Med. 2012, i, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Miwa, S.; Jow, H.; Baty, K.; Johnson, A.; Czapiewski, R.; Saretzki, G.; Treumann, A.; von Zglinicki, T. Low abundance of the matrix arm of complex I in mitochondria predicts longevity in mice. Nat. Commun. 2014, 5, 3837. [Google Scholar] [CrossRef] [PubMed]
- Vogel, R.O.; Janssen, R.J.; Ugalde, C.; Grovenstein, M.; Huijbens, R.J.; Visch, H.J.; van den Heuvel, L.P.; Willems, P.H.; Zeviani, M.; Smeitink, J.A.; et al. Human mitochondrial complex I assembly is mediated by NDUFAF1. FEBS J. 2005, 272, 5317–5326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papa, S.; De Rasmo, D. Complex I deficiencies in neurological disorders. Trends Mol. Med. 2013, 19, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.C.; Yanos, M.E.; Kayser, E.B.; Quintana, A.; Sangesland, M.; Castanza, A.; Uhde, L.; Hui, J.; Wall, V.Z.; Gagnidze, A.; et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 2013, 342, 1524–1528. [Google Scholar] [CrossRef] [PubMed]
- Boffoli, D.; Scacco, S.C.; Vergari, R.; Persio, M.T.; Solarino, G.; Laforgia, R.; Papa, S. Ageing is associated in females with a decline in the content and activity on the b-c1 complex in skeletal muscle mitochondria. Biochim. Biophys. Acta 1996, 1315, 66–72. [Google Scholar] [CrossRef]
- Alexander, C.; Votruba, M.; Pesch, U.E.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef]
- Opalińska, M.; Jańska, H. AAA Proteases: Guardians of Mitochondrial Function and Homeostasis. Cells 2018, 7, 163. [Google Scholar] [CrossRef]
- Schapira, A.H.; Tolosa, E. Molecular and clinical prodrome of Parkinson disease: Implications for treatment. Nat. Rev. Neurol. 2010, 6, 309–317. [Google Scholar] [CrossRef]
- Piccoli, C.; Sardanelli, A.; Scrima, R.; Ripoli, M.; Quarato, G.; D’Aprile, A.; Bellomo, F.; Scacco, S.; De Michele, G.; Filla, A.; et al. Mitochondrial respiratory dysfunction in familiar parkinsonism associated with PINK1 mutation. Neurochem. Res. 2008, 33, 2565–2574. [Google Scholar] [CrossRef]
- Pacelli, C.; De Rasmo, D.; Signorile, A.; Grattagliano, I.; di Tullio, G.; D’Orazio, A.; Nico, B.; Comi, G.P.; Ronchi, D.; Ferranini, E.; et al. Mitochondrial defect and PGC-1α dysfunction in parkin-associated familial Parkinson’s disease. Biochim. Biophys. Acta 2011, 1812, 1041–1053. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Austin, G.L.; Young, L.E.A.; Johnson, L.A.; Sun, R. Mitochondrial Metabolism in Major Neurological Diseases. Cells 2018, 7, 229. [Google Scholar] [CrossRef]
- Lachén-Montes, M.; González-Morales, A.; Zelaya, M.V.; Pérez-Valderrama, E.; Ausín, K.; Ferrer, I.; Fernández-Irigoyen, J.; Santamaría, E. Olfactory bulb neuroproteomics reveals a chronological perturbation of survival routes and a disruption of prohibitin complex during Alzheimer’s disease progression. Sci. Rep. 2017, 7, 9115. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. Endosome function and dysfunction in Alzheimer’s disease and other neurodegenerative diseases. Neurobiol. Aging. 2005, 26, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, G.; Farrace, M.G.; Mastroberardino, P.G.; Viti, I.; Fimia, G.M.; Van Beeumen, J.; Devreese, B.; Melino, G.; Molinaro, G.; Busceti, C.L.; et al. Transglutaminase 2 ablation leads to defective function of mitochondrial respiratory complex I affecting neuronal vulnerability in experimental models of extrapyramidal disorders. J. Neurochem. 2007, 100, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emma, F.; Montini, G.; Parikh, S.M.; Salviati, L. Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat. Rev. Nephrol. 2016, 12, 267–280. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, P.; Keegan, J.; Schaefer, H. Mechanism of aminonucleoside-induced nephrosis in the rat. III. Kidney mitochondrial phosphorylation and dephosphorylation activity. Proc. Soc. Exp. Biol. Med. 1963, 112, 96–101. [Google Scholar] [CrossRef]
- Town, M.; Jean, G.; Cherqui, S.; Attard, M.; Forestier, L.; Whitmore, S.A.; Callen, D.F.; Gribouval, O.; Broyer, M.; Bates, G.P.; et al. A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat. Genet. 1998, 18, 319–324. [Google Scholar] [CrossRef]
- Gahl, W.A.; Bashan, N.; Tietze, F.; Bernardini, I.; Schulman, J.D. Cystine transport is defective in isolated leukocyte lysosomes from patients with cystinosis. Science 1982, 217, 1263–1265. [Google Scholar] [CrossRef]
- Cherqui, S.; Courtoy, P.J. The renal Fanconi syndrome in cystinosis: Pathogenic insights and therapeutic perspectives. Nat. Rev. Nephrol. 2017, 13, 115–131. [Google Scholar] [CrossRef]
- Festa, B.P.; Chen, Z.; Berquez, M.; Debaix, H.; Tokonami, N.; Prange, J.A.; Hoek, G.V.; Alessio, C.; Raimondi, A.; Nevo, N.; et al. Impaired autophagy bridges lysosomal storage disease and epithelial dysfunction in the kidney. Nat. Commun. 2018, 9, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellomo, F.; Signorile, A.; Tamma, G.; Ranieri, M.; Emma, F.; De Rasmo, D. Impact of atypical mitochondrial cyclic-AMP level in nephropathic cystinosis. Cell. Mol. Life Sci. 2018, 75, 3411–3422. [Google Scholar] [CrossRef] [PubMed]
- Vásquez-Trincado, C.; García-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 2016, 594, 509–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, D.; Tangutur, A.D.; Khatua, T.N.; Saxena, P.; Banerjee, S.K.; Bhadra, M.P. A proteomic view of isoproterenol induced cardiac hypertrophy: Prohibitin identified as a potential biomarker in rats. J. Transl. Med. 2013, 11, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Long, B.; Zhou, L.Y.; Liu, F.; Zhou, Q.Y.; Liu, C.Y.; Fan, Y.Y.; Li, P.F. CARL lncRNA inhibits anoxia-induced mitochondrial fission and apoptosis in cardiomyocytes by impairing miR-539-dependent PHB2 downregulation. Nat. Commun. 2014, 5, 3596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhang, L.; Liang, J. Unraveling the Expression Profiles of Long Noncoding RNAs in Rat Cardiac Hypertrophy and Functions of lncRNA BC088254 in Cardiac Hypertrophy Induced by Transverse Aortic Constriction. Cardiology 2016, 134, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Moncunill-Massaguer, C.; Saura-Esteller, J.; Pérez-Perarnau, A.; Palmeri, C.M.; Núñez-Vázquez, S.; Cosialls, A.M.; González-Gironès, D.M.; Pomares, H.; Korwitz, A.; Preciado, S.; et al. A novel prohibitin-binding compound induces the mitochondrial apoptotic pathway through NOXA and BIM upregulation. Oncotarget 2015, 6, 41750–41765. [Google Scholar] [CrossRef] [Green Version]
- Sievers, C.; Billig, G.; Gottschalk, K.; Rudel, T. Prohibitins are required for cancer cell proliferation and adhesion. PLoS ONE 2010, 5, e12735. [Google Scholar] [CrossRef]
- Kang, X.; Zhang, L.; Sun, J.; Ni, Z.; Ma, Y.; Chen, X.; Sheng, X.; Chen, T. Prohibitin: A potential biomarker for tissue-based detection of gastric cancer. J. Gastroenterol. 2008, 43, 618–625. [Google Scholar] [CrossRef]
- Ren, H.Z.; Wang, J.S.; Wang, P.; Pan, G.Q.; Wen, J.F.; Fu, H.; Shan, X.Z. Increased expression of prohibitin and its relationship with poor prognosis in esophageal squamous cell carcinoma. Pathol. Oncol. Res. 2010, 16, 515–522. [Google Scholar] [CrossRef]
- Chen, D.; Chen, F.; Lu, X.; Yang, X.; Xu, Z.; Pan, J.; Huang, Y.; Lin, H.; Chi, P. Identification of prohibitin as a potential biomarker for colorectal carcinoma based on proteomics technology. Int. J. Oncol. 2010, 37, 355–365. [Google Scholar] [PubMed]
- Ummanni, R.; Junker, H.; Zimmermann, U.; Venz, S.; Teller, S.; Giebel, J.; Scharf, C.; Woenckhaus, C.; Dombrowski, F.; Walther, R. Prohibitin identified by proteomic analysis of prostate biopsies distinguishes hyperplasia and cancer. Cancer Lett. 2008, 266, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Webster, L.R.; Provan, P.J.; Graham, D.J.; Byth, K.; Walker, R.L.; Davis, S.; Salisbury, E.L.; Morey, A.L.; Ward, R.L.; Hawkins, N.J.; et al. Prohibitin expression is associated with high grade breast cancer but is not a driver of amplification at 17q21.33. Pathology 2013, 45, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.F.; Wu, H.; Wang, Y.W.; Chang, T.Y.; Chan, S.H.; Lin, Y.P.; Liu, H.S.; Chow, N.H. Prohibitin in the pathogenesis of transitional cell bladder cancer. Anticancer Res. 2007, 27, 895–900. [Google Scholar] [PubMed]
- Franzoni, A.; Dima, M.; D’Agostino, M.; Puppin, C.; Fabbro, D.; Loreto, C.D.; Pandolfi, M.; Puxeddu, E.; Moretti, S.; Celano, M.; et al. Prohibitin is overexpressed in papillary thyroid carcinomas bearing the BRAF(V600E) mutation. Thyroid 2009, 19, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.A.; Robles-Escajeda, E.; Oaxaca, D.M.; Padilla, D.L.; Kirken, R.A. The prohibitin protein complex promotes mitochondrial stabilization and cell survival in hematologic malignancies. Oncotarget 2017, 8, 65445–65456. [Google Scholar] [CrossRef] [PubMed]
- Kakehashi, A.; Ishii, N.; Shibata, T.; Wei, M.; Okazaki, E.; Tachibana, T.; Fukushima, S.; Wanibuchi, H. Mitochondrial prohibitins and septin 9 are implicated in the onset of rat hepatocarcinogenesis. Toxicol. Sci. 2011, 119, 61–72. [Google Scholar] [CrossRef]
- Xu, Z.; Wu, J.; Zha, X. Up-regulation of prohibitin 1 is involved in the proliferation and migration of liver cancer cells. SCI China Life Sci. 2011, 54, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Signorile, S.; De Rasmo, D.; Cormio, A.; Musicco, C.; Rossi, R.; Fortarezza, F.; Palese, L.L.; Loizzi, V.; Resta, L.; Scillitani, G.; et al. Altered mitochondrial function, biogenesis and structure in ovarian cancer: Involvement of cAMP pathway, SIRT3, OPA1, DRP1 and PHB2 proteins. Manuscript in preparation.
- Chiu, C.F.; Ho, M.Y.; Peng, J.M.; Hung, S.W.; Lee, W.H.; Liang, C.M.; Liang, S.M. Raf activation by Ras and promotion of cellular metastasis require phosphorylation of prohibitin in the raft domain of the plasma membrane. Oncogene 2013, 32, 777–787. [Google Scholar] [CrossRef]
- Polier, G.; Neumann, J.; Thuaud, F.; Ribeiro, N.; Gelhaus, C.; Schmidt, H.; Giaisi, M.; Köhler, R.; Müller, W.W.; Proksch, P.; et al. The natural anticancer compounds rocaglamides inhibit the Raf-MEK-ERK pathway by targeting prohibitin 1 and 2. Chem. Biol. 2012, 19, 1093–1104. [Google Scholar] [CrossRef]
- Yurugi, H.; Marini, F.; Weber, C.; David, K.; Zhao, Q.; Binder, H.; Désaubry, L.; Rajalingam, K. Targeting prohibitins with chemical ligands inhibits KRAS-mediated lung tumours. Oncogene 2017, 36, 5914. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xu, L.; Yang, Y.; Dong, L.; Zhao, B.; Lu, J.; Zhang, T.; Zhao, Y. A novel prognostic marker and immunogenic membrane antigen: Prohibitin (PHB) in pancreatic cancer. Clin. Transl. Gastroenterol. 2018, 9, 178. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Signorile, A.; Sgaramella, G.; Bellomo, F.; De Rasmo, D. Prohibitins: A Critical Role in Mitochondrial Functions and Implication in Diseases. Cells 2019, 8, 71. https://doi.org/10.3390/cells8010071
Signorile A, Sgaramella G, Bellomo F, De Rasmo D. Prohibitins: A Critical Role in Mitochondrial Functions and Implication in Diseases. Cells. 2019; 8(1):71. https://doi.org/10.3390/cells8010071
Chicago/Turabian StyleSignorile, Anna, Giuseppe Sgaramella, Francesco Bellomo, and Domenico De Rasmo. 2019. "Prohibitins: A Critical Role in Mitochondrial Functions and Implication in Diseases" Cells 8, no. 1: 71. https://doi.org/10.3390/cells8010071
APA StyleSignorile, A., Sgaramella, G., Bellomo, F., & De Rasmo, D. (2019). Prohibitins: A Critical Role in Mitochondrial Functions and Implication in Diseases. Cells, 8(1), 71. https://doi.org/10.3390/cells8010071