A Diet Induced Maladaptive Increase in Hepatic Mitochondrial DNA Precedes OXPHOS Defects and May Contribute to Non-Alcoholic Fatty Liver Disease


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Model and Diet
2.2. Liver Histology
2.3. Mitochondrial DNA Quantification
2.4. Proteomics
2.5. Statistical Analysis
3. Results
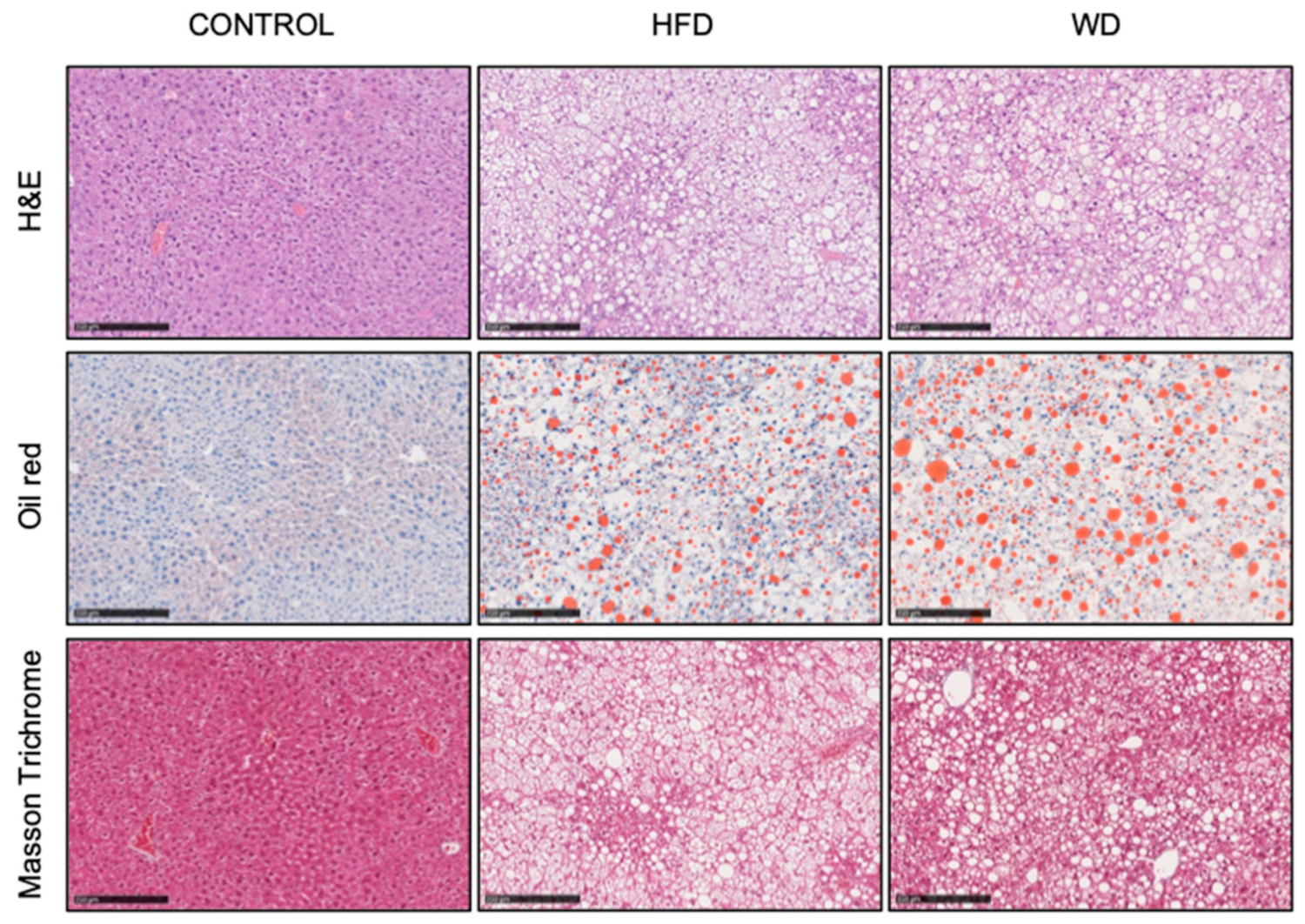
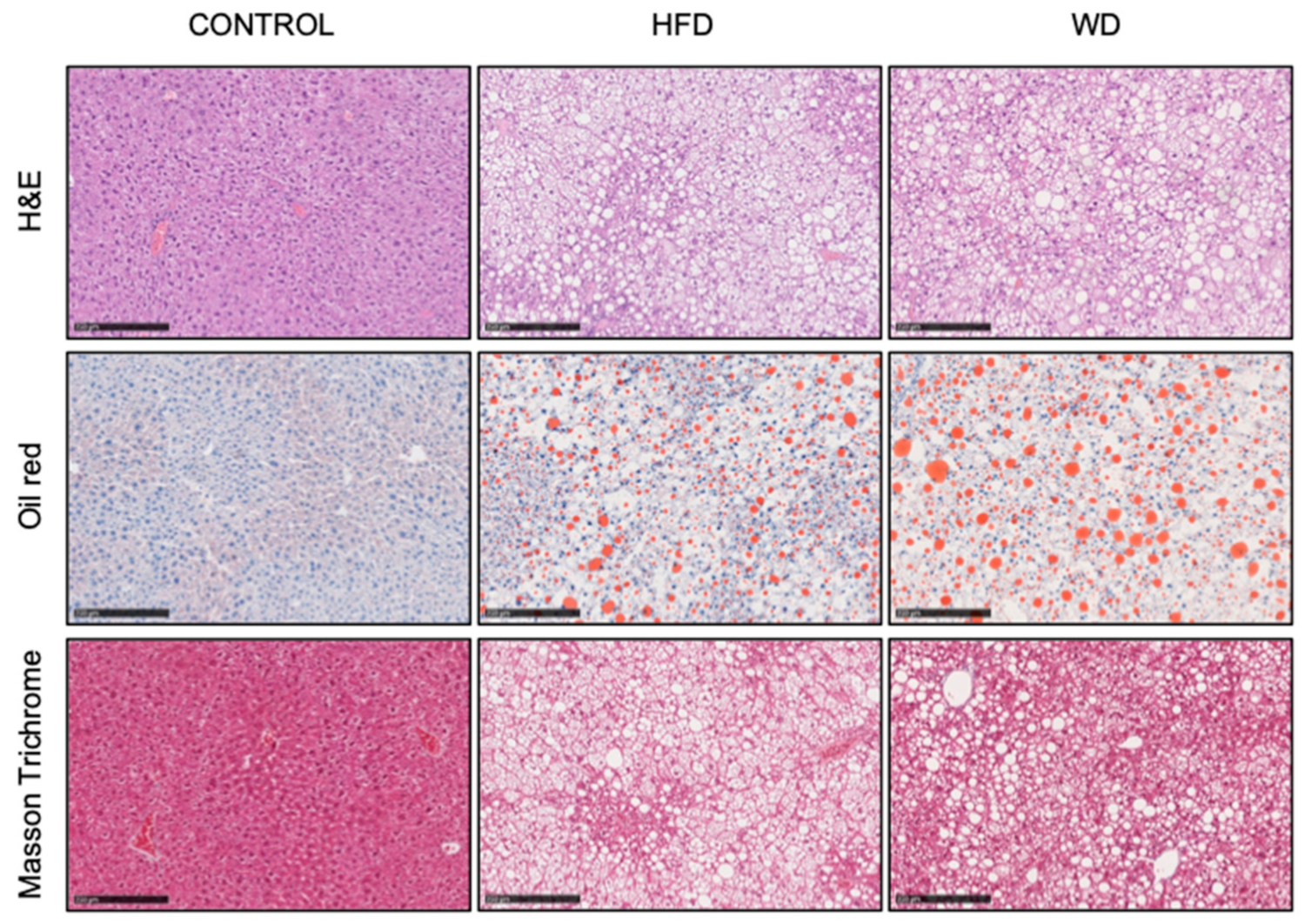
3.1. Development of Diet Induced Liver Steatosis
3.2. Mitochondrial DNA Content in the Control Mouse Liver
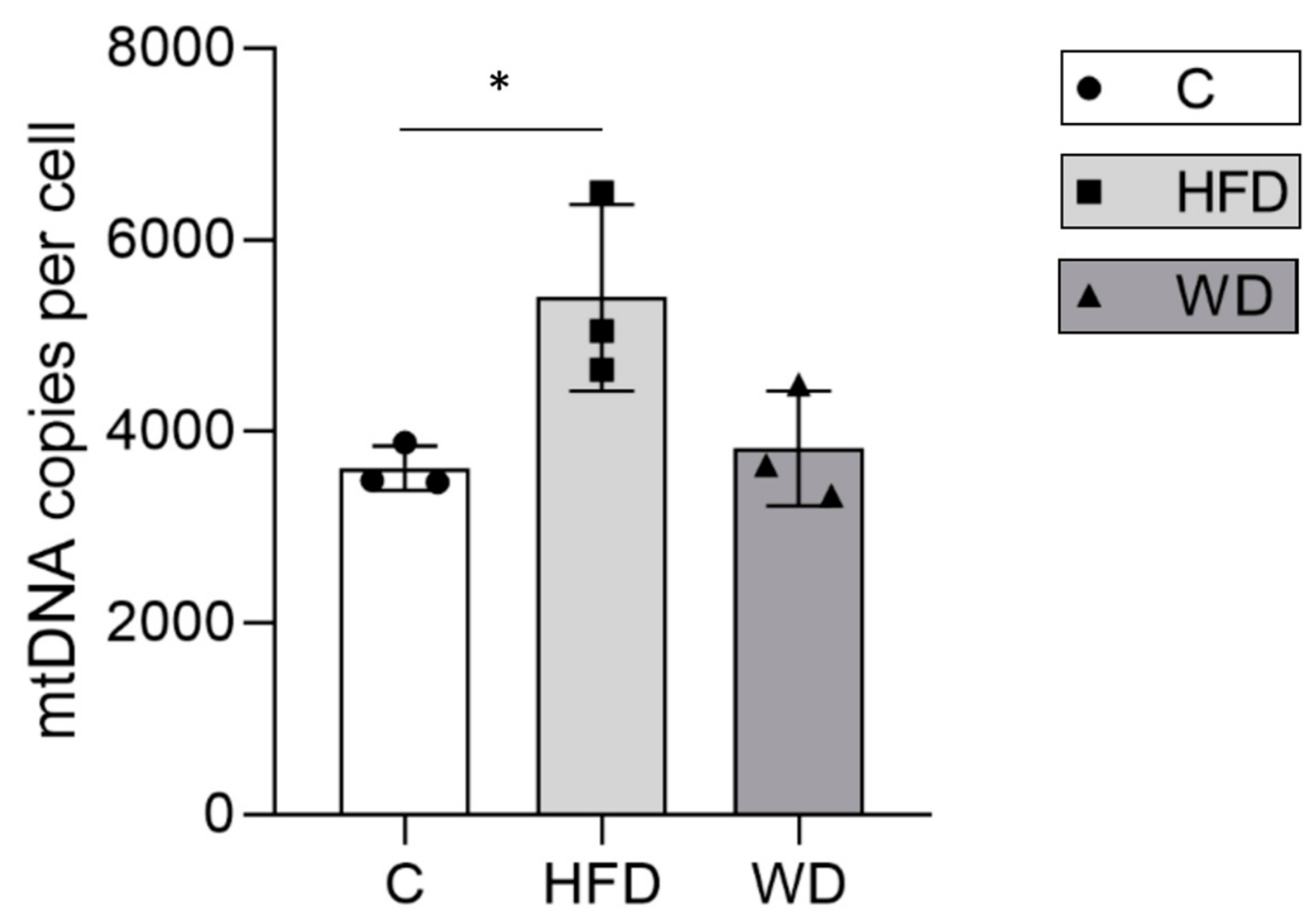
3.3. Hepatic Mitochondrial DNA Content Increased in Parallel with Steatosis
3.4. Alterations in Hepatic Mitochondrial Proteins Suggest Reduced OXPHOS, Reduced Mitochondrial Biogenesis, and Increased Inflammation
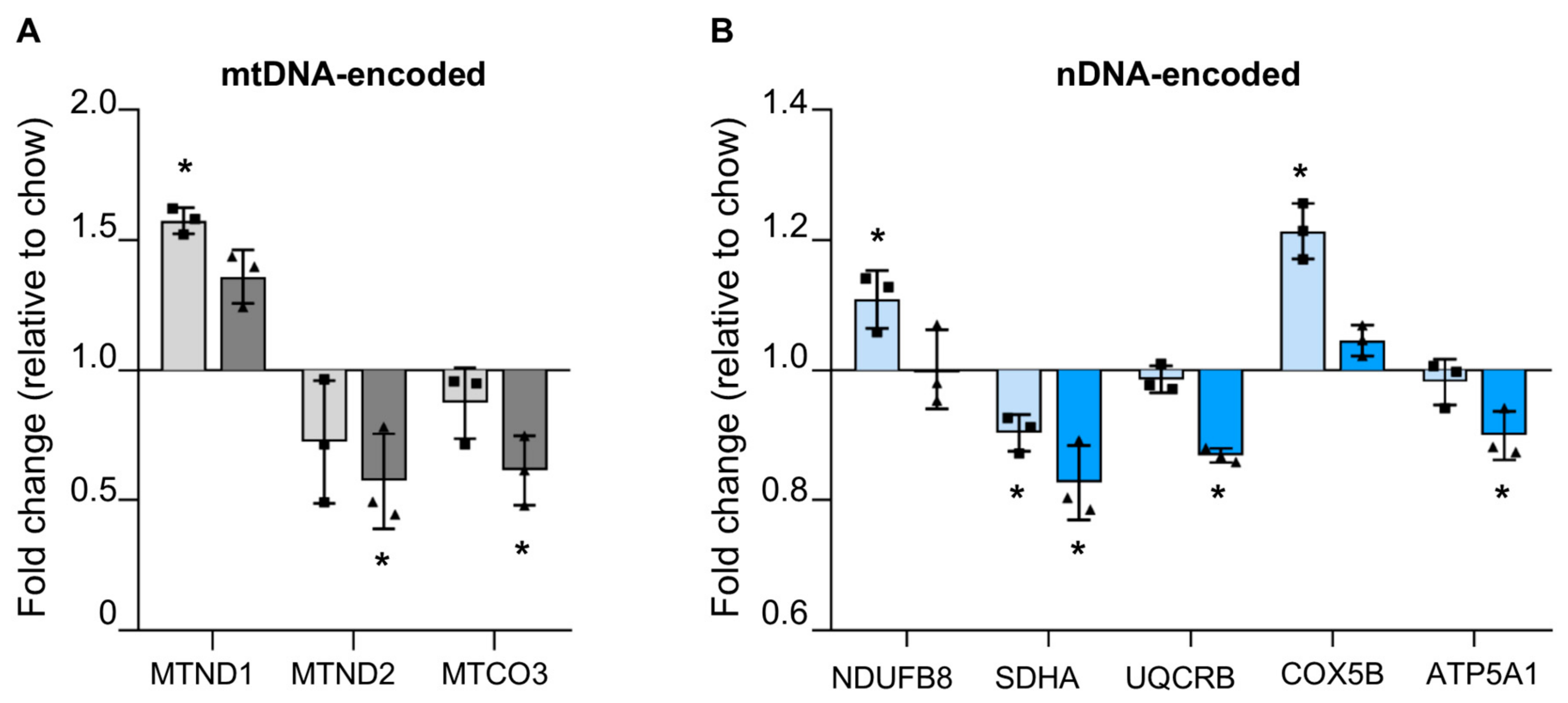
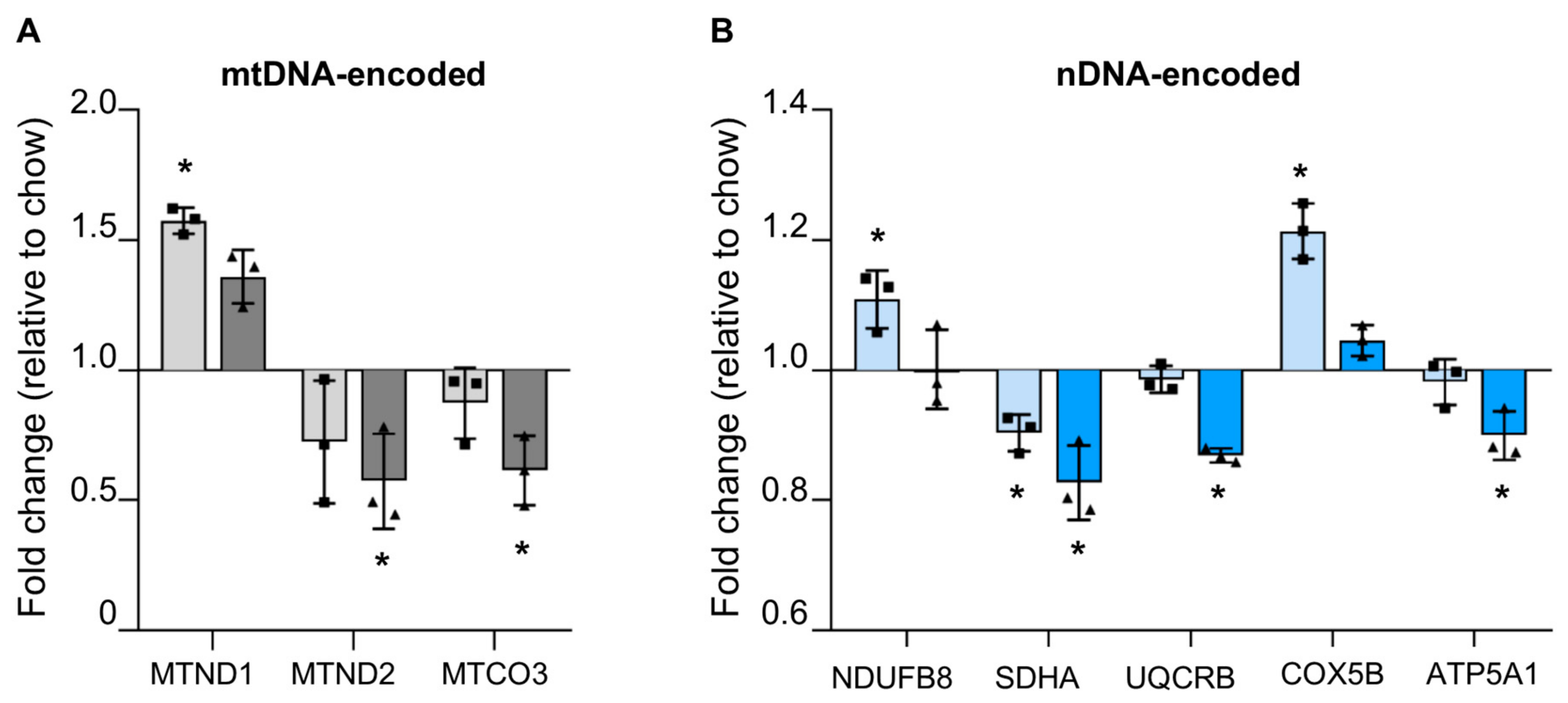
3.4.1. Reduced mtDNA Encoded OXPHOS Proteins in Steatotic Liver
3.4.2. Reduced Nuclear Encoded OXPHOS Proteins in Steatotic Liver
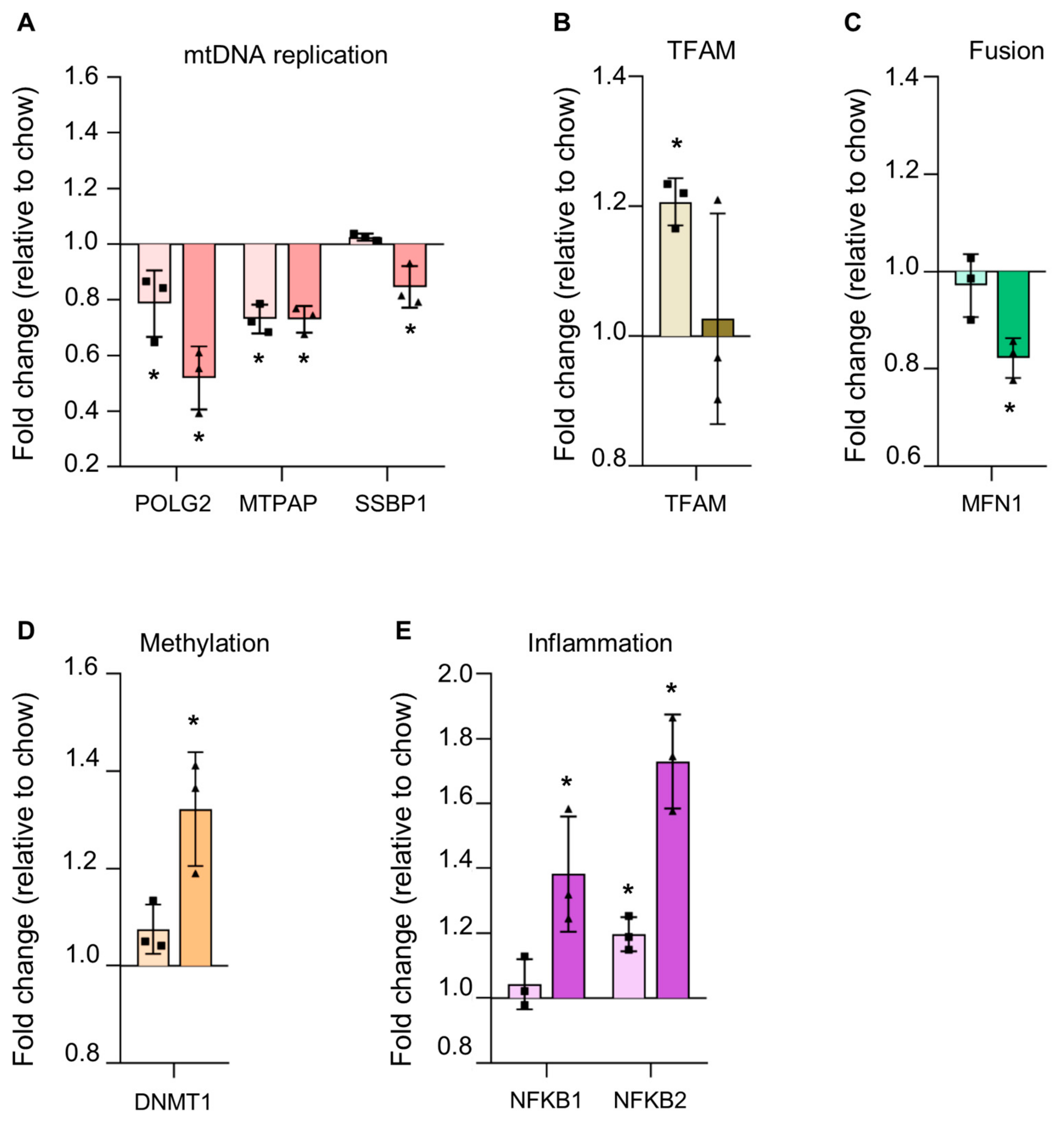
3.4.3. Reduction in Proteins Involved in mtDNA Replication in Steatosis
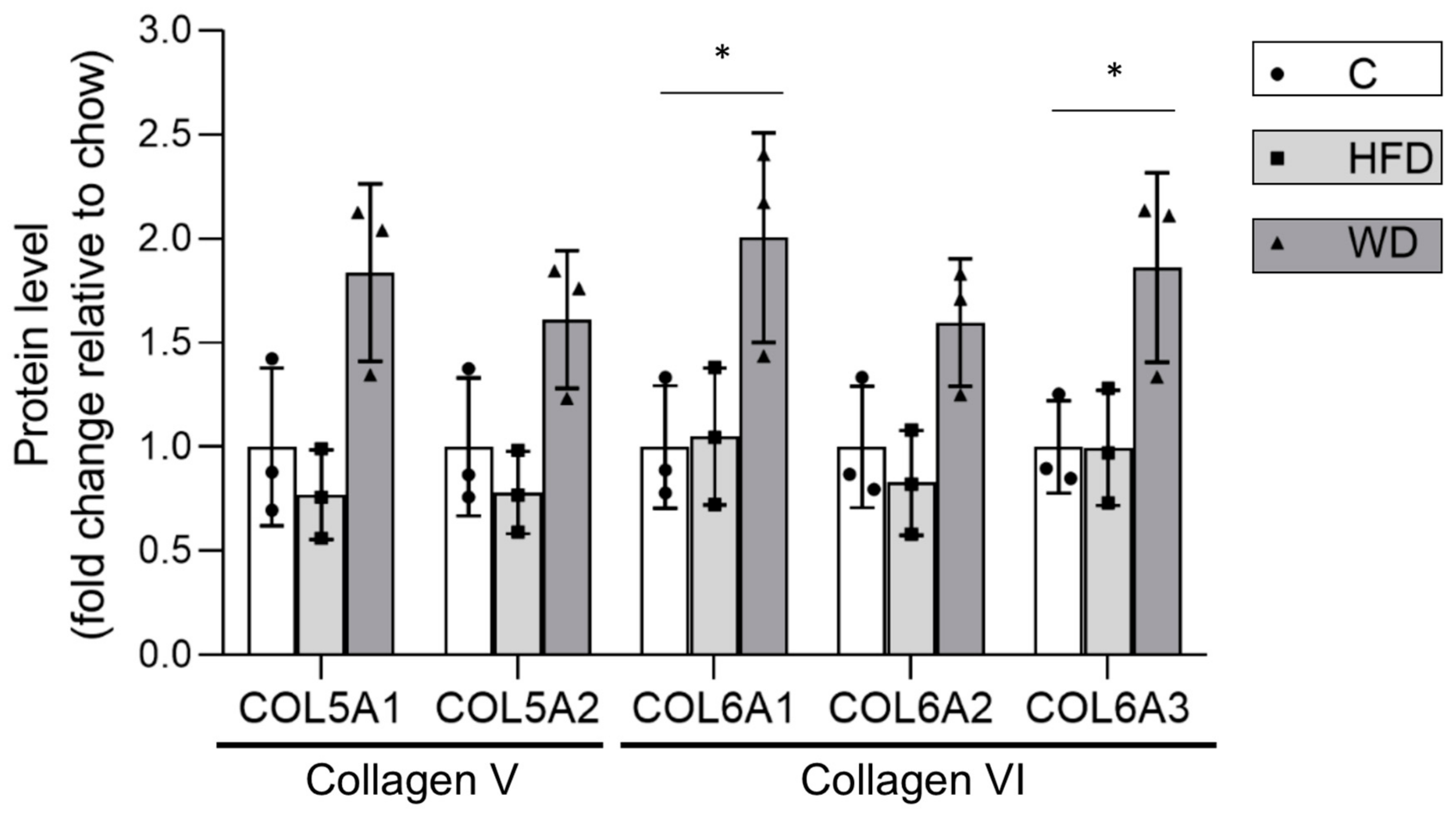
3.4.4. Alterations in Steatotic Livers Proteins Suggests Altered Nuclear Genome Methylation and Increased Inflammation
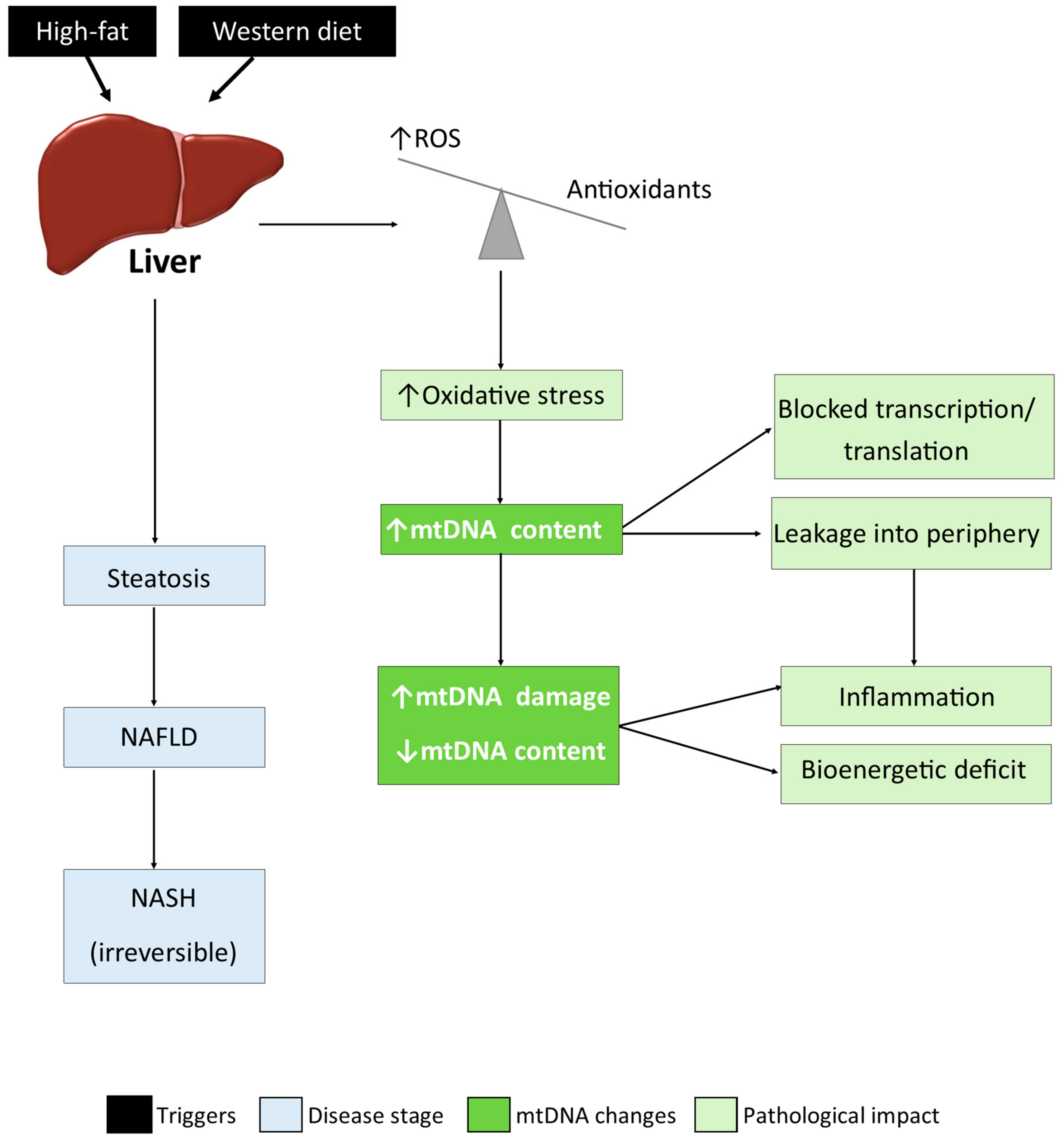
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Petta, S.; Dalle Grave, R. Diet, weight loss, and liver health in nonalcoholic fatty liver disease: Pathophysiology, evidence, and practice. Hepatology 2016, 63, 2032–2043. [Google Scholar] [CrossRef] [PubMed]
- Trauner, M.; Arrese, M.; Wagner, M. Fatty liver and lipotoxicity. Biochim. et Biophys. acta 2010, 1801, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Glen, J.; Floros, L.; Day, C.; Pryke, R. Non-alcoholic fatty liver disease (NAFLD): Summary of NICE guidance. BMJ 2016, 354, 4428. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinella, M.; Charlton, M. The globalization of nonalcoholic fatty liver disease: Prevalence and impact on world health. Hepatology 2016, 64, 19–22. [Google Scholar] [CrossRef] [Green Version]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pr. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyer, T.D.; Wright, T.L.; Manns, M.P.; Zakim, D. Zakim and Boyer’s Hepatology: A Textbook of Liver Disease, 5th ed.; Elsevier Saunders: Philadelphia, PA, USA, 2006. [Google Scholar]
- Freitas-Lopes, M.A.; Mafra, K.; David, B.A.; Carvalho-Gontijo, R.; Menezes, G.B. Differential Location and Distribution of Hepatic Immune Cells. Cells 2017, 6, 48. [Google Scholar] [CrossRef]
- Giri, S.; Bader, A. Shortcut route for generation of functional hepatocyte cells from human skin allogenically for autologous treatment of chronic liver diseases. J. Clin. Exp. Hepatol. 2014, 4, 74–78. [Google Scholar] [CrossRef]
- Simões, I.C.; Fontes, A.; Pinton, P.; Zischka, H.; Wieckowski, M.R. Mitochondria in non-alcoholic fatty liver disease. Int. J. Biochem. Cell Boil. 2018, 95, 93–99. [Google Scholar] [CrossRef]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; De Bruijn, M.H.L.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Lightowlers, R.N.; Chinnery, P.F.; Turnbull, D.M.; Howell, N. Mammalian mitochondrial genetics: Heredity, heteroplasmy and disease. Trends Genet. 1997, 13, 450–455. [Google Scholar] [CrossRef]
- Malik, A.N.; Czajka, A. Is mitochondrial DNA content a potential biomarker of mitochondrial dysfunction? Mitochondrion 2013, 13, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.N.; Shahni, R.; Iqbal, M.M. Increased peripheral blood mitochondrial DNA in type 2 diabetic patients with nephropathy. Diabetes Res. Clin. Pr. 2009, 86, e22–e24. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.N.; Shahni, R.; Rodriguez-De-Ledesma, A.; Laftah, A.; Cunningham, P. Mitochondrial DNA as a non-invasive biomarker: Accurate quantification using real time quantitative PCR without co-amplification of pseudogenes and dilution bias. Biochem. Biophys. Res. Commun. 2011, 412, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.N.; Czajka, A.; Cunningham, P. Accurate quantification of mouse mitochondrial DNA without co-amplification of nuclear mitochondrial insertion sequences. Mitochondrion 2016, 29, 59–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirola, C.J.; Scian, R.; Gianotti, T.F.; Dopazo, H.; Rohr, C.; Martino, J.S.; Castaño, G.O.; Sookoian, S. Epigenetic modifications in the biology of nonalcoholic fatty liver disease: The role of DNA hydroxymethylation and tet proteins. Medicine 2015, 94, e1480. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Rosselli, M.S.; Gemma, C.; Burgueno, A.L.; Fernandez Gianotti, T.; Castano, G.O.; Pirola, C.J. Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: Impact of liver methylation of the peroxisome proliferator-activated receptor gamma coactivator 1alpha promoter. Hepatology 2010, 52, 1992–2000. [Google Scholar] [CrossRef]
- Szymańska-Dębińska, T.; Karkucińska-Więckowska, A.; Piekutowska-Abramczuk, D.; Jurkiewicz, E.; Iwanicka-Pronicka, K.; Rokicki, D.; Pronicki, M. Leigh disease due to SCO2 mutations revealed at extended autopsy. J. Clin. Pathol. 2015, 68, 397–399. [Google Scholar] [CrossRef]
- Czajka, A.; Ajaz, S.; Gnudi, L.; Parsade, C.K.; Jones, P.; Reid, F.; Malik, A.N. Altered Mitochondrial Function, Mitochondrial DNA and Reduced Metabolic Flexibility in Patients With Diabetic Nephropathy. EBioMedicine 2015, 2, 499–512. [Google Scholar] [CrossRef] [Green Version]
- Malik, A.N.; Parsade, C.K.; Ajaz, S.; Crosby-Nwaobi, R.; Gnudi, L.; Czajka, A.; Sivaprasad, S. Altered circulating mitochondrial DNA and increased inflammation in patients with diabetic retinopathy. Diabetes Res. Clin. Pr. 2015, 110, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.N.; Rosa, H.S. Potential Mechanisms of Mitochondrial DNA Mediated Acquired Mitochondrial Disease. In Mitochondrial Biology and Experimental Therapeutics; Springer: Cham, Switzerland, 2018; pp. 297–315. [Google Scholar]
- Rocher, C.; Taanman, J.-W.; Pierron, D.; Faustin, B.; Bénard, G.; Rossignol, R.; Malgat, M.; Pedespan, L.; Letellier, T. Influence of mitochondrial DNA level on cellular energy metabolism: Implications for mitochondrial diseases. J. Bioenerg. Biomembr. 2008, 40, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, P.L.; Greenawalt, J.W.; Reynafarje, B.; Hullihen, J.; Decker, G.L.; Soper, J.W.; Bustamente, E. Chapter 26 Preparation and Characterization of Mitochondria and Submitochondrial Particles of Rat Liver and Liver-Derived Tissues. Methods Cell Biol. 1978, 20, 411–481. [Google Scholar] [PubMed]
- McClave, S.A.; Snider, H.L. Dissecting the energy needs of the body. Curr. Opin. Clin. Nutr. Metab. Care 2001, 4, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ying, Z.; Bosy-Westphal, A.; Zhang, J.; Heller, M.; Later, W.; Heymsfield, S.B.; Müller, M.J. Evaluation of specific metabolic rates of major organs and tissues: Comparison between men and women. Am. J. Hum. Biol. 2011, 23, 333–338. [Google Scholar] [CrossRef]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.-E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of Hepatic Mitochondrial Function in Humans with Non-Alcoholic Fatty Liver Is Lost in Steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, C.G.; Wenceslau, C.F.; Goulopoulou, S.; Ogbi, S.; Baban, B.; Sullivan, J.C.; Matsumoto, T.; Webb, R.C. Circulating mitochondrial DNA and Toll-like receptor 9 are associated with vascular dysfunction in spontaneously hypertensive rats. Cardiovasc. Res. 2015, 107, 119–130. [Google Scholar] [CrossRef]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef]
- Chiappini, F.; Barrier, A.; Saffroy, R.; Domart, M.C.; Dagues, N.; Azoulay, D.; Sebagh, M.; Franc, B.; Chevalier, S.; Debuire, B.; et al. Exploration of global gene expression in human liver steatosis by high-density oligonucleotide microarray. Lab. Investig. 2006, 86, 154–165. [Google Scholar] [CrossRef]
- Kamfar, S.; Alavian, S.M.; Houshmand, M.; Yadegarazari, R.; Zarei, B.S.; Khalaj, A.; Shabab, N.; Saidijam, M. Liver Mitochondrial DNA Copy Number and Deletion Levels May Contribute to Nonalcoholic Fatty Liver Disease Susceptibility. Zahedan J. Res. Med. Sci. 2016, 16, 40774. [Google Scholar] [CrossRef] [PubMed]
- Abdelmalek, M.F.; Suzuki, A.; Guy, C.; Unalp-Arida, A.; Colvin, R.; Johnson, R.J.; Diehl, A.M. Nonalcoholic Steatohepatitis Clinical Research Network Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Cydylo, M.A.; Davis, A.T.; Kavanagh, K. Fatty liver promotes fibrosis in monkeys consuming high fructose. Obesity 2017, 25, 290–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grattagliano, I.; Montezinho, L.P.; Oliveira, P.J.; Frühbeck, G.; Gómez-Ambrosi, J.; Montecucco, F.; Carbone, F.; Wieckowski, M.R.; Wang, D.Q.-H.; Portincasa, P. Targeting mitochondria to oppose the progression of nonalcoholic fatty liver disease. Biochem. Pharmacol. 2019, 160, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Martinez, I.; Santoro, N.; Chen, Y.; Hoque, R.; Ouyang, X.; Caprio, S.; Shlomchik, M.J.; Coffman, R.L.; Candia, A.; Mehal, W.Z. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J. Clin. Investig. 2016, 126, 859–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sookoian, S.; Flichman, D.; Scian, R.; Rohr, C.; Dopazo, H.; Gianotti, T.F.; Martino, J.S.; Castaño, G.O.; Pirola, C.J. Mitochondrial genome architecture in non-alcoholic fatty liver disease. J. Pathol. 2016, 240, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Ou, Z.; Cai, C.; Li, P.; Gong, J.; Ruan, X.Z.; He, K. Fatty acid activates NLRP3 inflammasomes in mouse Kupffer cells through mitochondrial DNA release. Cell. Immunol. 2018, 332, 111–120. [Google Scholar] [CrossRef]
- Handa, P.; Vemulakonda, A.; Kowdley, K.V.; Uribe, M.; Méndez-Sánchez, N. Mitochondrial DNA from hepatocytes as a ligand for TLR9: Drivers of nonalcoholic steatohepatitis? World J. Gastroenterol. 2016, 22, 6965–6971. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diet Group | Animal | mtDNA Copies | B2m Copies | MtN | mtDNA Content Per Cell | Group mtDNA Content |
|---|---|---|---|---|---|---|
| Control | 1 | 25,937,328 (1,366,548) | 13,353 (979) | 1942 | 3885 | 3617 (233) |
| 2 | 9,627,784 (308,494) | 5512 (67) | 1747 | 3493 | ||
| 3 | 23,219,751 (1,539,598) | 13,377 (1,034) | 1736 | 3472 | ||
| HFD | 4 | 41,352,736 (2,450,499) | 17,816 (704) | 2321 | 4642 | 5395 (975) |
| 5 | 21,168,369 (1,697,161) | 6518 (353) | 3248 | 6496 | ||
| 6 | 52,091,806 (833,201) | 20,641 (798) | 2524 | 5047 | ||
| WD | 7 | 23,516,887 (576,445) | 12,888 (594) | 1825 | 3650 | 3822 (600) |
| 8 | 14,047,757 (169,535) | 8446 (163) | 1663 | 3327 | ||
| 9 | 23,185,769 (93,493) | 10,329 (461) | 2245 | 4490 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malik, A.N.; Simões, I.C.M.; Rosa, H.S.; Khan, S.; Karkucinska-Wieckowska, A.; Wieckowski, M.R. A Diet Induced Maladaptive Increase in Hepatic Mitochondrial DNA Precedes OXPHOS Defects and May Contribute to Non-Alcoholic Fatty Liver Disease. Cells 2019, 8, 1222. https://doi.org/10.3390/cells8101222
Malik AN, Simões ICM, Rosa HS, Khan S, Karkucinska-Wieckowska A, Wieckowski MR. A Diet Induced Maladaptive Increase in Hepatic Mitochondrial DNA Precedes OXPHOS Defects and May Contribute to Non-Alcoholic Fatty Liver Disease. Cells. 2019; 8(10):1222. https://doi.org/10.3390/cells8101222
Chicago/Turabian StyleMalik, Afshan N, Inês C.M. Simões, Hannah S. Rosa, Safa Khan, Agnieszka Karkucinska-Wieckowska, and Mariusz R. Wieckowski. 2019. "A Diet Induced Maladaptive Increase in Hepatic Mitochondrial DNA Precedes OXPHOS Defects and May Contribute to Non-Alcoholic Fatty Liver Disease" Cells 8, no. 10: 1222. https://doi.org/10.3390/cells8101222
APA StyleMalik, A. N., Simões, I. C. M., Rosa, H. S., Khan, S., Karkucinska-Wieckowska, A., & Wieckowski, M. R. (2019). A Diet Induced Maladaptive Increase in Hepatic Mitochondrial DNA Precedes OXPHOS Defects and May Contribute to Non-Alcoholic Fatty Liver Disease. Cells, 8(10), 1222. https://doi.org/10.3390/cells8101222