NUP214 in Leukemia: It’s More than Transport


Abstract
:1. Introduction
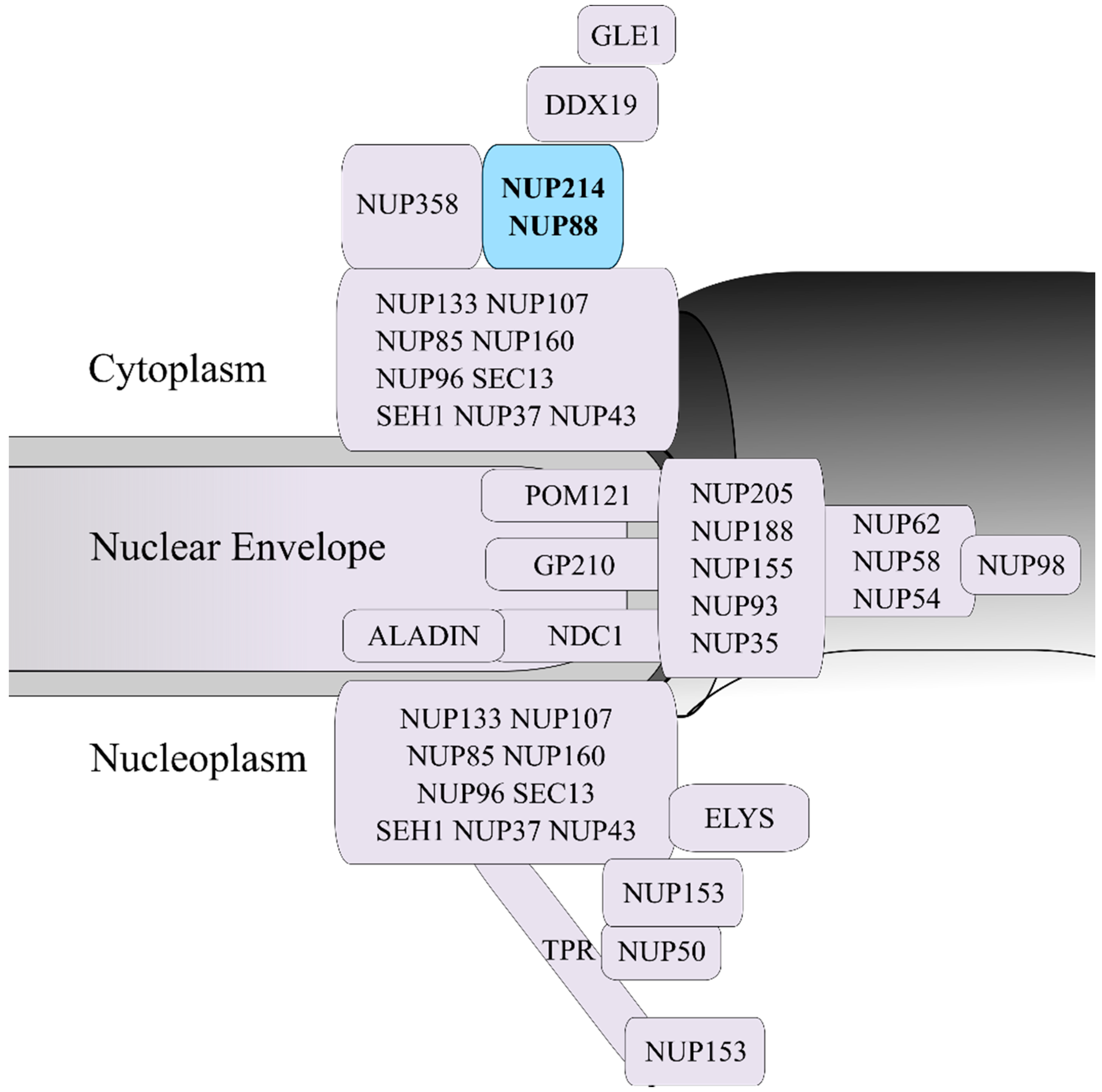
2. NUP214 Is Critical for Nucleocytoplasmic Transport
3. NUP214 Is a Recurrent Player in Acute Leukemia
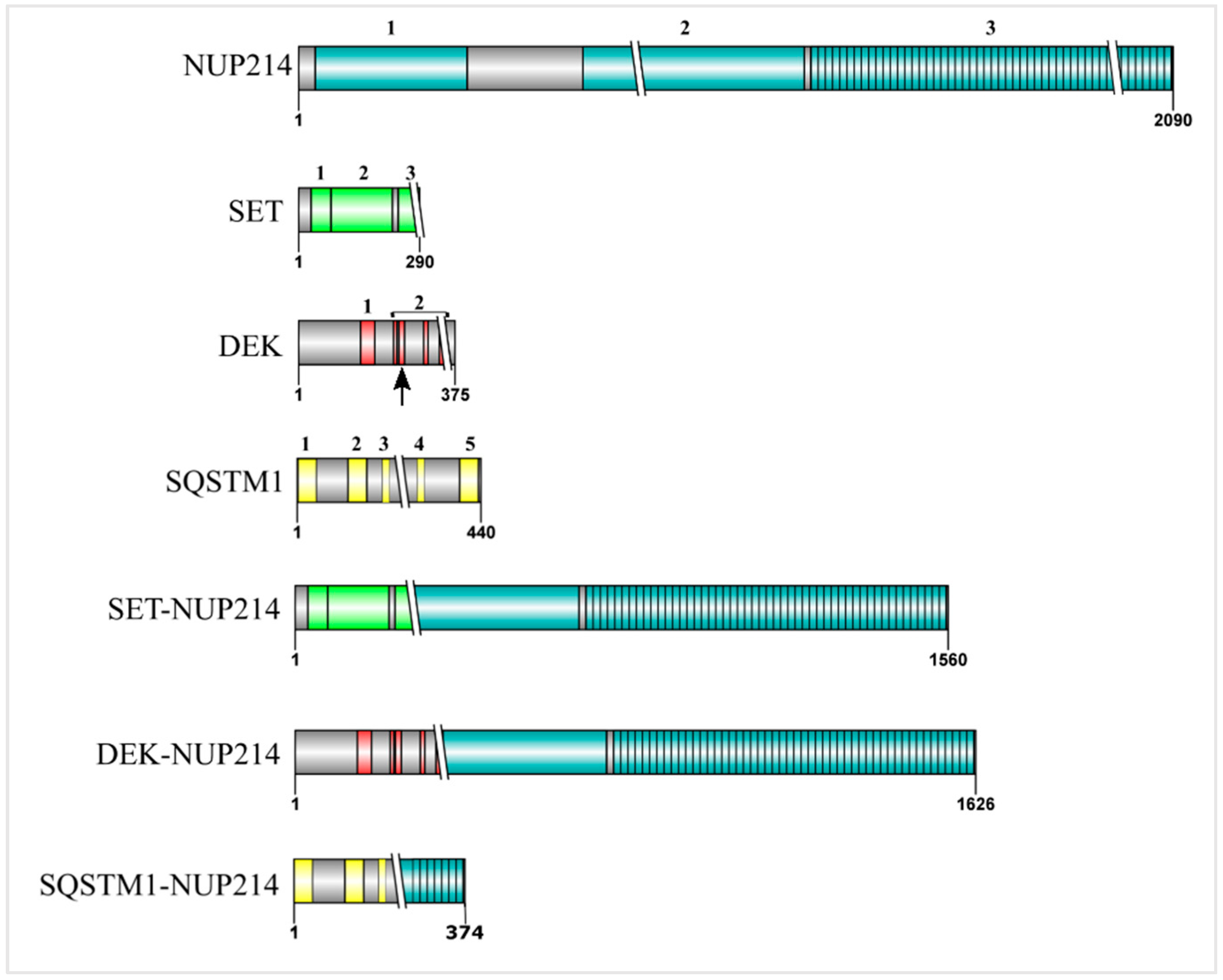
4. SET-NUP214 and DEK-NUP214: Same, Same, but Different
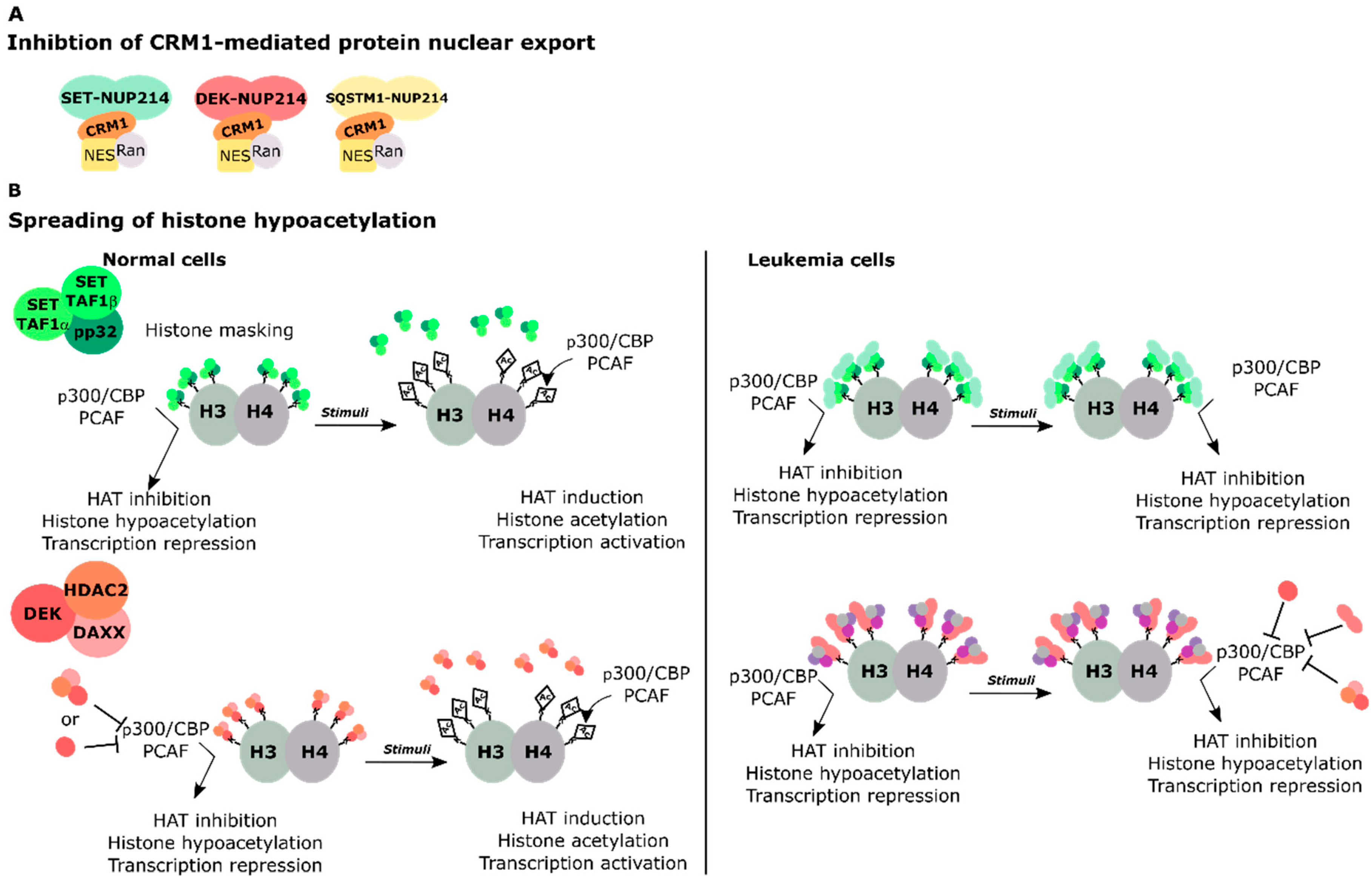
4.1. SET-NUP214 and DEK-NUP214 Act via Shared Molecular Mechanisms
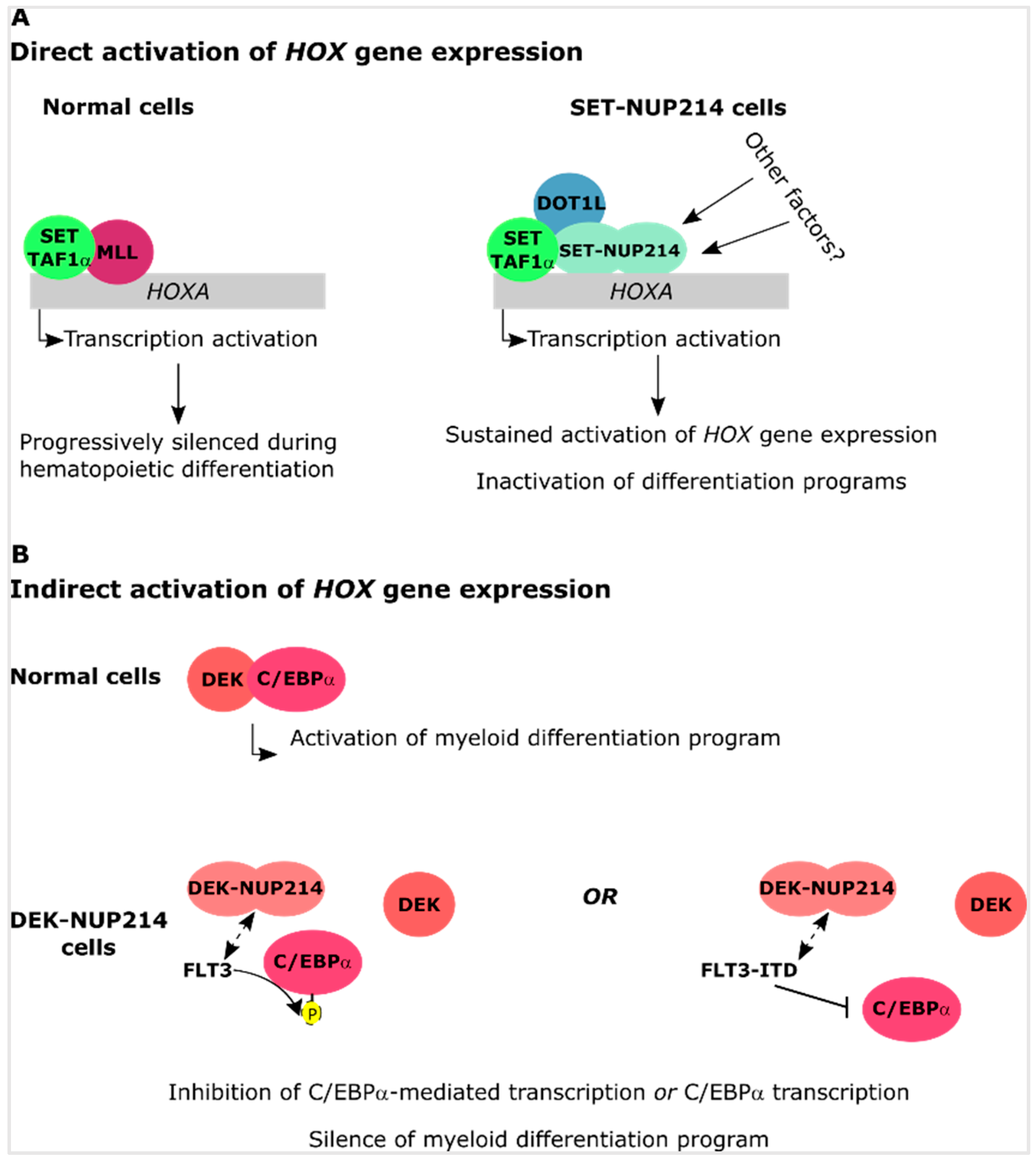
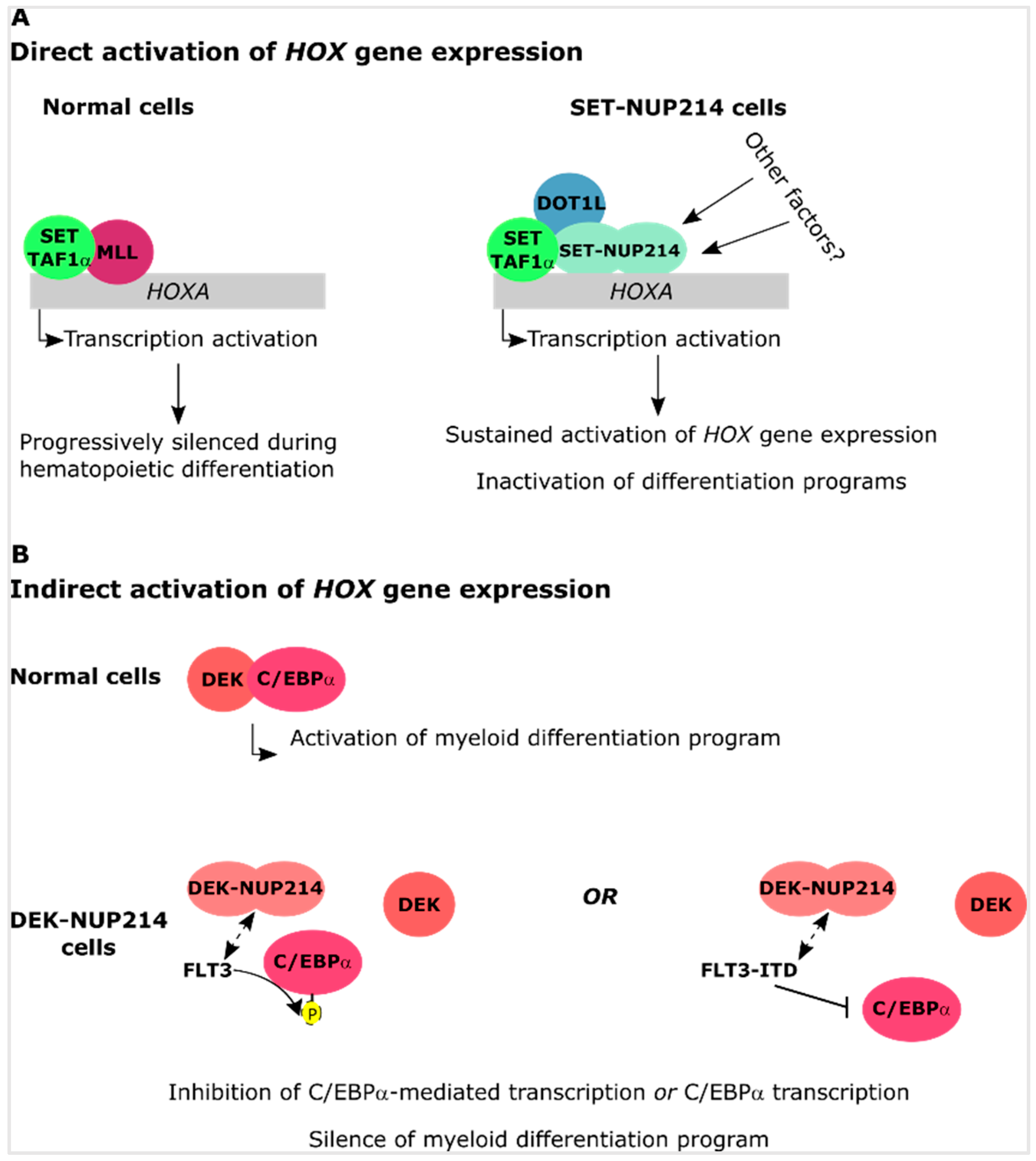
4.2. SET-NUP214 and DEK-NUP214 Associate with Distinct Gene Expression Profiles
5. The SET Protein
6. The DEK Protein
7. SQSTM1-NUP214: Playing with Autophagy
8. Conclusions
Funding
Conflicts of Interest
References
- Ferlay, J.S.I.; Ervik, M.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11. Available online: http://globocan.iarc.fr (accessed on 24 November 2018).
- The National Cancer Institute. Available online: www.cancer.gov (accessed on 24 November 2018).
- Cancer Research, UK. Available online: http://www.cancerresearchuk.org (accessed on 24 November 2018).
- Leukemia & Lymphoma Society. Available online: http://www.lls.org/ (accessed on 5 November 2018).
- Behrmann, L.; Wellbrock, J.; Fiedler, W. Acute Myeloid Leukemia and the Bone Marrow Niche-Take a Closer Look. Front. Oncol. 2018, 8, 444. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, T.U. Modularity within the architecture of the nuclear pore complex. Curr. Opin. Struct. Biol. 2005, 15, 221–226. [Google Scholar] [CrossRef]
- Devos, D.; Dokudovskaya, S.; Williams, R.; Alber, F.; Eswar, N.; Chait, B.T.; Rout, M.P.; Sali, A. Simple fold composition and modular architecture of the nuclear pore complex. Proc. Natl. Acad. Sci. USA 2006, 103, 2172–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alber, F.; Dokudovskaya, S.; Veenhoff, L.M.; Zhang, W.; Kipper, J.; Devos, D.; Suprapto, A.; Karni-Schmidt, O.; Williams, R.; Chait, B.T.; et al. The molecular architecture of the nuclear pore complex. Nature 2007, 450, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Lim, R.Y.; Aebi, U.; Fahrenkrog, B. Towards reconciling structure and function in the nuclear pore complex. Histochem. Cell Biol. 2008, 129, 105–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depping, R.; Steinhoff, A.; Schindler, S.G.; Friedrich, B.; Fagerlund, R.; Metzen, E.; Hartmann, E.; Kohler, M. Nuclear translocation of hypoxia-inducible factors (HIFs): Involvement of the classical importin alpha/beta pathway. Biochim. Biophys. Acta 2008, 1783, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Kose, S.; Imamoto, N. Nucleocytoplasmic transport under stress conditions and its role in HSP70 chaperone systems. Biochim. Biophys. Acta 2014, 1840, 2953–2960. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, A.; Hetzer, M.W. Nuclear pore proteins and the control of genome functions. Genes Dev. 2015, 29, 337–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, R.Y.; Huang, N.P.; Koser, J.; Deng, J.; Lau, K.H.; Schwarz-Herion, K.; Fahrenkrog, B.; Aebi, U. Flexible phenylalanine-glycine nucleoporins as entropic barriers to nucleocytoplasmic transport. Proc. Natl. Acad. Sci. USA 2006, 103, 9512–9517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denning, D.P.; Patel, S.S.; Uversky, V.; Fink, A.L.; Rexach, M. Disorder in the nuclear pore complex: The FG repeat regions of nucleoporins are natively unfolded. Proc. Natl. Acad. Sci. USA 2003, 100, 2450–2455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.S.; Belmont, B.J.; Sante, J.M.; Rexach, M.F. Natively unfolded nucleoporins gate protein diffusion across the nuclear pore complex. Cell 2007, 129, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Rexach, M.; Blobel, G. Protein import into nuclei: Association and dissociation reactions involving transport substrate, transport factors, and nucleoporins. Cell 1995, 83, 683–692. [Google Scholar] [CrossRef]
- Yang, W. ‘Natively unfolded’ nucleoporins in nucleocytoplasmic transport: Clustered or evenly distributed? Nucleus 2011, 2, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Lim, R.Y.; Huang, B.; Kapinos, L.E. How to operate a nuclear pore complex by Kap-centric control. Nucleus 2015, 6, 366–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, P.S.; Aramburu, I.V.; Mercadante, D.; Tyagi, S.; Chowdhury, A.; Spitz, D.; Shammas, S.L.; Grater, F.; Lemke, E.A. Two Differential Binding Mechanisms of FG-Nucleoporins and Nuclear Transport Receptors. Cell Rep. 2018, 22, 3660–3671. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.; Asano, S.; Nakamura, T.; Adachi, M.; Yoshida, M.; Yanagida, M.; Nishida, E. CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature 1997, 390, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Askjaer, P.; Bachi, A.; Wilm, M.; Bischoff, F.R.; Weeks, D.L.; Ogniewski, V.; Ohno, M.; Niehrs, C.; Kjems, J.; Mattaj, I.W.; et al. RanGTP-regulated interactions of CRM1 with nucleoporins and a shuttling DEAD-box helicase. Mol. Cell. Biol. 1999, 19, 6276–6285. [Google Scholar] [CrossRef]
- Thakar, K.; Karaca, S.; Port, S.A.; Urlaub, H.; Kehlenbach, R.H. Identification of CRM1-dependent Nuclear Export Cargos Using Quantitative Mass Spectrometry. Mol. Cell. Proteom. MCP 2013, 12, 664–678. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yamada, M.; Mabuchi, N.; Shida, H. Cellular requirements for CRM1 import and export. J. Biochem. 2003, 134, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, F.R.; Gorlich, D. RanBP1 is crucial for the release of RanGTP from importin beta-related nuclear transport factors. FEBS Lett. 1997, 419, 249–254. [Google Scholar] [CrossRef]
- Kehlenbach, R.H.; Dickmanns, A.; Kehlenbach, A.; Guan, T.; Gerace, L. A role for RanBP1 in the release of CRM1 from the nuclear pore complex in a terminal step of nuclear export. J. Cell Biol. 1999, 145, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Ribbeck, K.; Lipowsky, G.; Kent, H.M.; Stewart, M.; Gorlich, D. NTF2 mediates nuclear import of Ran. EMBO J. 1998, 17, 6587–6598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernad, R.; Engelsma, D.; Sanderson, H.; Pickersgill, H.; Fornerod, M. Nup214-Nup88 nucleoporin subcomplex is required for CRM1-mediated 60 S preribosomal nuclear export. J. Biol. Chem. 2006, 281, 19378–19386. [Google Scholar] [CrossRef] [PubMed]
- Hutten, S.; Kehlenbach, R.H. Nup214 is required for CRM1-dependent nuclear protein export in vivo. Mol. Cell. Biol. 2006, 26, 6772–6785. [Google Scholar] [CrossRef]
- Iwamoto, M.; Asakawa, H.; Hiraoka, Y.; Haraguchi, T. Nucleoporin Nup98: A gatekeeper in the eukaryotic kingdoms. Genes Cells Devoted Mol. Cell. Mech. 2010, 15, 661–669. [Google Scholar] [CrossRef]
- Oka, M.; Asally, M.; Yasuda, Y.; Ogawa, Y.; Tachibana, T.; Yoneda, Y. The mobile FG nucleoporin Nup98 is a cofactor for Crm1-dependent protein export. Mol. Biol. Cell 2010, 21, 1885–1896. [Google Scholar] [CrossRef]
- Roloff, S.; Spillner, C.; Kehlenbach, R.H. Several phenylalanine-glycine motives in the nucleoporin Nup214 are essential for binding of the nuclear export receptor CRM1. J. Biol. Chem. 2013, 288, 3952–3963. [Google Scholar] [CrossRef]
- van Deursen, J.; Boer, J.; Kasper, L.; Grosveld, G. G2 arrest and impaired nucleocytoplasmic transport in mouse embryos lacking the proto-oncogene CAN/Nup214. EMBO J. 1996, 15, 5574–5583. [Google Scholar] [CrossRef]
- Boer, J.; Bonten-Surtel, J.; Grosveld, G. Overexpression of the nucleoporin CAN/NUP214 induces growth arrest, nucleocytoplasmic transport defects, and apoptosis. Mol. Cell. Biol. 1998, 18, 1236–1247. [Google Scholar] [CrossRef]
- Chatel, G.; Fahrenkrog, B. Nucleoporins: Leaving the nuclear pore complex for a successful mitosis. Cell. Signal. 2011, 23, 1555–1562. [Google Scholar] [CrossRef]
- Chatel, G.; Fahrenkrog, B. Dynamics and diverse functions of nuclear pore complex proteins. Nucleus 2012, 3, 162–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, S.; Zhao, R.; Barsotti, A.M.; Ouwehand, A.; Fazollahi, M.; Coutavas, E.; Breuhahn, K.; Neumann, O.; Longerich, T.; Pusterla, T.; et al. Nuclear pore component Nup98 is a potential tumor suppressor and regulates posttranscriptional expression of select p53 target genes. Mol. Cell 2012, 48, 799–810. [Google Scholar] [CrossRef] [PubMed]
- Kalverda, B.; Pickersgill, H.; Shloma, V.V.; Fornerod, M. Nucleoporins directly stimulate expression of developmental and cell-cycle genes inside the nucleoplasm. Cell 2010, 140, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Capelson, M.; Liang, Y.; Schulte, R.; Mair, W.; Wagner, U.; Hetzer, M.W. Chromatin-bound nuclear pore components regulate gene expression in higher eukaryotes. Cell 2010, 140, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Fahrenkrog, B. Nucleoporin Gene Fusions and Hematopoietic Malignancies. New J. Sci. 2014, 2014, 18. [Google Scholar] [CrossRef]
- Martins, N.; Mendes, A.; Fahrenkrog, B. On the Effects of Leukemogenic Nucleoporin Fusion Proteins on Nucleocytoplasmic Transport and Gene Expression. In Nuclear-Cytoplasmic Transport; Yang, W., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 223–248. [Google Scholar] [CrossRef]
- Gough, S.M.; Slape, C.I.; Aplan, P.D. NUP98 gene fusions and hematopoietic malignancies: Common themes and new biologic insights. Blood 2011, 118, 6247–6257. [Google Scholar] [CrossRef]
- Lam, D.H.; Aplan, P.D. NUP98 gene fusions in hematologic malignancies. Leukemia 2001, 15, 1689–1695. [Google Scholar] [CrossRef] [Green Version]
- Slape, C.; Aplan, P.D. The role of NUP98 gene fusions in hematologic malignancy. Leukemia Lymphoma 2004, 45, 1341–1350. [Google Scholar] [CrossRef]
- Nakamura, T. NUP98 fusion in human leukemia: Dysregulation of the nuclear pore and homeodomain proteins. Int. J. Hematol. 2005, 82, 21–27. [Google Scholar] [CrossRef]
- Shima, Y.; Yumoto, M.; Katsumoto, T.; Kitabayashi, I. MLL is essential for NUP98-HOXA9-induced leukemia. Leukemia 2017, 31, 2200–2210. [Google Scholar] [CrossRef]
- Ahuja, H.G.; Popplewell, L.; Tcheurekdjian, L.; Slovak, M.L. NUP98 gene rearrangements and the clonal evolution of chronic myelogenous leukemia. Genes Chromosomes Cancer 2001, 30, 410–415. [Google Scholar] [CrossRef]
- Moore, M.A.; Chung, K.Y.; Plasilova, M.; Schuringa, J.J.; Shieh, J.H.; Zhou, P.; Morrone, G. NUP98 dysregulation in myeloid leukemogenesis. Ann. N. Y. Acad. Sci. 2007, 1106, 114–142. [Google Scholar] [CrossRef] [PubMed]
- Tosic, N.; Stojiljkovic, M.; Colovic, N.; Colovic, M.; Pavlovic, S. Acute myeloid leukemia with NUP98-HOXC13 fusion and FLT3 internal tandem duplication mutation: Case report and literature review. Cancer Genet. Cytogenet. 2009, 193, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.T.; Hess, J.L. Deregulation of the HOXA9/MEIS1 axis in acute leukemia. Curr. Opin Hematol. 2016, 23, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Sandahl, J.D.; Coenen, E.A.; Forestier, E.; Harbott, J.; Johansson, B.; Kerndrup, G.; Adachi, S.; Auvrignon, A.; Beverloo, H.B.; Cayuela, J.M.; et al. t(6;9)(p22;q34)/DEK-NUP214-rearranged pediatric myeloid leukemia: An international study of 62 patients. Haematologica 2014, 99, 865–872. [Google Scholar] [CrossRef]
- Ommen, H.B.; Touzart, A.; MacIntyre, E.; Kern, W.; Haferlach, T.; Haferlach, C.; Tobal, K.; Hokland, P.; Schnittger, S. The kinetics of relapse in DEK-NUP214-positive acute myeloid leukemia patients. Eur. J. Haematol. 2015, 95, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Slovak, M.L.; Gundacker, H.; Bloomfield, C.D.; Dewald, G.; Appelbaum, F.R.; Larson, R.A.; Tallman, M.S.; Bennett, J.M.; Stirewalt, D.L.; Meshinchi, S.; et al. A retrospective study of 69 patients with t(6;9)(p23;q34) AML emphasizes the need for a prospective, multicenter initiative for rare ’poor prognosis’ myeloid malignancies. Leukemia 2006, 20, 1295–1297. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Gao, L.; Jing, Y.; Xu, Y.Y.; Ding, Y.; Zhou, M.H.; Ma, C.; Li, M.Y.; Sun, J.Z.; Wang, L.L.; et al. Detection and clinical significance of gene rearrangements in Chinese patients with adult acute lymphoblastic leukemia. Leukemia lymphoma 2013, 54, 1521–1526. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.H.; Zhao, X.S.; Qin, Y.Z.; Lai, Y.Y.; Jiang, H. B-cell acute lymphoblastic leukemia associated with SET-NUP214 rearrangement: A case report and review of the literature. Oncol. Lett. 2016, 11, 2644–2650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulillo, S.M.; Powers, M.A.; Ullman, K.S.; Fahrenkrog, A.B. Changes in Nucleoporin Domain Topology in Response to Chemical Effectors. J. Mol. Biol. 2006, 363, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Gaik, M.; Flemming, D.; von Appen, A.; Kastritis, P.; Mucke, N.; Fischer, J.; Stelter, P.; Ori, A.; Bui, K.H.; Bassler, J.; et al. Structural basis for assembly and function of the Nup82 complex in the nuclear pore scaffold. J. Cell Biol. 2015, 208, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Bui, K.H.; von Appen, A.; DiGuilio, A.L.; Ori, A.; Sparks, L.; Mackmull, M.T.; Bock, T.; Hagen, W.; Andres-Pons, A.; Glavy, J.S.; et al. Integrated structural analysis of the human nuclear pore complex scaffold. Cell 2013, 155, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Von Moeller, H.; Basquin, C.; Conti, E. The mRNA export protein DBP5 binds RNA and the cytoplasmic nucleoporin NUP214 in a mutually exclusive manner. Nat. Struct. Mol. Biol. 2009, 16, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Katahira, J.; Strasser, K.; Podtelejnikov, A.; Mann, M.; Jung, J.U.; Hurt, E. The Mex67p-mediated nuclear mRNA export pathway is conserved from yeast to human. EMBO J. 1999, 18, 2593–2609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Port, S.A.; Monecke, T.; Dickmanns, A.; Spillner, C.; Hofele, R.; Urlaub, H.; Ficner, R.; Kehlenbach, R.H. Structural and Functional Characterization of CRM1-Nup214 Interactions Reveals Multiple FG-Binding Sites Involved in Nuclear Export. Cell Rep. 2015, 13, 690–702. [Google Scholar] [CrossRef] [Green Version]
- Napetschnig, J.; Blobel, G.; Hoelz, A. Crystal structure of the N-terminal domain of the human protooncogene Nup214/CAN. Proc. Natl. Acad. Sci. USA 2007, 104, 1783–1788. [Google Scholar] [CrossRef] [PubMed]
- Napetschnig, J.; Kassube, S.A.; Debler, E.W.; Wong, R.W.; Blobel, G.; Hoelz, A. Structural and functional analysis of the interaction between the nucleoporin Nup214 and the DEAD-box helicase Ddx19. Proc. Natl. Acad. Sci. USA 2009, 106, 3089–3094. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.H.; Correia, A.R.; Cai, S.W.; Huber, F.M.; Jette, C.A.; Hoelz, A. Structural and functional analysis of mRNA export regulation by the nuclear pore complex. Nat. Commun. 2018, 9, 2319. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjya, S.; Roy, K.S.; Ganguly, A.; Sarkar, S.; Panda, C.K.; Bhattacharyya, D.; Bhattacharyya, N.P.; Roychoudhury, S. Inhibition of nucleoporin member Nup214 expression by miR-133b perturbs mitotic timing and leads to cell death. Mol. Cancer 2015, 14, 42. [Google Scholar] [CrossRef] [PubMed]
- von Lindern, M.; Poustka, A.; Lerach, H.; Grosveld, G. The (6;9) chromosome translocation, associated with a specific subtype of acute nonlymphocytic leukemia, leads to aberrant transcription of a target gene on 9q34. Mol. Cell. Biol. 1990, 10, 4016–4026. [Google Scholar] [CrossRef] [PubMed]
- von Lindern, M.; Breems, D.; van Baal, S.; Adriaansen, H.; Grosveld, G. Characterization of the translocation breakpoint sequences of two DEK-CAN fusion genes present in t(6;9) acute myeloid leukemia and a SET-CAN fusion gene found in a case of acute undifferentiated leukemia. Genes Chromosomes Cancer 1992, 5, 227–234. [Google Scholar] [CrossRef] [PubMed]
- von Lindern, M.; Fornerod, M.; van Baal, S.; Jaegle, M.; de Wit, T.; Buijs, A.; Grosveld, G. The translocation (6;9), associated with a specific subtype of acute myeloid leukemia, results in the fusion of two genes, dek and can, and the expression of a chimeric, leukemia-specific dek-can mRNA. Mol. Cell. Biol. 1992, 12, 1687–1697. [Google Scholar] [CrossRef] [PubMed]
- Graux, C.; Cools, J.; Melotte, C.; Quentmeier, H.; Ferrando, A.; Levine, R.; Vermeesch, J.R.; Stul, M.; Dutta, B.; Boeckx, N.; et al. Fusion of NUP214 to ABL1 on amplified episomes in T-cell acute lymphoblastic leukemia. Nature genetics 2004, 36, 1084–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorello, P.; La Starza, R.; Di Giacomo, D.; Messina, M.; Puzzolo, M.C.; Crescenzi, B.; Santoro, A.; Chiaretti, S.; Mecucci, C. SQSTM1-NUP214: A new gene fusion in adult T-cell acute lymphoblastic leukemia. Haematologica 2010, 95, 2161–2163. [Google Scholar] [CrossRef] [PubMed]
- Levin, J.Z.; Berger, M.F.; Adiconis, X.; Rogov, P.; Melnikov, A.; Fennell, T.; Nusbaum, C.; Garraway, L.A.; Gnirke, A. Targeted next-generation sequencing of a cancer transcriptome enhances detection of sequence variants and novel fusion transcripts. Genome Biol. 2009, 10, R115. [Google Scholar] [CrossRef] [PubMed]
- Abe, A.; Yamamoto, Y.; Iba, S.; Okamoto, A.; Tokuda, M.; Inaguma, Y.; Yanada, M.; Morishima, S.; Kanie, T.; Tsuzuki, M.; et al. NUP214-RAC1 and RAC1-COL12A1 Fusion in Complex Variant Translocations Involving Chromosomes 6, 7 and 9 in an Acute Myeloid Leukemia Case with DEK-NUP214. Cytogenet. Genome Res. 2015, 146, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Von Lindern, M.; van Baal, S.; Wiegant, J.; Raap, A.; Hagemeijer, A.; Grosveld, G. Can, a putative oncogene associated with myeloid leukemogenesis, may be activated by fusion of its 3’ half to different genes: Characterization of the set gene. Mol. Cell. Biol. 1992, 12, 3346–3355. [Google Scholar] [CrossRef]
- Port, S.A.; Mendes, A.; Valkova, C.; Spillner, C.; Fahrenkrog, B.; Kaether, C.; Kehlenbach, R.H. The Oncogenic Fusion Proteins SET-Nup214 and Sequestosome-1 (SQSTM1)-Nup214 Form Dynamic Nuclear Bodies and Differentially Affect Nuclear Protein and Poly(A)+ RNA Export. J. Biol. Chem. 2016, 291, 23068–23083. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Cigdem, S.; Okuwaki, M.; Nagata, K. Leukemia-Associated Nup214 Fusion Proteins Disturb the XPO1-Mediated Nuclear-Cytoplasmic Transport Pathway and Thereby the NF-kappaB Signaling Pathway. Mol. Cell. Biol. 2016, 36, 1820–1835. [Google Scholar] [CrossRef]
- Zhou, M.H.; Yang, Q.M. NUP214 fusion genes in acute leukemia (Review). Oncol. Lett. 2014, 8, 959–962. [Google Scholar] [CrossRef] [Green Version]
- Van Vlierberghe, P.; van Grotel, M.; Tchinda, J.; Lee, C.; Beverloo, H.B.; van der Spek, P.J.; Stubbs, A.; Cools, J.; Nagata, K.; Fornerod, M.; et al. The recurrent SET-NUP214 fusion as a new HOXA activation mechanism in pediatric T-cell acute lymphoblastic leukemia. Blood 2008, 111, 4668–4680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oka, M.; Mura, S.; Yamada, K.; Sangel, P.; Hirata, S.; Maehara, K.; Kawakami, K.; Tachibana, T.; Ohkawa, Y.; Kimura, H.; et al. Chromatin-prebound Crm1 recruits Nup98-HoxA9 fusion to induce aberrant expression of Hox cluster genes. eLife 2016, 5, e09540. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.B.; McNamara, P.; Heo, S.; Turner, A.; Lane, W.S.; Chakravarti, D. Regulation of histone acetylation and transcription by INHAT, a human cellular complex containing the set oncoprotein. Cell 2001, 104, 119–130. [Google Scholar] [CrossRef]
- Kutney, S.N.; Hong, R.; Macfarlan, T.; Chakravarti, D. A signaling role of histone-binding proteins and INHAT subunits pp32 and Set/TAF-Ibeta in integrating chromatin hypoacetylation and transcriptional repression. J. Biol. Chem. 2004, 279, 30850–30855. [Google Scholar] [CrossRef]
- Gamble, M.J.; Erdjument-Bromage, H.; Tempst, P.; Freedman, L.P.; Fisher, R.P. The histone chaperone TAF-I/SET/INHAT is required for transcription in vitro of chromatin templates. Mol. Cell. Biol. 2005, 25, 797–807. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, K.B.; Son, H.J.; Chae, Y.C.; Oh, S.T.; Kim, D.W.; Pak, J.H.; Seo, S.B. H3K27 methylation and H3S28 phosphorylation-dependent transcriptional regulation by INHAT subunit SET/TAF-Ibeta. FEBS Lett. 2012, 586, 3159–3165. [Google Scholar] [CrossRef]
- Hollenbach, A.D.; McPherson, C.J.; Mientjes, E.J.; Iyengar, R.; Grosveld, G. Daxx and histone deacetylase II associate with chromatin through an interaction with core histones and the chromatin-associated protein Dek. J. Cell Sci. 2002, 115, 3319–3330. [Google Scholar]
- Waldmann, T.; Scholten, I.; Kappes, F.; Hu, H.G.; Knippers, R. The DEK protein—An abundant and ubiquitous constituent of mammalian chromatin. Gene 2004, 343, 1–9. [Google Scholar] [CrossRef]
- Ko, S.I.; Lee, I.S.; Kim, J.Y.; Kim, S.M.; Kim, D.W.; Lee, K.S.; Woo, K.M.; Baek, J.H.; Choo, J.K.; Seo, S.B. Regulation of histone acetyltransferase activity of p300 and PCAF by proto-oncogene protein DEK. FEBS Lett. 2006, 580, 3217–3222. [Google Scholar] [CrossRef] [Green Version]
- Privette Vinnedge, L.M.; Kappes, F.; Nassar, N.; Wells, S.I. Stacking the DEK: From chromatin topology to cancer stem cells. Cell Cycle 2013, 12, 51–66. [Google Scholar] [CrossRef] [Green Version]
- Sanden, C.; Jarvstrat, L.; Lennartsson, A.; Brattas, P.L.; Nilsson, B.; Gullberg, U. The DEK oncoprotein binds to highly and ubiquitously expressed genes with a dual role in their transcriptional regulation. Mol. Cancer 2014, 13, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervoni, N.; Detich, N.; Seo, S.B.; Chakravarti, D.; Szyf, M. The oncoprotein Set/TAF-1beta, an inhibitor of histone acetyltransferase, inhibits active demethylation of DNA, integrating DNA methylation and transcriptional silencing. J. Biol. Chem. 2002, 277, 25026–25031. [Google Scholar] [CrossRef] [PubMed]
- Muto, S.; Senda, M.; Akai, Y.; Sato, L.; Suzuki, T.; Nagai, R.; Senda, T.; Horikoshi, M. Relationship between the structure of SET/TAF-Ibeta/INHAT and its histone chaperone activity. Proc. Natl. Acad. Sci. USA 2007, 104, 4285–4290. [Google Scholar] [CrossRef] [PubMed]
- Koleva, R.I.; Ficarro, S.B.; Radomska, H.S.; Carrasco-Alfonso, M.J.; Alberta, J.A.; Webber, J.T.; Luckey, C.J.; Marcucci, G.; Tenen, D.G.; Marto, J.A. C/EBPalpha and DEK coordinately regulate myeloid differentiation. Blood 2012, 119, 4878–4888. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Malek, S.; Cowell, J.K.; Ren, M. Transformation of human CD34+ hematopoietic progenitor cells with DEK-NUP214 induces AML in an immunocompromised mouse model. Oncogene 2016, 35, 5686–5691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, S.; Campo, E.; Harris, N.; Jaffe, E.; Pileri, S.; Stein, H. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; IARC: Lyon, France, 2008; Volume 2. [Google Scholar]
- Ozbek, U.; Kandilci, A.; van Baal, S.; Bonten, J.; Boyd, K.; Franken, P.; Fodde, R.; Grosveld, G.C. SET-CAN, the product of the t(9;9) in acute undifferentiated leukemia, causes expansion of early hematopoietic progenitors and hyperproliferation of stomach mucosa in transgenic mice. Am. J. Pathol. 2007, 171, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Nouno, K.; Shimizu, R.; Yamamoto, M.; Nagata, K. Impairment of erythroid and megakaryocytic differentiation by a leukemia-associated and t(9;9)-derived fusion gene product, SET/TAF-Ibeta-CAN/Nup214. J. Cell. Physiol. 2008, 214, 322–333. [Google Scholar] [CrossRef]
- Oancea, C.; Ruster, B.; Henschler, R.; Puccetti, E.; Ruthardt, M. The t(6;9) associated DEK/CAN fusion protein targets a population of long-term repopulating hematopoietic stem cells for leukemogenic transformation. Leukemia 2010, 24, 1910–1919. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Jiang, L.; Zhong, M.L.; Li, J.F.; Li, B.S.; Peng, L.J.; Dai, Y.T.; Cui, B.W.; Yan, T.Q.; Zhang, W.N.; et al. Identification of fusion genes and characterization of transcriptome features in T-cell acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 2018, 115, 373–378. [Google Scholar] [CrossRef]
- Wang, Q.; Qiu, H.; Jiang, H.; Wu, L.; Dong, S.; Pan, J.; Wang, W.; Ping, N.; Xia, J.; Sun, A.; et al. Mutations of PHF6 are associated with mutations of NOTCH1, JAK1 and rearrangement of SET-NUP214 in T-cell acute lymphoblastic leukemia. Haematologica 2011, 96, 1808–1814. [Google Scholar] [CrossRef] [Green Version]
- Meng, Y.S.; Khoury, H.; Dick, J.E.; Minden, M.D. Oncogenic potential of the transcription factor LYL1 in acute myeloblastic leukemia. Leukemia 2005, 19, 1941–1947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, F.C.; Still, E.; Koche, R.P.; Yim, C.Y.; Takao, S.; Cifani, P.; Reed, C.; Gunasekera, S.; Ficarro, S.B.; Romanienko, P.; et al. MEF2C Phosphorylation Is Required for Chemotherapy Resistance in Acute Myeloid Leukemia. Cancer Discov. 2018, 8, 478–497. [Google Scholar] [CrossRef]
- Loven, M.A.; Muster, N.; Yates, J.R.; Nardulli, A.M. A novel estrogen receptor alpha-associated protein, template-activating factor Ibeta, inhibits acetylation and transactivation. Mol. Endocrinol. 2003, 17, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.B.; Macfarlan, T.; McNamara, P.; Hong, R.; Mukai, Y.; Heo, S.; Chakravarti, D. Regulation of histone acetylation and transcription by nuclear protein pp32, a subunit of the INHAT complex. J. Biol. Chem. 2002, 277, 14005–14010. [Google Scholar] [CrossRef] [PubMed]
- Schneider, R.; Bannister, A.J.; Weise, C.; Kouzarides, T. Direct binding of INHAT to H3 tails disrupted by modifications. J. Biol. Chem. 2004, 279, 23859–23862. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.S.; Oh, S.M.; Kim, S.M.; Lee, D.S.; Seo, S.B. Highly acidic C-terminal domain of pp32 is required for the interaction with histone chaperone, TAF-Ibeta. Biol. Pharm. Bull. 2006, 29, 2395–2398. [Google Scholar] [CrossRef] [PubMed]
- Ichijo, T.; Chrousos, G.P.; Kino, T. Activated glucocorticoid receptor interacts with the INHAT component Set/TAF-Ibeta and releases it from a glucocorticoid-responsive gene promoter, relieving repression: Implications for the pathogenesis of glucocorticoid resistance in acute undifferentiated leukemia with Set-Can translocation. Mol. Cell. Endocrinol. 2008, 283, 19–31. [Google Scholar] [CrossRef]
- Chae, Y.C.; Kim, K.B.; Kang, J.Y.; Kim, S.R.; Jung, H.S.; Seo, S.B. Inhibition of FoxO1 acetylation by INHAT subunit SET/TAF-Ibeta induces p21 transcription. FEBS Lett. 2014, 588, 2867–2873. [Google Scholar] [CrossRef]
- Wang, D.; Kon, N.; Lasso, G.; Jiang, L.; Leng, W.; Zhu, W.G.; Qin, J.; Honig, B.; Gu, W. Acetylation-regulated interaction between p53 and SET reveals a widespread regulatory mode. Nature 2016, 538, 118–122. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.B.; Kim, D.W.; Park, J.W.; Jeon, Y.J.; Kim, D.; Rhee, S.; Chae, J.I.; Seo, S.B. Inhibition of Ku70 acetylation by INHAT subunit SET/TAF-Ibeta regulates Ku70-mediated DNA damage response. Cell. Mol. Life Sci. CMLS 2014, 71, 2731–2745. [Google Scholar] [CrossRef]
- Inaba, H.; Pui, C.H. Glucocorticoid use in acute lymphoblastic leukaemia. Lancet Oncol. 2010, 11, 1096–1106. [Google Scholar] [CrossRef] [Green Version]
- Ben Abdelali, R.; Roggy, A.; Leguay, T.; Cieslak, A.; Renneville, A.; Touzart, A.; Banos, A.; Randriamalala, E.; Caillot, D.; Lioure, B.; et al. SET-NUP214 is a recurrent gammadelta lineage-specific fusion transcript associated with corticosteroid/chemotherapy resistance in adult T-ALL. Blood 2014, 123, 1860–1863. [Google Scholar] [CrossRef] [PubMed]
- Milne, T.A.; Briggs, S.D.; Brock, H.W.; Martin, M.E.; Gibbs, D.; Allis, C.D.; Hess, J.L. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol. Cell 2002, 10, 1107–1117. [Google Scholar] [CrossRef]
- Nakamura, T.; Mori, T.; Tada, S.; Krajewski, W.; Rozovskaia, T.; Wassell, R.; Dubois, G.; Mazo, A.; Croce, C.M.; Canaani, E. ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol. Cell 2002, 10, 1119–1128. [Google Scholar] [CrossRef]
- Okada, Y.; Feng, Q.; Lin, Y.; Jiang, Q.; Li, Y.; Coffield, V.M.; Su, L.; Xu, G.; Zhang, Y. hDOT1L links histone methylation to leukemogenesis. Cell 2005, 121, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Shimoyama, T.; Kato, K.; Miyaji-Yamaguchi, M.; Nagata, K. Synergistic action of MLL, a TRX protein with template activating factor-I, a histone chaperone. FEBS Lett. 2005, 579, 757–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kappes, F.; Burger, K.; Baack, M.; Fackelmayer, F.O.; Gruss, C. Subcellular localization of the human proto-oncogene protein DEK. J. Biol. Chem. 2001, 276, 26317–26323. [Google Scholar] [CrossRef]
- Bohm, F.; Kappes, F.; Scholten, I.; Richter, N.; Matsuo, H.; Knippers, R.; Waldmann, T. The SAF-box domain of chromatin protein DEK. Nucleic Acids Res. 2005, 33, 1101–1110. [Google Scholar] [CrossRef] [Green Version]
- Ivanauskiene, K.; Delbarre, E.; McGhie, J.D.; Kuntziger, T.; Wong, L.H.; Collas, P. The PML-associated protein DEK regulates the balance of H3.3 loading on chromatin and is important for telomere integrity. Genome Res. 2014, 24, 1584–1594. [Google Scholar] [CrossRef] [Green Version]
- Sanden, C.; Gullberg, U. The DEK oncoprotein and its emerging roles in gene regulation. Leukemia 2015, 29, 1632–1636. [Google Scholar] [CrossRef] [Green Version]
- Kappes, F.; Scholten, I.; Richter, N.; Gruss, C.; Waldmann, T. Functional domains of the ubiquitous chromatin protein DEK. Mol. Cell. Biol. 2004, 24, 6000–6010. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.A.; Krumpelbeck, E.F.; Jegga, A.G.; Prell, M.; Matrka, M.M.; Kappes, F.; Greis, K.D.; Ali, A.M.; Meetei, A.R.; Wells, S.I. The nuclear DEK interactome supports multi-functionality. Proteins 2018, 86, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Ageberg, M.; Drott, K.; Olofsson, T.; Gullberg, U.; Lindmark, A. Identification of a novel and myeloid specific role of the leukemia-associated fusion protein DEK-NUP214 leading to increased protein synthesis. Genes Chromosomes Cancer 2008, 47, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Sanden, C.; Ageberg, M.; Petersson, J.; Lennartsson, A.; Gullberg, U. Forced expression of the DEK-NUP214 fusion protein promotes proliferation dependent on upregulation of mTOR. BMC Cancer 2013, 13, 440. [Google Scholar] [CrossRef]
- Martinez-Useros, J.; Rodriguez-Remirez, M.; Borrero-Palacios, A.; Moreno, I.; Cebrian, A.; Gomez del Pulgar, T.; del Puerto-Nevado, L.; Vega-Bravo, R.; Puime-Otin, A.; Perez, N.; et al. DEK is a potential marker for aggressive phenotype and irinotecan-based therapy response in metastatic colorectal cancer. BMC Cancer 2014, 14, 965. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.C.; Liu, Y.L.; You, P.; Pan, J.S.; Zhou, J.Y.; Liu, Z.J.; Zhang, Z.Y. Overexpression of DEK gene is correlated with poor prognosis in hepatocellular carcinoma. Mol. Med. Rep. 2015, 11, 1318–1323. [Google Scholar] [CrossRef]
- Liu, X.; Qi, D.; Qi, J.; Mao, Z.; Li, X.; Zhang, J.; Li, J.; Gao, W. Significance of DEK overexpression for the prognostic evaluation of non-small cell lung carcinoma. Oncol. Rep. 2016, 35, 155–162. [Google Scholar] [CrossRef]
- Ou, Y.; Xia, R.; Kong, F.; Zhang, X.; Yu, S.; Jiang, L.; Zheng, L.; Lin, L. Overexpression of DEK is an indicator of poor prognosis in patients with gastric adenocarcinoma. Oncol. Lett. 2016, 11, 1823–1828. [Google Scholar] [CrossRef] [Green Version]
- Riveiro-Falkenbach, E.; Ruano, Y.; Garcia-Martin, R.M.; Lora, D.; Cifdaloz, M.; Acquadro, F.; Ballestin, C.; Ortiz-Romero, P.L.; Soengas, M.S.; Rodriguez-Peralto, J.L. DEK oncogene is overexpressed during melanoma progression. Pigment Cell Melanoma Res. 2017, 30, 194–202. [Google Scholar] [CrossRef]
- Sun, J.; Bi, F.; Yang, Y.; Zhang, Y.; Jin, A.; Li, J.; Lin, Z. DEK protein overexpression predicts poor prognosis in pancreatic ductal adenocarcinoma. Oncol. Rep. 2017, 37, 857–864. [Google Scholar] [CrossRef]
- Casas, S.; Nagy, B.; Elonen, E.; Aventin, A.; Larramendy, M.L.; Sierra, J.; Ruutu, T.; Knuutila, S. Aberrant expression of HOXA9, DEK, CBL and CSF1R in acute myeloid leukemia. Leukemia Lymphoma 2003, 44, 1935–1941. [Google Scholar] [CrossRef] [PubMed]
- Savli, H.; Sirma, S.; Nagy, B.; Aktan, M.; Dincol, G.; Salcioglu, Z.; Sarper, N.; Ozbek, U. Real-Time PCR analysis of af4 and dek genes expression in acute promyelocytic leukemia t (15;17) patients. Exp. Mol. Med. 2004, 36, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Logan, G.E.; Mor-Vaknin, N.; Braunschweig, T.; Jost, E.; Schmidt, P.V.; Markovitz, D.M.; Mills, K.I.; Kappes, F.; Percy, M.J. DEK oncogene expression during normal hematopoiesis and in Acute Myeloid Leukemia (AML). Blood Cells Mol. Dis. 2015, 54, 123–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broxmeyer, H.E.; Kappes, F.; Mor-Vaknin, N.; Legendre, M.; Kinzfogl, J.; Cooper, S.; Hangoc, G.; Markovitz, D.M. DEK regulates hematopoietic stem engraftment and progenitor cell proliferation. Stem Cells Dev. 2012, 21, 1449–1454. [Google Scholar] [CrossRef] [PubMed]
- Broxmeyer, H.E.; Mor-Vaknin, N.; Kappes, F.; Legendre, M.; Saha, A.K.; Ou, X.; O’Leary, H.; Capitano, M.; Cooper, S.; Markovitz, D.M. Concise review: Role of DEK in stem/progenitor cell biology. Stem Cells 2013, 31, 1447–1453. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.; Friedman, A.D.; Levis, M.; Li, L.; Weir, E.G.; Small, D. Internal tandem duplication mutation of FLT3 blocks myeloid differentiation through suppression of C/EBPalpha expression. Blood 2004, 103, 1883–1890. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.A.; Gole, B.; Willis, N.A.; Soria, R.; Starnes, L.M.; Krumpelbeck, E.F.; Jegga, A.G.; Ali, A.M.; Guo, H.; Meetei, A.R.; et al. DEK is required for homologous recombination repair of DNA breaks. Sci. Rep. 2017, 7, 44662. [Google Scholar] [CrossRef] [Green Version]
- Kavanaugh, G.M.; Wise-Draper, T.M.; Morreale, R.J.; Morrison, M.A.; Gole, B.; Schwemberger, S.; Tichy, E.D.; Lu, L.; Babcock, G.F.; Wells, J.M.; et al. The human DEK oncogene regulates DNA damage response signaling and repair. Nucleic Acids Res. 2011, 39, 7465–7476. [Google Scholar] [CrossRef] [Green Version]
- Burma, S.; Chen, B.P.; Murphy, M.; Kurimasa, A.; Chen, D.J. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001, 276, 42462–42467. [Google Scholar] [CrossRef]
- Deutzmann, A.; Ganz, M.; Schonenberger, F.; Vervoorts, J.; Kappes, F.; Ferrando-May, E. The human oncoprotein and chromatin architectural factor DEK counteracts DNA replication stress. Oncogene 2015, 34, 4270–4277. [Google Scholar] [CrossRef]
- Brown, F.C.; Cifani, P.; Drill, E.; He, J.; Still, E.; Zhong, S.; Balasubramanian, S.; Pavlick, D.; Yilmazel, B.; Knapp, K.M.; et al. Genomics of primary chemoresistance and remission induction failure in paediatric and adult acute myeloid leukaemia. Br. J. Haematol. 2017, 176, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Seibenhener, M.L.; Babu, J.R.; Geetha, T.; Wong, H.C.; Krishna, N.R.; Wooten, M.W. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol. Cell. Biol. 2004, 24, 8055–8068. [Google Scholar] [CrossRef] [PubMed]
- Babu, J.R.; Geetha, T.; Wooten, M.W. Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J. Neurochem. 2005, 94, 192–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjorkoy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moscat, J.; Diaz-Meco, M.T. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 2009, 137, 1001–1004. [Google Scholar] [CrossRef] [PubMed]
- Clausen, T.H.; Lamark, T.; Isakson, P.; Finley, K.; Larsen, K.B.; Brech, A.; Overvatn, A.; Stenmark, H.; Bjorkoy, G.; Simonsen, A.; et al. p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy 2010, 6, 330–344. [Google Scholar] [CrossRef] [Green Version]
- Cabe, M.; Rademacher, D.J.; Karlsson, A.B.; Cherukuri, S.; Bakowska, J.C. PB1 and UBA domains of p62 are essential for aggresome-like induced structure formation. Biochem. Biophys. Res. Commun. 2018, 503, 2306–2311. [Google Scholar] [CrossRef] [PubMed]
- Moscat, J.; Karin, M.; Diaz-Meco, M.T. p62 in Cancer: Signaling Adaptor Beyond Autophagy. Cell 2016, 167, 606–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wooten, M.W.; Geetha, T.; Seibenhener, M.L.; Babu, J.R.; Diaz-Meco, M.T.; Moscat, J. The p62 scaffold regulates nerve growth factor-induced NF-kappaB activation by influencing TRAF6 polyubiquitination. J. Biol. Chem. 2005, 280, 35625–35629. [Google Scholar] [CrossRef] [PubMed]
- Duran, A.; Linares, J.F.; Galvez, A.S.; Wikenheiser, K.; Flores, J.M.; Diaz-Meco, M.T.; Moscat, J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell 2008, 13, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Kordes, U.; Krappmann, D.; Heissmeyer, V.; Ludwig, W.D.; Scheidereit, C. Transcription factor NF-kappaB is constitutively activated in acute lymphoblastic leukemia cells. Leukemia 2000, 14, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Bhatlekar, S.; Fields, J.Z.; Boman, B.M. Role of HOX Genes in Stem Cell Differentiation and Cancer. Stem Cells Int. 2018, 2018, 3569493. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosomal Translocation | Fusion Protein | Leukemia Subtype | Refs. |
|---|---|---|---|
| del9(9q34.11; 9q34.13) t(6;9) (q21;q34.1) | SET-NUP214 | T-ALL, ETP-ALL, AUL, AML | [65,66,67,72] |
| t(6;9) (p23;q34) | DEK-NUP214 | AML | [65,66,67] |
| der(5) t(5;9) (q35;q34) | SQSTM1-NUP214 | T-ALL | [69] |
| 9q34 episome amplification | NUP214-ABL1 | T-ALL | [68] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendes, A.; Fahrenkrog, B. NUP214 in Leukemia: It’s More than Transport. Cells 2019, 8, 76. https://doi.org/10.3390/cells8010076
Mendes A, Fahrenkrog B. NUP214 in Leukemia: It’s More than Transport. Cells. 2019; 8(1):76. https://doi.org/10.3390/cells8010076
Chicago/Turabian StyleMendes, Adélia, and Birthe Fahrenkrog. 2019. "NUP214 in Leukemia: It’s More than Transport" Cells 8, no. 1: 76. https://doi.org/10.3390/cells8010076
APA StyleMendes, A., & Fahrenkrog, B. (2019). NUP214 in Leukemia: It’s More than Transport. Cells, 8(1), 76. https://doi.org/10.3390/cells8010076