Update on the Role of the Non-Canonical Wnt/Planar Cell Polarity Pathway in Neural Tube Defects

Abstract
:1. Introduction
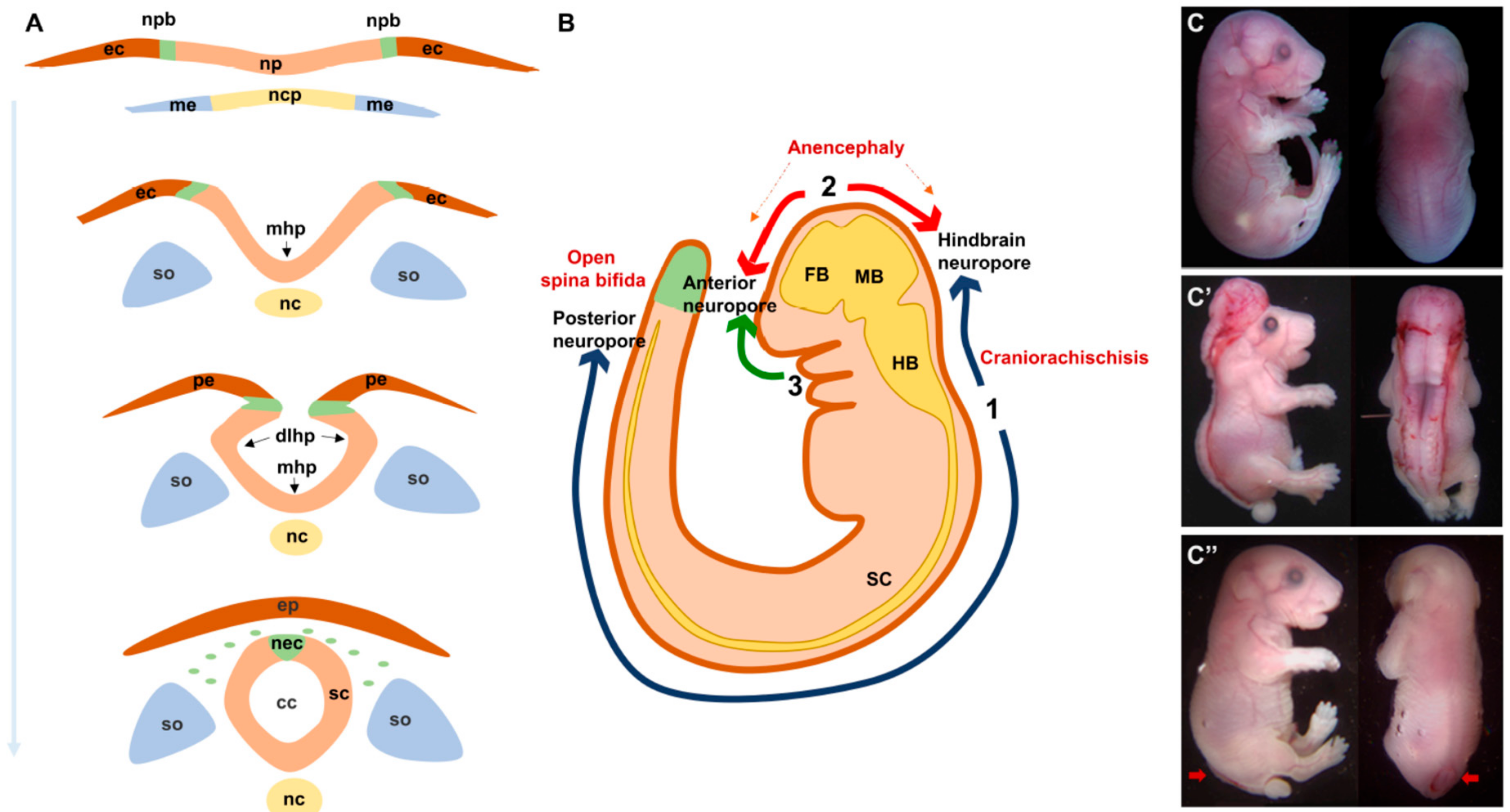
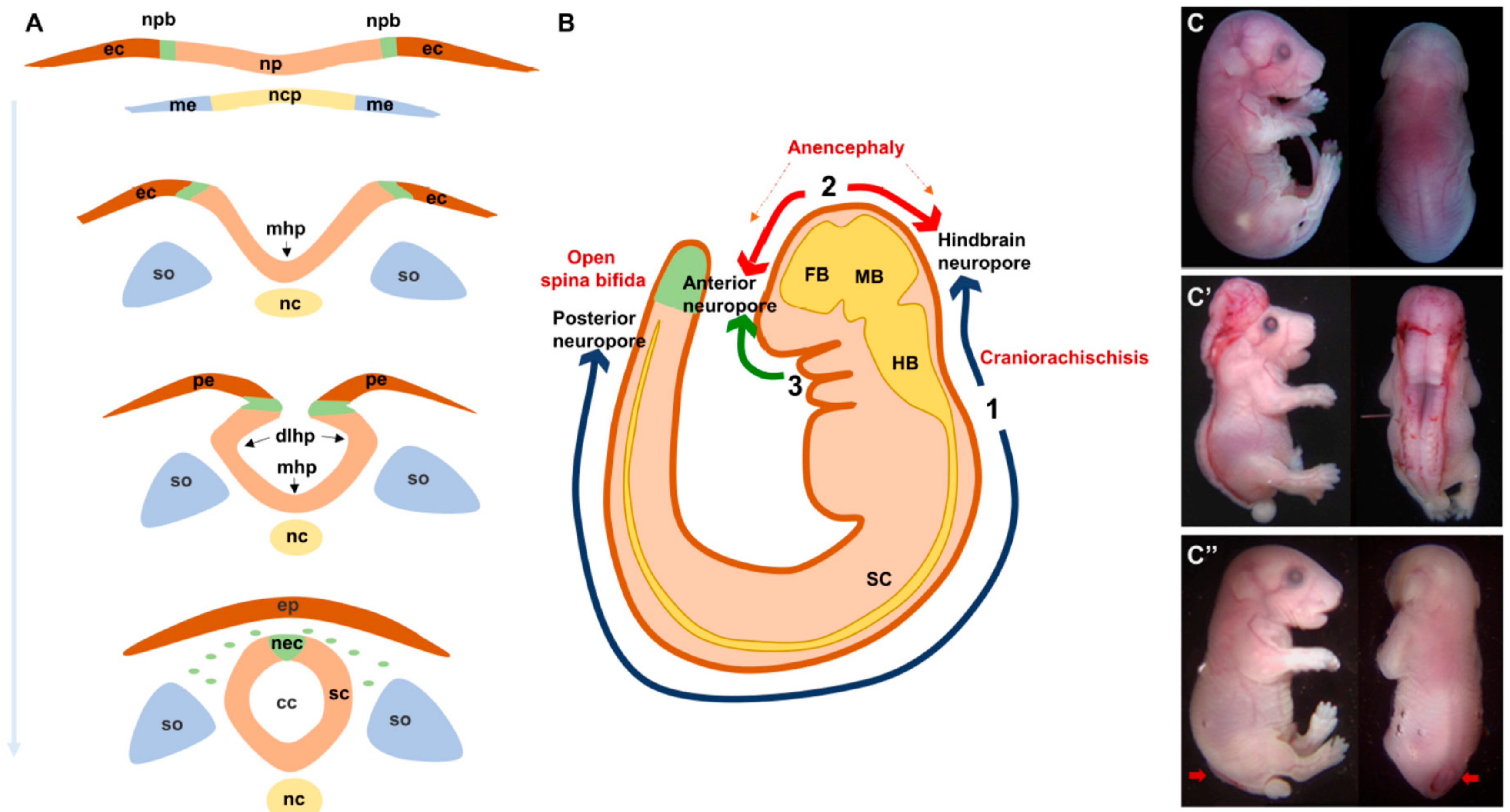
2. Neural Tube Formation: A Rapid, Multi-Step and Complex Process
3. Neural Tube Defects: What Could Go Wrong?
4. PCP Signaling: A Non-Canonical Branch of Wnt Signaling with Unresolved Mysteries
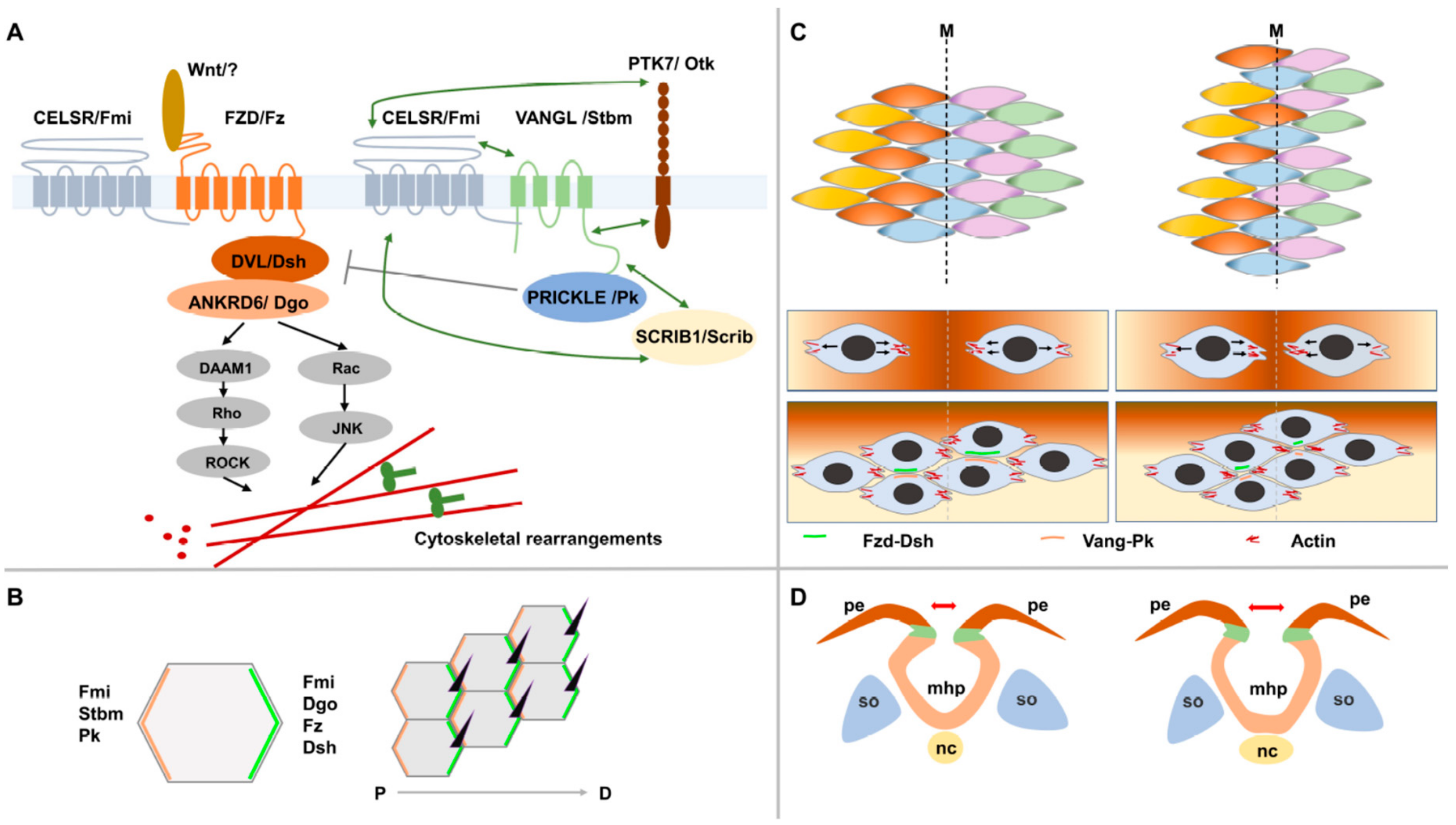
4.1. Non-Canonical Wnt/PCP Signaling: From Flies to Vertebrates
4.2. PCP and CE during Neural Tube Formation: Strong Evidence from Animal Models
4.3. PCP: Molecular Crosstalks to Other Processes and Pathways during Neurulation
5. PCP Signaling Genes in Human NTDs: What do We Know So Far?
5.1. Genetic Studies of PCP Signaling in Human NTDs
5.2. Are PCP Genes Major Culprits and/or Accomplices in the Complex Etiology of Human NTDs?
6. Conclusions and Challenges
Author Contributions
Funding
Conflicts of Interest
References
- Rossi, A.; Biancheri, R.; Cama, A.; Piatelli, G.; Ravegnani, M.; Tortori-Donati, P. Imaging in spine and spinal cord malformations. Eur. J. Radiol. 2004, 50, 177–200. [Google Scholar] [CrossRef] [PubMed]
- Copp, A.J.; Stanier, P.; Greene, N.D.E. Neural tube defects: Recent advances, unsolved questions, and controversies. Lancet Neurol. 2013, 12, 799–810. [Google Scholar] [CrossRef]
- Prevention of neural tube defects: Results of the Medical Research Council Vitamin Study. MRC Vitamin Study Research Group. Lancet 1991, 338, 131–137. [CrossRef]
- Blom, H.J.; Shaw, G.M.; den Heijer, M.; Finnell, R.H. Neural tube defects and folate: Case far from closed. Nat. Rev. Neurosci. 2006, 7, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Blencowe, H.; Kancherla, V.; Moorthie, S.; Darlison, M.W.; Modell, B. Estimates of global and regional prevalence of neural tube defects for 2015: A systematic analysis. Ann. N. Y. Acad. Sci. 2018, 1414, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Mosley, B.S.; Cleves, M.A.; Siega-Riz, A.M.; Shaw, G.M.; Canfield, M.A.; Waller, D.K.; Werler, M.M.; Hobbs, C.A. National Birth Defects Prevention Study Neural tube defects and maternal folate intake among pregnancies conceived after folic acid fortification in the United States. Am. J. Epidemiol. 2009, 169, 9–17. [Google Scholar] [CrossRef]
- Greene, N.D.E.; Leung, K.-Y.; Gay, V.; Burren, K.; Mills, K.; Chitty, L.S.; Copp, A.J. Inositol for the prevention of neural tube defects: A pilot randomised controlled trial. Br. J. Nutr. 2016, 115, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Greene, N.D.E.; Leung, K.-Y.; Copp, A.J. Inositol, neural tube closure and the prevention of neural tube defects. Birth Defects Res. 2017, 109, 68–80. [Google Scholar] [CrossRef]
- Bassuk, A.G.; Kibar, Z. Genetic basis of neural tube defects. Semin. Pediatr. Neurol. 2009, 16, 101–110. [Google Scholar] [CrossRef]
- Avagliano, L.; Massa, V.; George, T.M.; Qureshy, S.; Bulfamante, G.P.; Finnell, R.H. Overview on neural tube defects: From development to physical characteristics. Birth Defects Res. 2018. [Google Scholar] [CrossRef]
- Detrait, E.R.; George, T.M.; Etchevers, H.C.; Gilbert, J.R.; Vekemans, M.; Speer, M.C. Human neural tube defects: Developmental biology, epidemiology, and genetics. Neurotoxicol. Teratol. 2005, 27, 515–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, S.A. Non-multifactorial neural tube defects. Am. J. Med. Genet. C Semin Med. Genet. 2005, 135C, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Massarwa, R.; Ray, H.J.; Niswander, L. Morphogenetic movements in the neural plate and neural tube: Mouse. Wiley Interdiscip. Rev. Dev. Biol. 2014, 3, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Nikolopoulou, E.; Galea, G.L.; Rolo, A.; Greene, N.D.E.; Copp, A.J. Neural tube closure: Cellular, molecular and biomechanical mechanisms. Development 2017, 144, 552–566. [Google Scholar] [CrossRef] [PubMed]
- Ybot-Gonzalez, P.; Cogram, P.; Gerrelli, D.; Copp, A.J. Sonic hedgehog and the molecular regulation of mouse neural tube closure. Development 2002, 129, 2507–2517. [Google Scholar] [PubMed]
- Ybot-Gonzalez, P.; Gaston-Massuet, C.; Girdler, G.; Klingensmith, J.; Arkell, R.; Greene, N.D.E.; Copp, A.J. Neural plate morphogenesis during mouse neurulation is regulated by antagonism of Bmp signalling. Development 2007, 134, 3203–3211. [Google Scholar] [CrossRef] [Green Version]
- Haigo, S.L.; Hildebrand, J.D.; Harland, R.M.; Wallingford, J.B. Shroom induces apical constriction and is required for hingepoint formation during neural tube closure. Curr. Biol. 2003, 13, 2125–2137. [Google Scholar] [CrossRef]
- Martin, P. Morphogenesis: Shroom in to close the neural tube. Curr. Biol. 2004, 14, 150–151. [Google Scholar] [CrossRef]
- Sawyer, J.M.; Harrell, J.R.; Shemer, G.; Sullivan-Brown, J.; Roh-Johnson, M.; Goldstein, B. Apical constriction: A cell shape change that can drive morphogenesis. Dev. Biol. 2010, 341, 5–19. [Google Scholar] [CrossRef] [Green Version]
- Pai, Y.-J.; Abdullah, N.L.; Mohd-Zin, S.W.; Mohammed, R.S.; Rolo, A.; Greene, N.D.E.; Abdul-Aziz, N.M.; Copp, A.J. Epithelial fusion during neural tube morphogenesis. Birth Defects Res. Part. A Clin. Mol. Teratol. 2012, 94, 817–823. [Google Scholar] [CrossRef] [Green Version]
- Camerer, E.; Barker, A.; Duong, D.N.; Ganesan, R.; Kataoka, H.; Cornelissen, I.; Darragh, M.R.; Hussain, A.; Zheng, Y.-W.; Srinivasan, Y.; et al. Local protease signaling contributes to neural tube closure in the mouse embryo. Dev. Cell 2010, 18, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Pyrgaki, C.; Liu, A.; Niswander, L. Grainyhead-like 2 regulates neural tube closure and adhesion molecule expression during neural fold fusion. Dev. Biol. 2011, 353, 38–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolo, A.; Savery, D.; Escuin, S.; de Castro, S.C.; Armer, H.E.J.; Munro, P.M.G.; Molè, M.A.; Greene, N.D.E.; Copp, A.J. Regulation of cell protrusions by small GTPases during fusion of the neural folds. Elife 2016. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Aziz, N.M.; Turmaine, M.; Greene, N.D.E.; Copp, A.J. EphrinA-EphA receptor interactions in mouse spinal neurulation: Implications for neural fold fusion. Int. J. Dev. Biol. 2009, 53, 559–568. [Google Scholar] [CrossRef]
- Van Allen, M.I.; Kalousek, D.K.; Chernoff, G.F.; Juriloff, D.; Harris, M.; McGillivray, B.C.; Yong, S.L.; Langlois, S.; MacLeod, P.M.; Chitayat, D. Evidence for multi-site closure of the neural tube in humans. Am. J. Med. Genet. 1993, 47, 723–743. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, M.I. Multisite neural tube closure in humans. Birth Defects Orig. Artic. Ser. 1996, 30, 203–225. [Google Scholar] [PubMed]
- De Bakker, B.S.; Driessen, S.; Boukens, B.J.D.; van den Hoff, M.J.B.; Oostra, R.-J. Single-site neural tube closure in human embryos revisited. Clin. Anat. 2017, 30, 988–999. [Google Scholar] [CrossRef]
- Catala, M. Genetic control of caudal development. Clin. Genet. 2002, 61, 89–96. [Google Scholar] [CrossRef]
- Harris, M.J.; Juriloff, D.M. An update to the list of mouse mutants with neural tube closure defects and advances toward a complete genetic perspective of neural tube closure. Birth Defects Res. Part. A Clin. Mol. Teratol. 2010, 88, 653–669. [Google Scholar] [CrossRef]
- Nusse, R.; Varmus, H.E. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell 1982, 31, 99–109. [Google Scholar] [CrossRef]
- Nusse, R.; Varmus, H. Three decades of Wnts: A personal perspective on how a scientific field developed. EMBO J. 2012, 31, 2670–2684. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Anastas, J.N.; Moon, R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.T.; Wallingford, J.B. Planar cell polarity in development and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Maung, S.M.T.W.; Jenny, A. Planar cell polarity in Drosophila. Organogenesis 2011, 7, 165–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Mlodzik, M. Wnt-Frizzled/planar cell polarity signaling: Cellular orientation by facing the wind (Wnt). Annu. Rev. Cell Dev. Biol. 2015, 31, 623–646. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Borchers, A.G.M.; Jolicoeur, C.; Rayburn, H.; Baker, J.C.; Tessier-Lavigne, M. PTK7/CCK-4 is a novel regulator of planar cell polarity in vertebrates. Nature 2004, 430, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Minami, Y.; Oishi, I.; Endo, M.; Nishita, M. Ror-family receptor tyrosine kinases in noncanonical Wnt signaling: Their implications in developmental morphogenesis and human diseases. Dev. Dyn. 2010, 239, 1–15. [Google Scholar] [CrossRef]
- Macheda, M.L.; Sun, W.W.; Kugathasan, K.; Hogan, B.M.; Bower, N.I.; Halford, M.M.; Zhang, Y.F.; Jacques, B.E.; Lieschke, G.J.; Dabdoub, A.; et al. The Wnt receptor Ryk plays a role in mammalian planar cell polarity signaling. J. Biol. Chem. 2012, 287, 29312–29323. [Google Scholar] [CrossRef]
- Murdoch, J.N.; Henderson, D.J.; Doudney, K.; Gaston-Massuet, C.; Phillips, H.M.; Paternotte, C.; Arkell, R.; Stanier, P.; Copp, A.J. Disruption of scribble (Scrb1) causes severe neural tube defects in the circletail mouse. Hum. Mol. Genet. 2003, 12, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Barrow, J.R. Wnt/PCP signaling: A veritable polar star in establishing patterns of polarity in embryonic tissues. Semin. Cell Dev. Biol. 2006, 17, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Vladar, E.K.; Antic, D.; Axelrod, J.D. Planar cell polarity signaling: The developing cell’s compass. Cold Spring Harb. Perspect. Biol. 2009. [Google Scholar] [CrossRef] [PubMed]
- McNeill, H. Planar cell polarity: Keeping hairs straight is not so simple. Cold Spring Harb. Perspect. Biol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Sebbagh, M.; Borg, J.-P. Insight into planar cell polarity. Exp. Cell Res. 2014, 328, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Henderson, D.J.; Long, D.A.; Dean, C.H. Planar cell polarity in organ formation. Curr. Opin. Cell Biol. 2018, 55, 96–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphries, A.C.; Mlodzik, M. From instruction to output: Wnt/PCP signaling in development and cancer. Curr. Opin. Cell Biol. 2018, 51, 110–116. [Google Scholar] [CrossRef] [PubMed]
- VanderVorst, K.; Dreyer, C.A.; Konopelski, S.E.; Lee, H.; Ho, H.-Y.H.; Carraway, K.L. Wnt/PCP Signaling Contribution to Carcinoma Collective Cell Migration and Metastasis. Cancer Res. 2019, 79, 1719–1729. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Nathans, J. Tissue/planar cell polarity in vertebrates: New insights and new questions. Development 2007, 134, 647–658. [Google Scholar] [CrossRef]
- Roszko, I.; Sawada, A.; Solnica-Krezel, L. Regulation of convergence and extension movements during vertebrate gastrulation by the Wnt/PCP pathway. Semin. Cell Dev. Biol. 2009, 20, 986–997. [Google Scholar] [CrossRef] [Green Version]
- Huebner, R.J.; Wallingford, J.B. Coming to Consensus: A Unifying Model Emerges for Convergent Extension. Dev. Cell 2018, 46, 389–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallingford, J.B. Neural tube closure and neural tube defects: Studies in animal models reveal known knowns and known unknowns. Am. J. Med. Genet. C Semin Med. Genet. 2005, 135, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Tada, M.; Heisenberg, C.-P. Convergent extension: Using collective cell migration and cell intercalation to shape embryos. Development 2012, 139, 3897–3904. [Google Scholar] [CrossRef] [PubMed]
- Shindo, A. Models of convergent extension during morphogenesis. Wiley Interdiscip. Rev. Dev. Biol. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Juriloff, D.M.; Harris, M.J. A consideration of the evidence that genetic defects in planar cell polarity contribute to the etiology of human neural tube defects. Birth Defects Res. Part. A Clin. Mol. Teratol. 2012, 94, 824–840. [Google Scholar] [CrossRef] [PubMed]
- Kibar, Z.; Vogan, K.J.; Groulx, N.; Justice, M.J.; Underhill, D.A.; Gros, P. Ltap, a mammalian homolog of Drosophila Strabismus/Van Gogh, is altered in the mouse neural tube mutant Loop-tail. Nat. Genet. 2001, 28, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, J.N.; Doudney, K.; Paternotte, C.; Copp, A.J.; Stanier, P. Severe neural tube defects in the loop-tail mouse result from mutation of Lpp1, a novel gene involved in floor plate specification. Hum. Mol. Genet. 2001, 10, 2593–2601. [Google Scholar] [CrossRef]
- Ybot-Gonzalez, P.; Savery, D.; Gerrelli, D.; Signore, M.; Mitchell, C.E.; Faux, C.H.; Greene, N.D.E.; Copp, A.J. Convergent extension, planar-cell-polarity signalling and initiation of mouse neural tube closure. Development 2007, 134, 789–799. [Google Scholar] [CrossRef] [Green Version]
- Curtin, J.A.; Quint, E.; Tsipouri, V.; Arkell, R.M.; Cattanach, B.; Copp, A.J.; Henderson, D.J.; Spurr, N.; Stanier, P.; Fisher, E.M.; et al. Mutation of Celsr1 disrupts planar polarity of inner ear hair cells and causes severe neural tube defects in the mouse. Curr. Biol. 2003, 13, 1129–1133. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, N.; Nathans, J. The role of Frizzled3 and Frizzled6 in neural tube closure and in the planar polarity of inner-ear sensory hair cells. J. Neurosci. 2006, 26, 2147–2156. [Google Scholar] [CrossRef]
- Etheridge, S.L.; Ray, S.; Li, S.; Hamblet, N.S.; Lijam, N.; Tsang, M.; Greer, J.; Kardos, N.; Wang, J.; Sussman, D.J.; et al. Murine dishevelled 3 functions in redundant pathways with dishevelled 1 and 2 in normal cardiac outflow tract, cochlea, and neural tube development. PLoS Genet. 2008, 4, e1000259. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, J.N.; Damrau, C.; Paudyal, A.; Bogani, D.; Wells, S.; Greene, N.D.E.; Stanier, P.; Copp, A.J. Genetic interactions between planar cell polarity genes cause diverse neural tube defects in mice. Dis. Model. Mech. 2014, 7, 1153–1163. [Google Scholar] [CrossRef] [PubMed]
- Montcouquiol, M.; Rachel, R.A.; Lanford, P.J.; Copeland, N.G.; Jenkins, N.A.; Kelley, M.W. Identification of Vangl2 and Scrb1 as planar polarity genes in mammals. Nature 2003, 423, 173–177. [Google Scholar] [CrossRef] [PubMed]
- McGreevy, E.M.; Vijayraghavan, D.; Davidson, L.A.; Hildebrand, J.D. Shroom3 functions downstream of planar cell polarity to regulate myosin II distribution and cellular organization during neural tube closure. Biol. Open 2015, 4, 186–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallingford, J.B.; Mitchell, B. Strange as it may seem: The many links between Wnt signaling, planar cell polarity, and cilia. Genes Dev. 2011, 25, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Assémat, E.; Bazellières, E.; Pallesi-Pocachard, E.; Le Bivic, A.; Massey-Harroche, D. Polarity complex proteins. Biochim. Biophys. Acta 2008, 1778, 614–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halaoui, R.; McCaffrey, L. Rewiring cell polarity signaling in cancer. Oncogene 2015, 34, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Kharfallah, F.; Guyot, M.C.; El Hassan, A.R.; Allache, R.; Merello, E.; De Marco, P.; Di Cristo, G.; Capra, V.; Kibar, Z. Scribble1 plays an important role in the pathogenesis of neural tube defects through its mediating effect of Par-3 and Vangl1/2 localization. Hum. Mol. Genet. 2017, 26, 2307–2320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Žigman, M.; Trinh, L.A.; Fraser, S.E.; Moens, C.B. Zebrafish Neural Tube Morphogenesis Requires Scribble-Dependent Oriented Cell Divisions. Curr. Biol. 2011, 21, 79–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milgrom-Hoffman, M.; Humbert, P.O. Regulation of cellular and PCP signalling by the Scribble polarity module. Semin. Cell Dev. Biol. 2018, 81, 33–45. [Google Scholar] [CrossRef]
- Weidinger, G.; Moon, R.T. When Wnts antagonize Wnts. J. Cell Biol. 2003, 162, 753–755. [Google Scholar] [CrossRef] [PubMed]
- Bryja, V.; Andersson, E.R.; Schambony, A.; Esner, M.; Bryjová, L.; Biris, K.K.; Hall, A.C.; Kraft, B.; Cajanek, L.; Yamaguchi, T.P.; et al. The extracellular domain of Lrp5/6 inhibits noncanonical Wnt signaling in vivo. Mol. Biol. Cell 2009, 20, 924–936. [Google Scholar] [CrossRef] [PubMed]
- Andersson, E.R.; Bryjova, L.; Biris, K.; Yamaguchi, T.P.; Arenas, E.; Bryja, V. Genetic interaction between Lrp6 and Wnt5a during mouse development. Dev. Dyn. 2010, 239, 237–245. [Google Scholar]
- Tahinci, E.; Thorne, C.A.; Franklin, J.L.; Salic, A.; Christian, K.M.; Lee, L.A.; Coffey, R.J.; Lee, E. Lrp6 is required for convergent extension during Xenopus gastrulation. Development 2007, 134, 4095–4106. [Google Scholar] [CrossRef] [PubMed]
- Pinson, K.I.; Brennan, J.; Monkley, S.; Avery, B.J.; Skarnes, W.C. An LDL-receptor-related protein mediates Wnt signalling in mice. Nature 2000, 407, 535–538. [Google Scholar] [CrossRef]
- Carter, M.; Chen, X.; Slowinska, B.; Minnerath, S.; Glickstein, S.; Shi, L.; Campagne, F.; Weinstein, H.; Ross, M.E. Crooked tail (Cd) model of human folate-responsive neural tube defects is mutated in Wnt coreceptor lipoprotein receptor-related protein 6. Proc. Natl. Acad. Sci. USA 2005, 102, 12843–12848. [Google Scholar] [CrossRef] [PubMed]
- Hayes, M.; Naito, M.; Daulat, A.; Angers, S.; Ciruna, B. Ptk7 promotes non-canonical Wnt/PCP-mediated morphogenesis and inhibits Wnt/β-catenin-dependent cell fate decisions during vertebrate development. Development 2013, 140, 1807–1818. [Google Scholar] [CrossRef]
- Peradziryi, H.; Kaplan, N.A.; Podleschny, M.; Liu, X.; Wehner, P.; Borchers, A.; Tolwinski, N.S. PTK7/Otk interacts with Wnts and inhibits canonical Wnt signalling. EMBO J. 2011, 30, 3729–3740. [Google Scholar] [CrossRef] [Green Version]
- Puppo, F.; Thomé, V.; Lhoumeau, A.-C.; Cibois, M.; Gangar, A.; Lembo, F.; Belotti, E.; Marchetto, S.; Lécine, P.; Prébet, T.; et al. Protein tyrosine kinase 7 has a conserved role in Wnt/β-catenin canonical signalling. EMBO Rep. 2011, 12, 43–49. [Google Scholar] [CrossRef]
- Bin-Nun, N.; Lichtig, H.; Malyarova, A.; Levy, M.; Elias, S.; Frank, D. PTK7 modulates Wnt signaling activity via LRP6. Development 2014, 141, 410–421. [Google Scholar] [CrossRef] [Green Version]
- Kibar, Z.; Torban, E.; McDearmid, J.R.; Reynolds, A.; Berghout, J.; Mathieu, M.; Kirillova, I.; De Marco, P.; Merello, E.; Hayes, J.M.; et al. Mutations in VANGL1 associated with neural-tube defects. N. Engl. J. Med. 2007, 356, 1432–1437. [Google Scholar] [CrossRef] [PubMed]
- Doudney, K.; Ybot-Gonzalez, P.; Paternotte, C.; Stevenson, R.E.; Greene, N.D.E.; Moore, G.E.; Copp, A.J.; Stanier, P. Analysis of the planar cell polarity gene Vangl2 and its co-expressed paralogue Vangl1 in neural tube defect patients. Am. J. Med. Genet. A 2005, 136, 90–92. [Google Scholar] [CrossRef] [PubMed]
- Kibar, Z.; Bosoi, C.M.; Kooistra, M.; Salem, S.; Finnell, R.H.; De Marco, P.; Merello, E.; Bassuk, A.G.; Capra, V.; Gros, P. Novel mutations in VANGL1 in neural tube defects. Hum. Mutat. 2009, 30, E706–E715. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.-P.; Zhang, T.; Li, H.; Wu, B.-L.; Jin, L.; Wang, H.-Y. VANGL2 mutations in human cranial neural-tube defects. N. Engl. J. Med. 2010, 362, 2232–2235. [Google Scholar] [CrossRef] [PubMed]
- Bosoi, C.M.; Capra, V.; Allache, R.; Trinh, V.Q.-H.; De Marco, P.; Merello, E.; Drapeau, P.; Bassuk, A.G.; Kibar, Z. Identification and characterization of novel rare mutations in the planar cell polarity gene PRICKLE1 in human neural tube defects. Hum. Mutat. 2011, 32, 1371–1375. [Google Scholar] [CrossRef] [PubMed]
- Kibar, Z.; Salem, S.; Bosoi, C.M.; Pauwels, E.; De Marco, P.; Merello, E.; Bassuk, A.G.; Capra, V.; Gros, P. Contribution of VANGL2 mutations to isolated neural tube defects. Clin. Genet. 2011, 80, 76–82. [Google Scholar] [CrossRef]
- Allache, R.; De Marco, P.; Merello, E.; Capra, V.; Kibar, Z. Role of the planar cell polarity gene CELSR1 in neural tube defects and caudal agenesis. Birth Defects Res. Part. A Clin. Mol. Teratol. 2012, 94, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, O.; Kirmes, I.; Thiede, A.; Lechno, S.; Gocan, H.; Florian, I.S.; Haaf, T.; Zechner, U.; Sabova, L.; Horn, F. Novel VANGL1 Gene Mutations in 144 Slovakian, Romanian and German Patients with Neural Tube Defects. Mol. Syndromol. 2012, 3, 76–81. [Google Scholar] [CrossRef] [Green Version]
- De Marco, P.; Merello, E.; Rossi, A.; Piatelli, G.; Cama, A.; Kibar, Z.; Capra, V. FZD6 is a novel gene for human neural tube defects. Hum. Mutat. 2012, 33, 384–390. [Google Scholar] [CrossRef]
- Robinson, A.; Escuin, S.; Doudney, K.; Vekemans, M.; Stevenson, R.E.; Greene, N.D.E.; Copp, A.J.; Stanier, P. Mutations in the planar cell polarity genes CELSR1 and SCRIB are associated with the severe neural tube defect craniorachischisis. Hum. Mutat. 2012, 33, 440–447. [Google Scholar] [CrossRef]
- De Marco, P.; Merello, E.; Consales, A.; Piatelli, G.; Cama, A.; Kibar, Z.; Capra, V. Genetic analysis of disheveled 2 and disheveled 3 in human neural tube defects. J. Mol. Neurosci. 2013, 49, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Zhu, H.; Duhon, C.; Yang, W.; Ross, M.E.; Shaw, G.M.; Finnell, R.H. Mutations in planar cell polarity gene SCRIB are associated with spina bifida. PLoS ONE 2013, 8, e69262. [Google Scholar] [CrossRef] [PubMed]
- Allache, R.; Lachance, S.; Guyot, M.C.; De Marco, P.; Merello, E.; Justice, M.J.; Capra, V.; Kibar, Z. Novel mutations in Lrp6 orthologs in mouse and human neural tube defects affect a highly dosage-sensitive Wnt non-canonical planar cell polarity pathway. Hum. Mol. Genet. 2014, 23, 1687–1699. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Zhu, H.; Yang, W.; Ross, M.E.; Shaw, G.M.; Finnell, R.H. Identification of novel CELSR1 mutations in spina bifida. PLoS ONE 2014, 9, e92207. [Google Scholar] [CrossRef] [PubMed]
- Allache, R.; Wang, M.; De Marco, P.; Merello, E.; Capra, V.; Kibar, Z. Genetic studies of ANKRD6 as a molecular switch between Wnt signaling pathways in human neural tube defects. Birth Defects Res. Part. A Clin. Mol. Teratol. 2015, 103, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Fathe, K.; McCartney, D.; Zhu, H.; Yang, W.; Ross, M.E.; Shaw, G.M.; Finnell, R.H. Rare LRP6 variants identified in spina bifida patients. Hum. Mutat. 2015, 36, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Merello, E.; Mascelli, S.; Raso, A.; Piatelli, G.; Consales, A.; Cama, A.; Kibar, Z.; Capra, V.; Marco, P.D. Expanding the mutational spectrum associated to neural tube defects: Literature revision and description of novel VANGL1 mutations. Birth Defects Res. Part. A Clin. Mol. Teratol. 2015, 103, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; De Marco, P.; Merello, E.; Drapeau, P.; Capra, V.; Kibar, Z. Role of the planar cell polarity gene Protein tyrosine kinase 7 in neural tube defects in humans. Birth Defects Res. Part. A Clin. Mol. Teratol. 2015, 103, 1021–1027. [Google Scholar] [CrossRef]
- Qiao, X.; Liu, Y.; Li, P.; Chen, Z.; Li, H.; Yang, X.; Finnell, R.H.; Yang, Z.; Zhang, T.; Qiao, B.; et al. Genetic analysis of rare coding mutations in CELSR1-3 in Chinese Congenital Heart and Neural Tube Defects. Clin. Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Lei, Y.; Cao, X.; Zheng, Y.; Wang, F.; Bao, Y.; Peng, R.; Finnell, R.H.; Zhang, T.; Wang, H. Genetic analysis of Wnt/PCP genes in neural tube defects. BMC Med. Genomics 2018, 11, 38. [Google Scholar] [CrossRef]
- Wang, L.; Xiao, Y.; Tian, T.; Jin, L.; Lei, Y.; Finnell, R.H.; Ren, A. Digenic variants of planar cell polarity genes in human neural tube defect patients. Mol. Genet. Metab. 2018, 124, 94–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Y.; Kim, S.-E.; Chen, Z.; Cao, X.; Zhu, H.; Yang, W.; Shaw, G.M.; Zheng, Y.; Zhang, T.; Wang, H.-Y.; et al. Variants identified in PTK7 associated with neural tube defects. Mol. Genet. Genomic. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Shi, O.; Wang, B.; Chang, B.; Yang, R.; Wang, Y.; Wang, F.; Shen, C. Association between VANGL1 gene polymorphisms and neural tube defects. Neuropediatrics 2014, 45, 234–239. [Google Scholar] [PubMed]
- Reynolds, A.; McDearmid, J.R.; Lachance, S.; De Marco, P.; Merello, E.; Capra, V.; Gros, P.; Drapeau, P.; Kibar, Z. VANGL1 rare variants associated with neural tube defects affect convergent extension in zebrafish. Mech. Dev. 2010, 127, 385–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, M.; Gao, X.; Yu, L.X.; Paria, N.; Henkelman, R.M.; Wise, C.A.; Ciruna, B. ptk7 mutant zebrafish models of congenital and idiopathic scoliosis implicate dysregulated Wnt signalling in disease. Nat. Commun. 2014. [Google Scholar] [CrossRef] [PubMed]
- Lemay, P.; Guyot, M.-C.; Tremblay, É.; Dionne-Laporte, A.; Spiegelman, D.; Henrion, É.; Diallo, O.; De Marco, P.; Merello, E.; Massicotte, C.; et al. Loss-of-function de novo mutations play an important role in severe human neural tube defects. J. Med. Genet. 2015, 52, 493–497. [Google Scholar] [CrossRef] [Green Version]
- Veltman, J.A.; Brunner, H.G. De novo mutations in human genetic disease. Nat. Rev. Genet. 2012, 13, 565–575. [Google Scholar] [CrossRef]
- Wang, W.; Corominas, R.; Lin, G.N. De novo Mutations from Whole Exome Sequencing in Neurodevelopmental and Psychiatric Disorders: From Discovery to Application. Front. Genet. 2019. [Google Scholar] [CrossRef]
- Chen, Z.; Lei, Y.; Zheng, Y.; Aguiar-Pulido, V.; Ross, M.E.; Peng, R.; Jin, L.; Zhang, T.; Finnell, R.H.; Wang, H. Threshold for neural tube defect risk by accumulated singleton loss-of-function variants. Cell Res. 2018, 28, 1039–1041. [Google Scholar] [CrossRef]
- Boyle, E.A.; Li, Y.I.; Pritchard, J.K. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 2017, 169, 1177–1186. [Google Scholar] [CrossRef]
- Gibson, G. Rare and common variants: Twenty arguments. Nat. Rev. Genet. 2012, 13, 135–145. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Gene | NTD Cohort | Variant | Patient Description | Functional Validation | Reference |
|---|---|---|---|---|---|
| PCP core genes | |||||
| ANKRD6 | 391 Italians and 82 French Canadians (14 cranial, 246 MMC, and 213 closed spinal NTDs) | p.Pro548Leu p.Arg587Gln p.Arg632His | MC LipoMS and MC CA | p.Pro548Leu and p.Arg632His were proven to be partially loss of function in activation of PCP signaling and inhibition of canonical Wnt pathway | [95] |
| CELSR1 | 36 fetuses with CRS from US, France and England | p.Ala773Val p.Arg2438Gln p.Ser2964Leu p.Pro2983Ala | 1 CRS 1 CRS 2 CRS 2 CRS | All 4 variants significantly affected frequency of membrane localisation | [90] |
| 391 Italians and 82 French Canadians (same as above) | p.Arg541Trp p.Val551Met p.Gln834X p.Arg.836Cys p.Val1008Leu p.Asp1401Gly p.Thr1443Pro p.Arg1456Gln p.Arg1526Trp p.Arg1835Cys p.Arg2121Cys p.Ser2190Leu p.Ala2228Val p.Arg2359Cys p.Ser2963Thr2966del | MMC LMC MMC Lipoma TFT MMC LMMC CA MMC MMC LMMC Lipoma Lipoma LMC CA | Not conducted | [87] | |
| 192 American SB infants | p.Ala1023Gly p.Ile1124Met p.Thr1362Met p.Gly1410Arg Truncated protein (c.5050_5051 ins GT) p.Gly1825Ser Truncated protein (c.5719_5720 del TG) p.Gly2062Ser p.Arg2354Cys p.Arg2497Cys | All SB | Two truncated mutations disrupted CELSR1 membrane localization and its recruitment of Vangl2 to cell membrane. | [94] | |
| 352 Chinese patients (26 CRS, 73EC, 64AN, 3Ex, 255 SB, 1 unknown) | p.Pro870Leu | CRS | p.Pro870Leu was a gain-of-function variant in zebrafish overexpression and rescue experiments. It increased both PCP and canonical Wnt signalling | [99] | |
| 184 Chinese patients (36 AN,12 CRS, 32 EC, 2 Ex, 91 SB, 11 unknown) and 292 American patients (100% SB) | p.Arg714His p.Pro870Leu p.Thr875Ile p.Ala1019Ser p.Thr1086Met p.Arg1194His p.Gln1473Ter | AN, SB SB SB | Not conducted | [100] | |
| 510 Chinese patients (125 AN, 232 SB, 46 EC, 79 AN & SB, 1 AN & EC, 15 SB & EC, 4 AN & SB & EC, 8 unknown) | p. Arg769Trp p.Arg1057Cys p.Gly1122Ser p.Gln2924His | SB SB AN SB | Not conducted | [101] | |
| CELSR2 | 352 Chinese patients (same as above) | p.Ser628Gly p.Thr2026Met p.Arg2153Gly p.Arg2480Cys p.Arg2015Gly fs*22 p.Phe2397Lys fs*584 | AN, SB SB EC SB CRS SB | p.Thr2026Met downregulated PCP signaling in a luciferase assay | [99,100] |
| 184 Chinese patients (same as above) | p.Ser628Gly p.Arg1990His p.Arg2015Gly fs*23 p.Thr2026His p.Arg2153Gly p.Arg2480Cys p.Arg2626Cys | Not conducted | [100] | ||
| CELSR3 | 352 Chinese patients (same as above) | p.Gly194Val p.Val446Met p.Gly1754Asp | 1 AN, 1 SB CRS CRS | Not conducted | [99] |
| 184 Chinese patients (same as above) | p.Val446Met p.Ile1102Leu p.Arg1194His p.Arg1453Cys p.Gly1754Asp p.Gly1754Ser p.Val2584Gly p.Met2630Ile | Not conducted | [100] | ||
| DVL2 | 473 Italian and French Canadian patients (same as above) | p.Ala53Val p.Glu620X (c.1801_1802ins) p.Tyr667Cys | Lipoma CA and TC MMC | Not conducted | [91] |
| DVL3 | 184 Chinese patients (same as above) | p.Asp403Asn | SB | p.Asp403Asn disrupted DVL3 interaction with VANGL2, upregulated canonical Wnt signaling and down regulated non-PCP signaling | [100] |
| 510 Chinese patients (same as above) | p.Arg148Gln | AN | Not conducted | [101] | |
| FZD6 | 473 Italian and French Canadian patients (same as above) | p.Cys615Ter p.Arg405Gln p.Arg511Cys p.Arg511His | MC MMC MMC CA | Not conducted | [89] |
| PK1 | 810 patients: 421 Italian (11 cranial, 211 open spinal including 208 MMC, 199 closed spinal) and 389 Americans (4 cranial, 325 MMC, 60 closed spinal) | p.Ile69Thr p.Asn81His p.Thr275Met p.Val550Met p.Arg682Cys p.Ser739Phe p.Asp771Asn | DM MMC MMC MMC MMC MMC CA | p.Ile69Thr, p.Asn81His, p.Thr275Met and p.Arg682Cys acted as hypermorphic alleles in inducing CE defects in an overexpression assay in zebrafish; p.Arg682Cys antagonized the CE phenotype induced by the wild-type zpk1a in a dominant fashion | [85] |
| VANGL1 | 137 Italian patients (80 MMC, 57 closed spinal) 7 French fetuses with CRS | p.Val239Ile p.Arg274Gln p.Met328Thr | CA MMC MMC, TC | p.Val239Ile abrogated interaction between VANGL1 and all three Dvl proteins; p.Val239Ile and p.Met328Thr failed to induce CE defects in overexpression assays and to rescue MO-induced CE defects in zebrafish | [81,104] |
| 673 patients: 284 Italians (11 cranial, 131 open spinal including 128 MMC, 142 closed spinal) and 389 Americans (4 cranial, 325 MMC, 60 closed spinal) | p.Ser83Ile p.Phe153Ser p.Arg181Gln p.Leu202Phe p.Ala404Ser | 3 TFT and TC TFT and TC MMC MMC CA | Not conducted | [83] | |
| 144 patients with open or closed NTDs from Slovakia, Romania and Germany | p.Gly205Arg p.Arg186His p.Arg173His | MMC TC and lipoma unknown | Not conducted | [88] | |
| 53 Italian patients (9 MMC, 44 closed spinal) | p.Alal187Val p.Arg517His p.His350 His | LipoMS MMC Cephalocele | Not conducted | [97] | |
| VANGL2 | 66 English patients (21 CRS, 24 SB, 21 AN) | A 7 bp duplication detected 30 nucleotides into intron six (IVS6+30) | CRS | Not conducted | [82] |
| 163 Han Chinese fetuses (16 AN, 63 CRS, 8 EC, 4 HPS, 14 iniencephaly, 58 SB) | p.Ser84Phe p.Arg353Cys p.Phe437Ser | HPS AN with SB AN | p.Phe437Ser completely abrogated interaction with Dvl; p.Arg353Cys diminished but did not abolish this interaction | [84] | |
| 673 Italian and American patients (same as above) | p.Arg135Trp p.Arg177His p.Leu242Val p.Thr247Met p.Arg270His p.Arg482His | MMC DM 1MCS, 1MMC Lipoma Fibrolipoma CA and TC | Not conducted | [86] | |
| PCP mediators | |||||
| LRP6 | 285 Italian patients (6 closed cranial, 153 MMC, 126 closed spinal) | p.Tyr306His p.Thr373Cys p.Val1386Leu p.Thr1541Cys | MMC MMC EC CA | p.Tyr306His, p.Thr373Cys and p.Val1386Leu acted as hypomorphic alleles in activating canonical Wnt pathway and inhibiting PCP signaling | [93] |
| 192 American SB infants | p.Ala3Val p.Tyr544Cys p.Pro1482Leu p.Arg1574Leu | SB | p.Tyr544Cys lost its ability to bind MESD and its membrane localization and acted as a hypomorhic allele in activating canonical Wnt signalling; p.Arg1574Leu acted as a hypermorphic allele in activating canonical Wnt signaling; p.Pro1482Leu lost its ability to inhibit PCP signaling | [96] | |
| PTK7 | 473 Italian and French Canadian patients (same as above) | p.Ile121Met p.Val291Ile p.Pro345Leu p.Gly348Ser p.Pro545Arg | MMC LipoMS MC MMC MMC | p.Ile121Met, p.Pro345Leu and p.Pro545Arg could act in a hypomorphic manner; p.Gly348Ser acted in a hypermorphic manner in overexpression assays in zebrafish;p.Pro545Arg affected stability of protein | [98,105] |
| 343 Chinese patients (70 AN, 19 CRS, 80 EC, 3 EX, 170 SB, 1 unknown) and 192 American SB infants | p.Asn128Ser p.Thr186Met p.Ala560Thr p.Arg630Ser p.Pro706Arg p.Tyr725Phe p.Gly765Arg p.Val775Met p.Arg790Leu | EX SB SB SB 2 AN, CRS, 2 SB SB 2 SB SB 2 SB, EC, AN | p.Arg630Ser affected protein stability and increased interaction with Dvl2; p.Thr186Met decreased PTK7 interactions with Dvl2 | [102] | |
| 510 Chinese patients (same as above) | p.Pro642Arg | SB | Not conducted | [101] | |
| SCRIB1 | 52 fetuses with CRS (same as above) | p.Arg1535Gln | CRS | p.Arg1535Gln affected membrane localization of Scrib1 | [90] |
| 192 American SB infants | p.Ala366Thr p.Thr552Met p.Pro1043Leu p.Pro1332Leu p.Leu1520Arg | SB | p.Pro1043Leu, p.Pro1332Leu and p.Leu1520Arg significantly affected protein membrane localization | [92] | |
| 473 Italian and French Canadian patients (same as above) | p.Gly263Ser p.Pro649His p.Gln808His p.Arg1150Gln p.Thr1422Met | MMC CA MMC VS MMC | P.Gly263Ser and p.Gln808His significantly affected the subcellular localization of SCRIB1 and failed in rescuing the localization defect of Par-3 and Vangl1 caused by knockdown of Scrib1; P.Gln808His and p.Arg1150Gln abolished the interaction of Scrib1 with Vangl2. | [68] | |
| 510 Chinese patients (same as above) | p.Lys618Arg p.Gly644Val p.Arg1044Gln p.Gly1108Glu | SB SB AN SB | Not conducted | [101] | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; de Marco, P.; Capra, V.; Kibar, Z. Update on the Role of the Non-Canonical Wnt/Planar Cell Polarity Pathway in Neural Tube Defects. Cells 2019, 8, 1198. https://doi.org/10.3390/cells8101198
Wang M, de Marco P, Capra V, Kibar Z. Update on the Role of the Non-Canonical Wnt/Planar Cell Polarity Pathway in Neural Tube Defects. Cells. 2019; 8(10):1198. https://doi.org/10.3390/cells8101198
Chicago/Turabian StyleWang, Mingqin, Patrizia de Marco, Valeria Capra, and Zoha Kibar. 2019. "Update on the Role of the Non-Canonical Wnt/Planar Cell Polarity Pathway in Neural Tube Defects" Cells 8, no. 10: 1198. https://doi.org/10.3390/cells8101198
APA StyleWang, M., de Marco, P., Capra, V., & Kibar, Z. (2019). Update on the Role of the Non-Canonical Wnt/Planar Cell Polarity Pathway in Neural Tube Defects. Cells, 8(10), 1198. https://doi.org/10.3390/cells8101198