Transcriptional Regulation of Energy Metabolism in Cancer Cells

 ,
,
Abstract
:1. Introduction
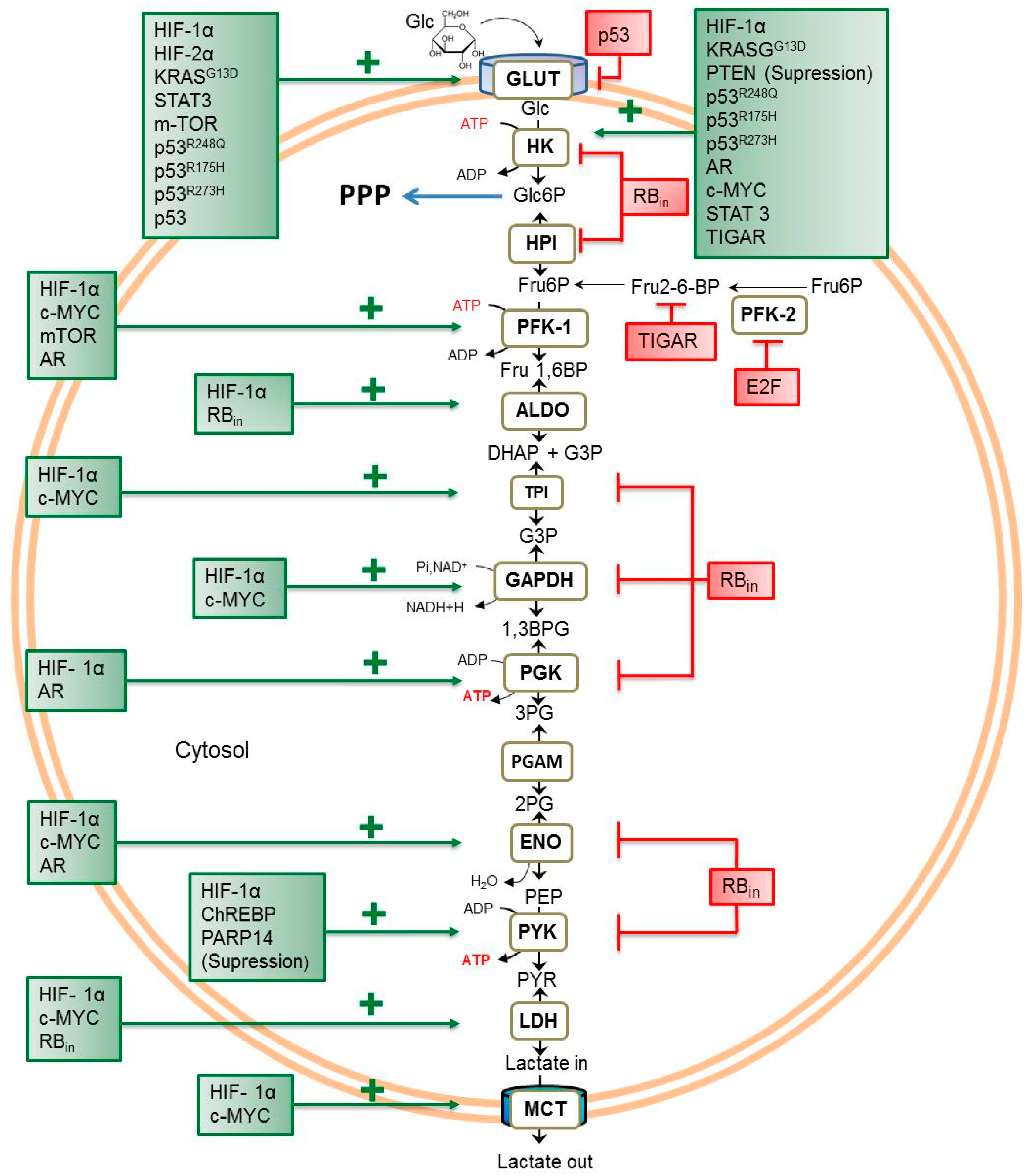
2. Transcriptional Regulators of Glycolytic Flux in Cancer Cells
2.1. Transcriptional Factors
2.1.1. Hypoxia-Inducible Factor-1 (HIF-1)
2.1.2. p53 Wild-Type and Mutant Isoforms
2.1.3. PGC1α
2.1.4. NF-κB
2.1.5. TFAM (Transcription Factor A, Mitochondrial)
2.1.6. STAT3
2.1.7. FOXO-1
2.1.8. E2F
2.1.9. AR (Androgen Receptor)
2.1.10. ChREBP
2.2. Oncogenes
2.2.1. c-MYC
2.2.2. RAS (HRAS and KRAS)
2.3. Tumor-Suppressor Genes
2.3.1. RB
2.3.2. PTEN
2.4. Protein Kinases
2.4.1. JNK
2.4.2. mTOR
3. Transcriptional Regulators of Tumor Oxidative Phosphorylation (OxPhos) in Cancer Cells
3.1. Transcription Factors
3.1.1. HIF-1α
3.1.2. p53, Wild-Type, and Mutant Isoforms
3.1.3. PGC1α
3.1.4. NRF-1
3.1.5. NF-κB
3.1.6. TFAM
3.1.7. STAT3
3.1.8. PTEN
3.1.9. FOXO3a
3.1.10. E2F
3.1.11. AR
3.1.12. ERs
3.1.13. PPARs
3.2. Oncogenes
3.2.1. c-MYC
3.2.2. RAS (HRAS and KRAS)
3.3. Tumor-Suppressor Genes
RB
3.4. Protein Kinases
3.4.1. JNK
3.4.2. mTOR
3.5. Plasma Membrane Receptors
Notch 1
4. Overview of TR Interplay and Action in Cancer Glycolysis and OxPhos
4.1. p53
4.2. c-MYC
4.3. mTOR
4.4. FOXO3a
4.5. RAS
4.6. STAT3
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Levine, M.; Tjian, R. Transcription regulation and animal diversity. Nature 2003, 424, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Y.; Muller, W.J. Oncogenes and tumor suppressor genes. Cold Spring Harb. Perspect. Biol. 2010, 2, a003236. [Google Scholar] [CrossRef] [PubMed]
- Ell, B.; Kang, Y. Transcriptional control of cancer metastasis. Trends Cell Biol. 2013, 23, 603–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [Green Version]
- Marín-Hernández, A.; Gallardo-Pérez, J.C.; Ralph, S.J.; Rodríguez-Enríquez, S.; Moreno-Sánchez, R. HIF-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini Rev. Med. Chem. 2009, 9, 1084–1101. [Google Scholar] [CrossRef]
- Philip, B.; Ito, K.; Moreno-Sánchez, R.; Ralph, S.J. HIF expression and the role of hypoxic microenvironments within primary tumours as protective sites driving cancer stem cell renewal and metastatic progression. Carcinogenesis 2013, 34, 1699–1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, J.P. Oxygen sensors in context. Biochim. Biophys. Acta 2008, 1777, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Carreau, A.; El Hafny-Rahbi, B.; Matejuk, A.; Grillon, C.; Kieda, C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J. Cell. Mol. Med. 2011, 15, 1239–1253. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.; De Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.A.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999, 59, 5830–5835. [Google Scholar]
- Monsef, N.; Helczynski, L.; Lundwall, A.; Pahlman, S.; Bjartell, A. Localization of immune reactive HIF-1alpha and HIF-2alpha in neuroendocrine cells of both benign and malignant prostate glands. Prostate 2007, 67, 1219–1229. [Google Scholar] [CrossRef]
- Koivunen, P.; Hirsilä, M.; Remes, A.M.; Hassinen, I.E.; Kivirikko, K.I.; Myllyharju, J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: Possible links between cell metabolism and stabilization of HIF. J. Biol. Chem. 2007, 282, 4524–4532. [Google Scholar] [CrossRef] [PubMed]
- Pollard, P.J.; Brière, J.J.; Alam, N.A.; Barwell, J.; Barclay, E.; Wortham, N.C.; Hunt, T.; Mitchell, M.; Olpin, S.; Moat, S.J.; et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum. Mol. Genet. 2005, 14, 2231–2239. [Google Scholar] [CrossRef]
- Sulkowski, P.L.; Sundaram, R.K.; Oeck, S.; Corso, C.D.; Liu, Y.; Noorbakhsh, S.; Niger, M.; Boeke, M.; Ueno, D.; Kalathil, A.N.; et al. Krebs-cycle-deficient hereditary cancer syndromes are defined by defects in homologous-recombination DNA repair. Nat. Genet. 2018, 50, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Ralph, S.J.; Nozuhur, S.; ALHulais, R.A.; Rodríguez-Enríquez, S.; Moreno-Sánchez, R. Repurposing drugs as pro-oxidant redox modifiers to eliminate cancer stem cells and improve the treatment of advanced stage cancers. Med. Res. Rev. 2019. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.N.; Xi, M.M.; Guo, Y.; Hai, C.X.; Yang, W.L.; Qin, X.J. NADPH oxidase-mitochondria axis-derived ROS mediate arsenite-induced HIF-1α stabilization by inhibiting prolyl hydroxylases activity. Toxicol. Lett. 2014, 224, 165–174. [Google Scholar] [CrossRef]
- Soni, S.; Padwad, Y.S. HIF-1 in cancer therapy: Two decade long story of a transcriptional factor. Acta Oncol. 2017, 56, 509–515. [Google Scholar] [CrossRef]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumor suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef]
- Bost, F.; Kaminski, L. The metabolic modulator PGC-1α in cancer. Am. J. Cancer Res. 2019, 9, 198–211. [Google Scholar]
- Zhou, Y.; Xu, Z.; Quan, D.; Zhang, F.; Zhang, H.; Xiao, T.; Hou, S.; Qiao, H.; Harismendy, O.; Wang, J.Y.J.; et al. Nuclear respiratory factor 1 promotes spheroid survival and mesenchymal transition in mammary epithelial cells. Oncogene 2018, 37, 6152–6165. [Google Scholar] [CrossRef]
- Xia, Y.; Shen, S.; Verma, I.M. NF-kB an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef]
- Yao, J.; Xu, F.; Zhang, D.; Yi, W.; Chen, X.; Chen, G.; Zhou, E. TP73-AS1 promotes breast cancer cell proliferation through miR-200a-mediated TFAM inhibition. J. Cell. Biochem. 2018, 119, 680–690. [Google Scholar] [CrossRef] [PubMed]
- You, W.; Tang, Q.; Zhang, C.; Wu, J.; Gu, C.; Wu, Z.; Li, X. IL-26 promotes the proliferation and survival of human gastric cancer cells by regulating the balance of STAT1 and STAT3 activation. PLoS ONE 2013, 8, e63588. [Google Scholar] [CrossRef] [PubMed]
- Xiong, A.; Yang, Z.; Shen, Y.; Zhou, J.; Shen, Q. Transcription factor STAT3 as a novel molecular target for cancer prevention. Cancers 2014, 6, 926–957. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk betweed cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Coomans de Brachene, A.; Demoulin, J.B. FOXO transcription factors in cancer development and therapy. Cell. Mol. Life Sci. 2016, 73, 1159–1172. [Google Scholar] [CrossRef] [PubMed]
- Schaal, C.; Pillais, S.; Chellappan, S.P. The RB-E2F transcriptional regulatory pathway in tumor angiogenesis and metastasis. Adv. Cancer Res. 2014, 121, 147–182. [Google Scholar] [CrossRef]
- Iizuka, K. The transcription factor carbohydrate-response element-binding protein (ChREBP): A possible link between metabolic disease and cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 474–485. [Google Scholar] [CrossRef]
- Davey, R.A.; Grossmann, M. Androgen Receptor Structure, Function and Biology: From Bench to Bedside. Clin. Biochem. Rev. 2016, 37, 3–15. [Google Scholar]
- Klinge, C.M. Estrogenic control of mitochondrial function and biogenesis. J. Cell. Biochem. 2008, 105, 1342–13451. [Google Scholar] [CrossRef]
- Tachibana, K.; Kenji-Ishimoto, Y.; Doi, T. The Role of PPARs in Cancer. PPAR Res. 2008, 2008, 102737. [Google Scholar] [CrossRef]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Yuan, X.; Wei, M.; Wu, J.; Qin, Z.H. The diverse role of TIGAR in cellular homeostasis and cancer. Free Radic. Res. 2018, 52, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Prochownik, E.V. c-Myc: Linking transformation and genomic instability. Curr. Mol. Med. 2008, 8, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Der, C.J.; Cox, A.D. The role of wild type RAS isoforms in cancer. Semin. Cell Dev. Biol. 2016, 58, 60–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega-Molina, A.; Serrano, M. PTEN in cancer, metabolism, and aging. Trends Endocrinol. Metab. 2013, 24, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Chen, F. JNK-induced apoptosis, compensatory growth, and cancer stem cells. Cancer Res. 2012, 72, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Harachi, M.; Masui, K.; Okamura, Y.; Tsukui, R.; Mischel, P.S.; Shibata, N. mTOR complexes as a nutrient sensor for driving cancer progression. Int. J. Mol. Sci. 2018, 19, 3267. [Google Scholar] [CrossRef] [PubMed]
- Henrique, D.; Schweisguth, F. Mechanisms of Notch signaling: A simple logic deployed in time and space. Development 2019, 146. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Liu, X.; Yan, N.; Li, S.; Cao, G.; Cheng, Q.; Xia, Q.; Wang, H. Hypoxia-inducible transcription factor-1alpha promotes hypoxia-induced A549 apoptosis via a mechanism that involves the glycolysis pathway. BMC Cancer 2006, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Enriquez, S.; Careño-Fuentes, L.; Gallardo-Pérez, J.C.; Saavedra, E.; Quezada, H.; Vega, A.; Marín-Hernández, A.; Olín-Sandoval, V.; Torres-Márquez, M.E.; Moreno-Sánchez, R. Oxidative phosphorylation is impaired by prolonged hypoxia in breast and possibly in cervix carcinoma. Int. J. Biochem. Cell Biol. 2010, 42, 1744–1751. [Google Scholar] [CrossRef]
- Pescador, N.; Villar, D.; Cifuentes, D.; Garcia-Rocha, M.; Ortiz-Barahona, A.; Vazquez, S.; Ordoñez, A.; Cuevas, Y.; Saez-Morales, D.; Garcia-Bermejo, M.L.; et al. Hypoxia promotes glycogen accumulation through hypoxia inducible factor (HIF)-mediated induction of glycogen synthase 1. PLoS ONE 2010, 5, e9644. [Google Scholar] [CrossRef] [PubMed]
- Favaro, E.; Bensaad, K.; Chong, M.G.; Tennant, D.A.; Ferguson, D.J.; Snell, C.; Steers, G.; Turley, H.; Li, J.L.; Günther, U.L.; et al. Glucose utilization via glycogen phosphorylase sustains proliferation and prevents premature senescence in cancer cells. Cell Metab. 2012, 16, 51–764. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.; Bellot, G.; Gounon, P.; Lacas-Gervais, S.; Pouysségur, J.; Mazure, N.M. Glycogen synthesis is induced in hypoxia by the hypoxia-inducible factor and promotes cancer cell survival. Front. Oncol. 2012, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.J.; Wang, L.Y.; Chodosh, L.A.; Keith, B.; Simon, M.C. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol. Cell. Biol. 2003, 23, 9361–9374. [Google Scholar] [CrossRef] [PubMed]
- Sowter, H.; Raval, R.; Moore, J.; Ratcliffe, P.; Harris, A. Predominant role of hypoxia-inducible transcription factor (Hif)-1alpha versus Hif-2alpha in regulation of the transcriptional response to hypoxia. Cancer Res. 2003, 63, 6130–6134. [Google Scholar] [PubMed]
- Duan, C. Hypoxia-inducible factor 3 biology: Complexities and emerging themes. Am. J. Physiol. Cell Physiol. 2016, 310, C260–C269. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Forbes, R.; Verma, A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J. Biol. Chem. 2002, 277, 23111–23115. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.; Pandey, A.; Semenza, G. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef]
- Higashimura, Y.; Nakajima, Y.; Yamaji, R.; Harada, N.; Shibasaki, F.; Nakano, Y.; Inui, H. Up-regulation of glyceraldehyde-3-phosphate dehydrogenase gene expression by HIF-1 activity depending on Sp1 in hypoxic breast cancer cells. Arch. Biochem. Biophys. 2011, 509, 1–8. [Google Scholar] [CrossRef]
- Hernández-Reséndiz, I.; Román-Rosales, A.; García-Villa, E.; López-Macay, A.; Pineda, E.; Saavedra, E.; Gallardo-Pérez, J.C.; Alvarez-Ríos, E.; Gariglio, P.; Moreno-Sánchez, R.; et al. Dual regulation of energy metabolism by p53 in human cervix and breast cancer cells. Biochim. Biophys. Acta 2015, 1853, 3266–3278. [Google Scholar] [CrossRef] [Green Version]
- Vousden, K.H.; Ryan, K.M. p53 and metabolism. Nat. Rev. Cancer 2009, 9, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Reséndiz, I.; Gallardo-Pérez, J.C.; López-Macay, A.; Robledo-Cadena, D.X.; García-Villa, E.; Gariglio, P.; Saavedra, E.; Moreno-Sánchez, R.; Rodríguez-Enríquez, S. Mutant p53(R248Q) downregulates oxidative phosphorylation and upregulates glycolysis under normoxia and hypoxia in human cervix cancer cells. J. Cell. Physiol. 2019, 234, 5524–5536. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, J.; Liang, Y.; Wu, R.; Zhao, Y.; Hong, X.; Lin, M.; Yu, H.; Liu, L.; Levine, A.J.; et al. Tumour-associated mutant p53 drives the Warburg effect. Nat. Commun. 2013, 4, 2935. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Wu, X.; Lan, L.; Shangguan, F.; Lin, X.; Chen, F.; Xu, S.; Zhang, Y.; Chen, Z.; Huang, K.; et al. Downregulation of TFAM inhibits the tumorigenesis of non-small cell lung cancer by activating ROS-mediated JNK/p38MAPK signaling and reducing cellular bioenergetics. Oncotarget 2016, 7, 11609–11624. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Jin, R.; Wang, W.; Zhang, T.; Sang, J.; Li, N.; Han, Q.; Zhao, W.; Li, C.; Liu, Z. STAT3 regulates glycolysis via targeting hexokinase 2 in hepatocellular carcinoma cells. Oncotarget 2017, 8, 24777–24784. [Google Scholar] [CrossRef] [Green Version]
- Camporeale, A.; Demaria, M.; Monteleone, E.; Giorgi, C.; Wieckowski, M.R.; Pinton, P.; Poli, V. STAT3 activities and energy metabolism: Dangerous liaisons. Cancers 2014, 6, 1579–1596. [Google Scholar] [CrossRef]
- Li, M.; Wang, W.; Jin, R.; Zhang, T.; Li, N.; Han, Q.; Wei, P.; Liu, Z. Differential association of STAT3 and HK-II expression in hepatitis B virus- and hepatitis C virus-related hepatocellular carcinoma. J. Med. Virol. 2016, 88, 1552–1559. [Google Scholar] [CrossRef]
- Darville, M.I.; Antoine, I.V.; Mertens-Strijthagen, J.R.; Dupriez, V.J.; Rousseau, G.G. An E2F-dependent late-serum-response promoter in a gene that controls glycolysis. Oncogene 1995, 11, 1509–1517. [Google Scholar]
- Audet-Walsh, É.; Yee, T.; McGuirk, S.; Vernier, M.; Ouellet, C.; St-Pierre, J.; Giguère, V. Androgen-dependent repression of ERRγ reprograms metabolism in prostate cancer. Cancer Res. 2017, 77, 378–389. [Google Scholar] [CrossRef]
- Audet-Walsh, É.; Dufour, C.R.; Yee, T.; Zouanat, F.Z.; Yan, M.; Kalloghlian, G.; Vernier, M.; Caron, M.; Bourque, G.; Scarlata, E. Nuclear mTOR acts as a transcriptional integrator of the androgen signaling pathway in prostate cancer. Genes Dev. 2017, 31, 1228–1242. [Google Scholar] [CrossRef] [Green Version]
- Jeong, Y.S.; Kim, D.; Lee, Y.S.; Kim, H.J.; Han, J.Y.; Im, S.S.; Chong, H.K.; Kwon, J.K.; Cho, Y.H.; Kim, W.K. Integrated expression profiling and genome-wide analysis of ChREBP targets reveals the dual role for ChREBP in glucose-regulated gene expression. PLoS ONE 2011, 6, e22544. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Zhao, F.; Mancuso, A.; Gruber, J.J.; Thompson, C.B. The glucose-responsive transcription factor ChREBP contributes to glucose-dependent anabolic synthesis and cell proliferation. Proc. Natl. Acad. Sci. USA 2009, 106, 21660–21665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, E.C.; Ludwig, R.L.; Vousden, K.H. Mitochondrial localization of TIGAR under hypoxia stimulates HK2 and lowers ROS and cell death. Proc. Natl. Acad. Sci. USA 2012, 109, 20491–20496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.W.; Zeller, K.I.; Wang, Y.; Jegga, A.G.; Aronow, B.J.; O’Donnell, K.A.; Dang, C.V. Evaluation of myc E-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol. Cell. Biol. 2004, 24, 5923–5936. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Gao, P.; Liu, Y.C.; Semenza, G.L.; Dang, C.V. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol. Cell. Biol. 2007, 27, 7381–7393. [Google Scholar] [CrossRef]
- Doherty, J.R.; Yang, C.; Scott, K.E.; Cameron, M.D.; Fallahi, M.; Li, W.; Hall, M.A.; Amelio, A.L.; Mishra, J.K.; Li, F.; et al. Blocking lactate export by inhibiting the Myc target MCT1 Disables glycolysis and glutathione synthesis. Cancer Res. 2014, 74, 908–920. [Google Scholar] [CrossRef]
- Doe, M.R.; Ascano, J.M.; Kaur, M.; Cole, M.D. Myc post-transcriptionally induces HIF1 protein and target gene expression in normal and cancer cells. Cancer Res. 2012, 72, 949–957. [Google Scholar] [CrossRef]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.; Markowitz, S.; Zhou, S.; et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009, 325, 1555–1559. [Google Scholar] [CrossRef]
- Iwamoto, M.; Kawada, K.; Nakamoto, Y.; Itatani, Y.; Inamoto, S.; Toda, K.; Kimura, H.; Sasazuki, T.; Shirasawa, S.; Okuyama, H.; et al. Regulation of 18F-FDG accumulation in colorectal cancer cells with mutated KRAS. J. Nucl. Med. 2014, 55, 2038–2044. [Google Scholar] [CrossRef]
- Dias, P.L.; Shanmuganathan, S.S.; Rajaratnam, M. Lactic dehydrogenase activity of aqueous humour in retinoblastoma. Br. J. Ophthalmol. 1971, 55, 130–132. [Google Scholar] [CrossRef]
- Beemer, F.A.; Vlug, A.M.; Rijksen, G.; Hamburg, A.; Staal, G.E. Characterization of some glycolytic enzymes from human retina and retinoblastoma. Cancer Res. 1982, 42, 4228–4232. [Google Scholar] [PubMed]
- Zhou, X.; Yang, X.; Sun, X.; Xu, X.; Li, X.; Guo, Y.; Wang, J.; Li, X.; Yao, L.; Wang, H.; et al. Effect of PTEN loss on metabolic reprogramming in prostate cancer cells. Oncol. Lett. 2019, 17, 2856–2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phadngam, S.; Castiglioni, A.; Ferraresi, A.; Morani, F.; Follo, C.; Isidoro, C. PTEN dephosphorylates AKT to prevent the expression of GLUT1 on plasma membrane and to limit glucose consumption in cancer cells. Oncotarget 2016, 7, 84999–85020. [Google Scholar] [CrossRef] [PubMed]
- Iansante, V.; Choy, P.M.; Fung, S.W.; Liu, Y.; Chai, J.G.; Dyson, J.; Del Rio, A.; D’Santos, C.D.; Williams, R.; Chokshi, S.; et al. PARP14 promotes the Warburg effect in hepatocellular carcinoma by inhibiting INK-dependent PKM2 phosphorylation and activation. Nat. Commun. 2015, 6, 7882. [Google Scholar] [CrossRef]
- Düvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Poulain, L.; Sujobert, P.; Zylbersztejn, F.; Barreau, S.; Stuani, L.; Lambert, M.; Palama, T.L.; Chesnais, V.; Birsen, R.; Vergez, F.; et al. High mTORC1 activity drives glycolysis addiction and sensitivity to G6PD inhibition in acute myeloid leukemia cells. Leukemia 2017, 31, 2326–2335. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; An, S. Role of p53 isoforms and aggregations in cancer. Medicine 2016, 95, e3993. [Google Scholar] [CrossRef]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef]
- Hammond, E.M.; Denko, N.C.; Dorie, M.J.; Abraham, R.T.; Giaccia, A.J. Hypoxia links ATR and p53 through replication arrest. Mol. Cell. Biol. 2002, 22, 1834–1843. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Sowter, H.; Ratcliffe, P.; Watson, P.; Greenberg, A.; Harris, A. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001, 61, 6669–6673. [Google Scholar] [PubMed]
- Kolobova, E.; Tuganova, A.; Boulatnikov, I.; Popov, K.M. Regulation of pyruvate dehydrogenase activity through phosphorylation at multiple sites. Biochem. J. 2001, 358, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Contractor, T.; Harris, C.R. p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer Res. 2012, 72, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Matoba, S.; Kang, J.G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. p53 regulates mitochondrial respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef] [Green Version]
- Stambolsky, P.; Weisz, L.; Shats, I.; Klein, Y.; Goldfinger, N.; Oren, M.; Rotter, V. Regulation of AIF expression by p53. Cell Death Differ. 2006, 13, 2140–2149. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Lin, M.; Wu, R.; Wang, X.; Yang, B.; Levine, A.J.; Hu, W.; Feng, Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc. Natl. Acad. Sci. USA 2011, 108, 16259–16264. [Google Scholar] [CrossRef] [Green Version]
- Torrano, V.; Valcarcel-Jimenez, L.; Cortazar, A.R.; Liu, X.; Urosevic, J.; Castillo-Martin, M.; Fernández-Ruiz, S.; Morciano, G.; Caro-Maldonado, A.; Guiu, M.; et al. The metabolic co-regulator PGC1α suppresses prostate cancer metastasis. Nat. Cell Biol. 2016, 18, 645–656. [Google Scholar] [CrossRef]
- Evans, M.J.; Scarpulla, R.C. NRF-1: A trans-activator of nuclear-encoded respiratory genes in animal cells. Genes Dev. 1990, 4, 1023–1034. [Google Scholar] [CrossRef]
- Chau, C.M.; Evans, M.J.; Scarpulla, R.C. Nuclear respiratory factor 1 activation sites in genes encoding the gamma-subunit of ATP synthase, eukaryotic initiation factor 2 alpha, and tyrosine aminotransferase. Specific interaction of purified NRF-1 with multiple target genes. J. Biol. Chem. 1992, 267, 6999–7006. [Google Scholar] [PubMed]
- Larsson, N.G.; Wang, J.; Wilhelmsson, H.; Oldfors, A.; Rustin, P.; Lewandoski, M.; Barsh, G.S.; Clayton, D.A. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 1998, 18, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.; Sun, S.; Bai, Y.; Chen, Y.; Chai, R.; Li, H. Reduced mtDNA copy number increases the sensitivity of tumor cells to chemotherapeutic drugs. Cell Death Dis. 2015, 6, e1710. [Google Scholar] [CrossRef] [PubMed]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- Wegrzyn, J.; Potla, R.; Chwae, Y.J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef]
- Ferber, E.C.; Peck, B.; Delpuech, O.; Bell, G.P.; East, P.; Schulze, A. FOXO3a regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression. Cell Death Differ. 2012, 19, 968–979. [Google Scholar] [CrossRef]
- Ambrus, A.M.; Islam, A.B.; Holmes, K.B.; Moon, N.S.; Lopez-Bigas, N.; Benevolenskaya, E.V.; Frolov, M.V. Loss of dE2F compromises mitochondrial function. Dev. Cell 2013, 27, 438–451. [Google Scholar] [CrossRef]
- Chen, J.Q.; Delannoy, M.; Cooke, C.; Yager, J.D. Mitochondrial localization of ER alpha and ER β in human MCF7 cells. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E1011–E1022. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhou, J.; Du, Y. Estrogen receptor alpha interacts with mitochondrial protein HADHB and affects β-oxidation activity. Mol. Cell. Proteom. 2012, 11. [Google Scholar] [CrossRef]
- Mattingly, K.A.; Ivanova, M.M.; Riggs, K.A.; Wickramasinghe, N.S.; Barch, M.J.; Klinge, C.M. Estradiol stimulates transcription of nuclear respiratory factor-1 and increases mitochondrial biogenesis. Mol. Endocrinol. 2008, 22, 609–622. [Google Scholar] [CrossRef]
- Wang, X.; Moraes, C.T. Increases in mitochondrial biogenesis impair carcinogenesis at multiple levels. Mol. Oncol. 2011, 5, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Wanka, C.; Steinbach, J.P.; Rieger, J. Tp53-induced glycolysis and apoptosis regulator (TIGAR) protects glioma cells from starvation-induced cell death by up-regulating respiration and improving cellular redox homeostasis. J. Biol. Chem. 2012, 287, 33436–33446. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wang, Y.; Zeller, K.I.; Potter, J.J.; Wonsey, D.R.; O’Donnell, K.A.; Kim, J.W.; Yustein, J.T.; Lee, L.A.; Dang, C.V. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol. Cell. Biol. 2005, 25, 6225–6234. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; De Berardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrish, F.; Noonan, J.; Perez-Olsen, C.; Gafken, P.R.; Fitzgibbon, M.; Kelleher, J.; VanGilst, M.; Hockenbery, D. Myc-dependent mitochondrial generation of acetyl-CoA contributes to fatty acid biosynthesis and histone acetylation during cell cycle entry. J. Biol. Chem. 2010, 285, 36267–36274. [Google Scholar] [CrossRef] [PubMed]
- Morrish, F.; Hockenbery, D. MYC and mitochondrial biogenesis. Cold Spring Harb. Perspect. Med. 2014, 4, a014225. [Google Scholar] [CrossRef]
- Peruzzo, P.; Comelli, M.; Di Giorgio, E.; Franforte, E.; Mavelli, I.; Brancolini, C. Transformation by different oncogenes relies on specific metabolic adaptations. Cell Cycle 2016, 15, 2656–2668. [Google Scholar] [CrossRef] [Green Version]
- Yao, C.H.; Wang, R.; Wang, Y.; Kung, C.P.; Weber, J.D.; Patti, G.J. Mitochondrial fusion supports increased oxidative phosphorylation during cell proliferation. Elife 2019, 8, e41351. [Google Scholar] [CrossRef]
- Yang, D.; Wang, M.T.; Tang, Y.; Chen, Y.; Jiang, H.; Jones, T.T.; Rao, K.; Brewer, G.J.; Singh, K.K.; Nie, D. Impairment of mitochondrial respiration in mouse fibroblasts by oncogenic H-RAS(Q61L). Cancer Biol. Ther. 2010, 9, 122–133. [Google Scholar] [CrossRef]
- Telang, S.; Lane, A.N.; Nelson, K.K.; Arumugam, S.; Chesney, J. The oncoprotein H-RasV12 increases mitochondrial metabolism. Mol. Cancer 2007, 6, 77. [Google Scholar] [CrossRef]
- Baracca, A.; Chiaradonna, F.; Sgarbi, G.; Solaini, G.; Alberghina, L.; Lenaz, G. Mitochondrial Complex I decrease is responsible for bioenergetic dysfunction in K-ras transformed cells. Biochim. Biophys. Acta 2010, 1797, 314–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Song, M.; Zeng, Z.L.; Zhu, C.F.; Lu, W.H.; Yang, J.; Ma, M.Z.; Huang, A.M.; Hu, Y.; Huang, P. Identification of NDUFAF1 in mediating K-Ras induced mitochondrial dysfunction by a proteomic screening approach. Oncotarget 2015, 6, 3947–3962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.A.; Robinson, T.J.; Liu, J.C.; Shrestha, M.; Voisin, V.; Ju, Y.; Chung, P.E.; Pellecchia, G.; Fell, V.L.; Bae, S.; et al. RB1 deficiency in triple-negative breast cancer induces mitochondrial protein translation. J. Clin. Investig. 2016, 126, 3739–3757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Guzzi Cassago, C.A.; Dias, M.M.; Pinheiro, M.P.; Pasquali, C.C.; Bastos, A.C.S.; Islam, Z.; Consonni, S.R.; de Oliveira, J.F.; Gomes, E.M.; Ascenção, C.F.R.; et al. Glutaminase affects the transcriptional activity of peroxisome proliferator-activated receptor γ (PPARγ) via direct interaction. Biochemistry 2018, 57, 6293–6307. [Google Scholar] [CrossRef] [PubMed]
- Goo, C.K.; Lim, H.Y.; Ho, Q.S.; Too, H.P.; Clement, M.V.; Wong, K.P. PTEN/Akt signaling controls mitochondrial respiratory capacity through 4E-BP1. PLoS ONE 2012, 7, e45806. [Google Scholar] [CrossRef] [PubMed]
- Comelli, M.; Pretis, I.; Buso, A.; Mavelli, I. Mitochondrial energy metabolism and signalling in human glioblastoma cell lines with different PTEN gene status. J. Bioenerg. Biomembr. 2018, 50, 33–52. [Google Scholar] [CrossRef]
- Lukey, M.J.; Greene, K.S.; Erickson, J.W.; Wilson, K.F.; Cerione, R.A. The oncogenic transcription factor c-Jun regulates glutaminase expression and sensitizes cells to glutaminase-targeted therapy. Nat. Commun. 2016, 7, 11321. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.W.; LoGrasso, P.V. Mitochondrial c-Jun N-terminal kinase (JNK) signaling initiates physiological changes resulting in amplification of reactive oxygen species generation. J. Biol. Chem. 2011, 286, 16052–16062. [Google Scholar] [CrossRef]
- Morita, M.; Gravel, S.P.; Chénard, V.; Sikström, K.; Zheng, L.; Alain, T.; Gandin, V.; Avizonis, D.; Arguello, M.; Zakaria, C.; et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013, 18, 698–711. [Google Scholar] [CrossRef]
- Cunningham, J.T.; Rodgers, J.T.; Arlow, D.H.; Vazquez, F.; Mootha, V.K.; Puigserver, P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 2007, 450, 736–740. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, F.; Sorrentino, C.; Ucar, D.A.; Peng, Y.; Matossian, M.; Wyczechowska, D.; Crabtree, J.; Zabaleta, J.; Morello, S.; Del Valle, L.; et al. Notch signaling regulates mitochondrial metabolism and NF-κB activity in triple-negative breast cancer cells via IKKα-dependent non-canonical pathways. Front. Oncol. 2018, 8, 575. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.B.; Cao, L.; Ma, Y.Q.; Chen, Q.; Liang, Y.; Yuang, F.L.; Li, X.L.; Cheng, N. TIGAR mediates the inhibitory role of hypoxia on ROS production and apoptosis in rat nucleus pulposus cells. Osteoarthr. Cartil. 2018, 26, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Zang, M.; Guo, W. AMPK as a metabolic tumor suppressor: Control of metabolism and cell growth. Future Oncol. 2010, 6, 457–470. [Google Scholar] [CrossRef]
- Won, K.Y.; Lim, S.J.; Kim, G.Y.; Kim, Y.W.; Han, S.A.; Song, J.Y.; Lee, D.K. Regulatory role of p53 in cancer metabolism via SCO2 and TIGAR in human breast cancer. Hum. Pathol. 2012, 43, 221–228. [Google Scholar] [CrossRef]
- Martínez-Redondo, V.; Pettersson, A.T.; Ruas, J.L. The hitchhiker’s guide to PGC-1α isoform structure and biological functions. Diabetologia 2015, 58, 1969–1977. [Google Scholar] [CrossRef] [PubMed]
- Tohme, S.; Yazdani, H.O.; Liu, Y.; Loughran, P.; van der Windt, D.J.; Huang, H.; Simmons, R.L.; Shiva, S.; Tai, S.; Tsung, A. Hypoxia mediates mitochondrial biogenesis in hepatocellular carcinoma to promote tumor growth through HMGB1 and TLR9 interaction. Hepatology 2017, 66, 182–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Summermatter, S.; Baum, O.; Santos, G.; Hoppeler, H.; Handschin, C. Peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) promotes skeletal muscle lipid refueling in vivo by activating de novo lipogenesis and the pentose phosphate pathway. J. Biol. Chem. 2010, 285, 32793–32800. [Google Scholar] [CrossRef]
- Jacobs, M.D.; Harrison, S.C. Structure of an IκBalpha/NF-κB complex. Cell 1998, 95, 749–758. [Google Scholar] [CrossRef]
- Zheng, C.; Yin, Q.; Wu, H. Structural studies of NF-κB signaling. Cell Res. 2011, 21, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Ravi, R.; Mookerjee, B.; van Hensbergen, Y.; Bedi, G.C.; Giordano, A.; El-Deiry, W.S.; Fuchs, E.J.; Bedi, A. p53-mediated repression of nuclear factor-κB RelA via the transcriptional integrator p300. Cancer Res. 1998, 58, 4531–4536. [Google Scholar] [PubMed]
- Chen, W.; Wang, X.; Bai, L.; Liang, X.; Zhuang, J.; Lin, Y. Blockage of NF-κB by IKKβ- or RelA-siRNA rather than the NF-κB super-suppressor IκB alpha mutant potentiates adriamycin-induced cytotoxicity in lung cancer cells. J. Cell. Biochem. 2008, 105, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. p53 regulates glucose metabolism through an IKK-NF-κB pathway and inhibits cell transformation. Nat. Cell Biol. 2008, 10, 611–618. [Google Scholar] [CrossRef]
- Ngo, H.B.; Lovely, G.A.; Phillips, R.; Chan, D.C. Distinct structural features of TFAM drive mitochondrial DNA packaging versus transcriptional activation. Nat. Commun. 2014, 5, 3077. [Google Scholar] [CrossRef]
- Bonawitz, N.D.; Clayton, D.A.; Shadel, G.S. Initiation and beyond: Multiple functions of the human mitochondrial transcription machinery. Mol. Cell 2006, 24, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Gutsaeva, D.R.; Carraway, M.S.; Suliman, H.B.; Demchenko, I.T.; Shitara, H.; Yonekawa, H.; Piantadosi, C.A. Transient hypoxia stimulates mitochondrial biogenesis in brain subcortex by a neuronal nitric oxide synthase-dependent mechanism. J. Neurosci. 2008, 28, 2015–2024. [Google Scholar] [CrossRef]
- Aigner, P.; Just, V.; Stoiber, D. STAT3 isoforms: Alternative fates in cancer? Cytokine 2019, 118, 27–34. [Google Scholar] [CrossRef]
- Levy, D.E.; Darnell, J.E., Jr. Stats: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef]
- Link, W.; Fernandez-Marcos, P.J. FOXO transcription factors at the interface of metabolism and cancer. Int. J. Cancer 2017, 141, 2379–2391. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Patil, S.; Chauhan, B.; Guo, S.; Powell, D.R.; Le, J.; Klotsas, A.; Matika, R.; Xiao, X.; Franks, R.; et al. FoxO1 regulates multiple metabolic pathways in the liver: Effects on gluconeogenic, glycolytic, and lipogenic gene expression. J. Biol. Chem. 2006, 281, 10105–10117. [Google Scholar] [CrossRef]
- Gross, D.N.; Wan, M.; Birnbaum, M.J. The role of FOXO in the regulation of metabolism. Curr. Diabetes Rep. 2009, 9, 208–214. [Google Scholar] [CrossRef]
- Adams, M.R.; Sears, R.; Nuckolls, F.; Leone, G.; Nevins, J.R. Complex transcriptional regulatory mechanisms control expression of the E2F3 locus. Mol. Cell. Biol. 2000, 20, 3633–3639. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Z.; Tsai, S.Y.; Leone, G. Emerging roles of E2Fs in cancer: An exit from cell cycle control. Nat. Rev. Cancer 2009, 9, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Fernández de Mattos, S.; Lam, E.W.; Tauler, A. An E2F-binding site mediates the activation of the proliferative isoform of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase by phosphatidylinositol 3-kinase. Biochem. J. 2002, 368, 283–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez-Panizo, A.; Perez, P.; Rojas, A.; Fuentes-Prior, P.; Estebanez-Perpiña, E. Non-canonical dimerization of the androgen receptor and other nuclear receptors: Implications for human disease. Endocr. Relat. Cancer 2019, 26, R479–R497. [Google Scholar] [CrossRef]
- Shih, H.M.; Liu, Z.; Towle, H.C. Two CACGTG motifs with proper spacing dictate the carbohydrate regulation of hepatic gene transcription. J. Biol. Chem. 1995, 270, 21991–21997. [Google Scholar] [CrossRef]
- Airley, R.E.; McHugh, P.; Evans, A.R.; Harris, B.; Winchester, L.; Buffa, F.M.; Al-Tameemi, W.; Leek, R.; Harris, A.L. Role of carbohydrate response element-binding protein (ChREBP) in generating an aerobic metabolic phenotype and in breast cancer progression. Br. J. Cancer 2014, 110, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Robinson, L.N.; Towle, H.C. ChREBP*Mlx is the principal mediator of glucose-induced gene expression in the liver. J. Biol. Chem. 2006, 281, 28721–28730. [Google Scholar] [CrossRef] [PubMed]
- Vita, M.; Henriksson, M. The Myc oncoprotein as a therapeutic target for human cancer. Semin. Cancer Biol. 2006, 16, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, H.; Endo, H.; Akashika, T.; Kato, K.; Inoue, M. Downregulation of c-MYC protein levels contributes to cancer cell survival under dual deficiency of oxygen and glucose. Cancer Res. 2010, 70, 10213–10223. [Google Scholar] [CrossRef] [PubMed]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [Green Version]
- Zeng, M.; Kikuchi, H.; Pino, M.S.; Chung, D.C. Hypoxia activates the K-ras proto-oncogene to stimulate angiogenesis and inhibit apoptosis in colon cancer cells. PLoS ONE 2010, 5, e10966. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.; Tayyari, F.; Gowda, G.A.; Raftery, D.; McLamore, E.S.; Porterfield, D.M.; Donkin, S.S.; Bequette, B.; Teegarden, D. Altered glucose metabolism in Harvey-ras transformed MCF10A cells. Mol. Carcinog. 2015, 54, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Won, H.S.; Lee, Y.M.; Choi, J.W.; Oh, T.I.; Jang, J.H.; Choi, D.K.; Lim, B.O.; Kim, Y.J.; Park, J.W.; et al. Oxidative dimerization of PHD2 is responsible for its inactivation and contributes to metabolic reprogramming via HIF-1α activation. Sci. Rep. 2016, 6, 18928. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Lu, W.; Chen, G.; Wang, P.; Chen, Z.; Zhou, Y.; Ogasawara, M.; Trachootham, D.; Feng, L.; Pelicano, H.; et al. K-rasG12V transformation leads to mitochondrial dysfunction and metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. 2012, 22, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Sheng, H.; Zhang, X.; Qi, Q.; Chan, C.B.; Li, L.; Shan, C.; Ye, K. Cellular energy stress induces AMPK-mediated regulation of glioblastoma cell proliferation by PIKE-A phosphorylation. Cell Death Dis. 2019, 10, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polager, S.; Ginsberg, D. p53 and E2f: Partners in life and death. Nat. Rev. 2009, 9, 738–748. [Google Scholar] [CrossRef]
- Marín-Hernández, A.; Gallardo-Pérez, J.C.; Rodríguez-Enríquez, S.; Encalada, R.; Moreno-Sánchez, R.; Saavedra, E. Modeling cancer glycolysis. Biochim. Biophys. Acta 2011, 1807, 755–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clem, B.F.; Chesney, J. Molecular pathways: Regulation of metabolism by RB. Clin. Cancer Res. 2012, 18, 6096–6100. [Google Scholar] [CrossRef]
- Lee, Y.R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Bazzichetto, C.; Conciatori, F.; Pallocca, M.; Falcone, I.; Fanciulli, M.; Cognetti, F.; Milella, M.; Ciuffreda, L. PTEN as a Prognostic/Predictive Biomarker in Cancer: An Unfulfilled Promise? Cancers 2019, 11, 435. [Google Scholar] [CrossRef] [PubMed]
- Correia, N.C.; Gírio, A.; Antunes, I.; Martins, L.R.; Barata, J.T. The multiple layers of non-genetic regulation of PTEN tumour suppressor activity. Eur. J. Cancer 2014, 50, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Chen, J.; He, L.; Stiles, B.L. PTEN: Tumor suppressor and metabolic regulator. Front. Endocrinol. 2018, 9, 338. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Cao, I.; Song, M.S.; Hobbs, R.M.; Laurent, G.; Giorgi, C.; de Boer, V.; Anastasiou, D.; Ito, K.; Sasaki, A.T.; Rameh, L.; et al. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell 2012, 149, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, V.; Ram, P.T. Network motifs in JNK signaling. Genes Cancer 2013, 4, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Choy, P.M.; Bubici, C. The ERK and JNK pathways in the regulation of metabolic reprogramming. Oncogene 2019, 38, 2223–2240. [Google Scholar] [CrossRef]
- Razquin Navas, P.; Thedieck, K. Differential control of ageing and lifespan by isoforms and splice variants across the mTOR network. Essays Biochem. 2017, 61, 349–368. [Google Scholar] [CrossRef]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [Green Version]
- Menon, S.; Manning, B.D. Common corruption of the mTOR signaling network in human tumors. Oncogene 2008, 27, S43-51. [Google Scholar] [CrossRef]
- Moreno-Sánchez, R.; Saavedra, E.; Gallardo-Pérez, J.C.; Rumjanek, F.D.; Rodríguez-Enríquez, S. Understanding the cancer cell phenotype beyond the limitations of current omics analyses. FEBS J. 2016, 283, 54–73. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Falco, M.M.; Bleda, M.; Carbonell-Caballero, J.; Dopazo, J. The pan-cancer pathological regulatory landscape. Sci. Rep. 2016, 6, 39709. [Google Scholar] [CrossRef] [PubMed]
- Bhawe, K.; Roy, D. Interplay between NRF1, E2F4 and MYC transcription factors regulating common target genes contributes to cancer development and progression. Cell. Oncol. 2018, 41, 465–484. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Cheng, H.; Mu, C.; Geng, G.; Zhao, T.; Luo, Q.; Ma, K.; Chang, R.; Liu, Q.; Gao, R.; et al. The SIAH2-NRF1 axis spatially regulates tumor microenvironment remodeling for tumor progression. Nat. Commun. 2019, 10, 1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakkar, N.; Ladner, K.; Canan, B.D.; Liyanarachchi, S.; Bal, N.C.; Pant, M.; Periasamy, M.; Li, Q.; Janssen, P.M.; Guttridge, D.C. IKKα and alternative NF-κB regulate PGC-1β to promote oxidative muscle metabolism. J. Cell Biol. 2012, 196, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Tammineni, P.; Anugula, C.; Mohammed, F.; Anjaneyulu, M.; Larner, A.C.; Sepuri, N.B. The import of the transcription factor STAT3 into mitochondria depends on GRIM-19, a component of the electron transport chain. J. Biol. Chem. 2013, 288, 4723–4732. [Google Scholar] [CrossRef] [PubMed]
- Szczepanek, K.; Chen, Q.; Derecka, M.; Salloum, F.N.; Zhang, Q.; Szelag, M.; Cichy, J.; Kukreja, R.C.; Dulak, J.; Lesnefsky, E.J.; et al. Mitochondrial-targeted signal transducer and activator of transcription 3 (STAT3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species. J. Biol. Chem. 2011, 286, 29610–29620. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.; Brunet, A.; Grenier, J.M.; Datta, S.R.; Fornace, A.J., Jr.; DiStefano, P.S.; Chiang, L.W.; Greenberg, M.E. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science 2002, 296, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Yaşar, P.; Ayaz, G.; User, S.D.; Güpür, G.; Muyan, M. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod. Med. Biol. 2016, 16, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Abbott, B.D.; Wood, C.R.; Watkins, A.M.; Das, K.P.; Lau, C.S. Peroxisome proliferator-activated receptors alpha, Β, and gamma mRNA and protein expression in human fetal tissues. PPAR Res. 2010, 2010, 690907. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Evans, R. PPARs and ERRs: Molecular mediators of mitochondrial metabolism. Curr. Opin. Cell Biol. 2015, 33, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Hagland, H.R.; Nilsson, L.I.; Burri, L.; Nikolaisen, J.; Berge, R.K.; Tronstad, K.J. Induction of mitochondrial biogenesis and respiration is associated with mTOR regulation in hepatocytes of rats treated with the pan-PPAR activator tetradecylthioacetic acid (TTA). Biochem. Biophys. Res. Commun. 2013, 430, 573–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Gao, P.; Fukuda, R.; Kumar, G.; Krishnamachary, B.; Zeller, K.I.; Dang, C.V.; Semenza, G.L. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 2007, 11, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Castellano, E.; Santos, E. Functional specificity of ras isoforms: So similar but so different. Genes Cancer 2011, 2, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, M.R.; Lane, A.N.; Robertson, B.; Kemp, S.; Liu, Y.; Hill, B.G.; Dean, D.C.; Clem, B.F. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene 2014, 33, 556–566. [Google Scholar] [CrossRef]
- Tracy, K.; Dibling, B.C.; Spike, B.T.; Knabb, J.R.; Schumacker, P.; Macleod, K.F. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol. Cell. Biol. 2007, 27, 6229–6242. [Google Scholar] [CrossRef]
- Park, J.H.; Ko, J.; Park, Y.S.; Park, J.; Hwang, J.; Koh, H.C. Clearance of damaged mitochondria through PINK1 stabilization by JNK and ERK MAPK signaling in chlorpyrifos-treated neuroblastoma cells. Mol. Neurobiol. 2017, 54, 1844–1857. [Google Scholar] [CrossRef]
- Wisdom, R.; Johnson, R.S.; Moore, C. c-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J. 1999, 18, 188–197. [Google Scholar] [CrossRef]
- Allenspach, E.J.; Maillard, I.; Aster, J.C.; Pear, W.S. Notch signaling in cancer. Cancer Biol. Ther. 2002, 1, 466–476. [Google Scholar] [CrossRef]
- Sherr, C.J.; McCormick, F. The RB and p53 pathways in cancer. Cancer Cell 2002, 2, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.; Yao, J.; Zhuang, L.; Wang, D.; Han, J.; Lam, E.W.; Gan, B. The FoxO-BNIP3 axis exerts a unique regulation of mTORC1 and cell survival under energy stress. Oncogene 2014, 33, 3183–3194. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Sánchez, R.; Marín-Hernández, A.; Saavedra, E.; Pardo, J.P.; Ralph, S.J.; Rodríguez-Enríquez, S. Who controls the ATP supply in cancer cells? Biochemistry lessons to understand cancer energy metabolism. Int. J. Biochem. Cell Biol. 2014, 50, 10–23. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Transcription Regulator (Protein) | Intracellular Localization | Canonical Cellular Process Target | Refs. |
|---|---|---|---|
| Transcription Factors | |||
| HIF-1α | Nuclei | Angiogenesis, erythropoiesis, cellular proliferation, survival, vascular remodeling, tumorigenesis, invasion, metastasis | [16] |
| p53 | Nuclei | Cell cycle inhibition, apoptosis onset, antioxidant response, DNA damage repair systems, senescence, mitophagy | [17] |
| PGC-1α | Nuclei | Mitochondrial biogenesis and oxidative metabolism | [18] |
| NRF-1 | Nuclei | Expression of nuclear genes required for mitochondrial metabolism | [19] |
| NF-κB | Nuclei | Immune response, proliferation, apoptosis and angiogenesis suppression, metastasis | [20] |
| TFAM | Mitochondria | Cell cycle regulator, metastasis progression | [21] |
| STAT3 | Nuclei | Inhibition of immune activation against tumor cells, cancer progression | [22,23,24] |
| FOXO | Nuclei | Regulator of cell proliferation, apoptosis, invasion, metastasis | [25] |
| E2F | Nuclei | Cell proliferation, angiogenesis | [26] |
| ChREBP | Nuclei | Regulator of glucose metabolism and lipogenesis | [27] |
| AR | Nuclei | Regulator of development and function of male reproductive system and male phenotype | [28] |
| ER | Nuclei | Regulator of development and function of female reproductive system and female phenotype | [29] |
| PPARs | Nuclei | Regulator of lipid metabolism | [30] |
| p53-Induced Phosphatase | |||
| TIGAR | Cytosol | Cancer chemoresistance | [31,32] |
| Oncogenes | |||
| c-MYC | Nuclei Cytosol | Cell cycle regulation, apoptosis, cellular transformation | [33] |
| HRAS and KRAS | Metastasis and aggressive phenotype | [34] | |
| Tumor Suppressor | |||
| PTEN | Nuclei and cytosol | PI3K/AKT pathway blocking | [35] |
| Protein Kinases | |||
| JNK | Cytosol | Cell proliferation, differentiation, development, inflammatory response, apoptosis, malignancy, tumorigenesis | [36] |
| mTOR | Cytosol | Energy metabolism reprogramming, nutrient sensor | [37] |
| Plasma Membrane Receptors | |||
| Notch1 | Nuclei | Regulator of gene expression | [38] |
| Transcription Regulator (Protein) | Cancer Cell | Target | Measured Parameter | Variation | Refs. |
|---|---|---|---|---|---|
| Transcription Factor | |||||
| HIF-1α | Human U87 glioma | GLUT3, ALDO-A | mRNA content | Up ~2 times | [47] |
| HIF-1α (Hypoxia) | Human cervix HeLa; human liver HepG3B; human lung A549; human breast MCF-7 and MDA-MB-231; human colon LS174 and BE; human renal clear cell RCCA carcinomas; human glioma U87; mouse HepaC1 and HepaC4 hepatomas | GLUT1, HKI, HKII, PFK-L, ALDO-A, TPI, GAPDH, PGK1, ENO, PYK-M2, LDH-A, MCT4, GS, PGM | mRNA content | Up ~1.1–30 times | [5,39,40,41,42,43,48,49] |
| Protein content | Up ~2–10 times | ||||
| Glycolysis flux | Up ~3–6 times | ||||
| Glycogen content | Up ~1.7–26 times | ||||
| HIF-2α (Hypoxia) | human breast cancer MDA-MB-231, MDA-MB-468 | PGK1, PGM-1, PYKM, LDH- | mRNA content | Unchanged | [44] |
| Human renal 786-0 carcinoma | GLUT1 | mRNA content | Up ~ 2 times | [45] | |
| Protein content | Up ~ 2 times | ||||
| p53 (Normoxia) | Human Saos-2 sarcoma, human cervix HeLa carcinoma | GLUT1, GLUT3, GLUT4 | Protein content | Down 40–70% | [50,51] |
| Glycolytic flux | Unchanged | ||||
| Mutant p53 R248Q cervix HeLa carcinoma | GLUT1, GLUT3, HKI, HKII | Protein content | Up ~2–3 times | [50] | |
| Glycolysis flux | Up ~2 times | ||||
| p53 (Hypoxia) | Human cervix HeLa carcinoma | GLUT1, GLUT3 | Protein content | Up ~1–2.5 times | [50] |
| Glycolytic flux | Down ~30% | ||||
| Mutant p53 R175H, R248Q and R273H human cervix HeLa carcinoma; human lung H1299 carcinoma | GLUT1, GLUT3, HKI, HKII | Protein content | Up ~1–2 times | [52,53] | |
| Glycolysis flux, ECAR | Up ~1.5–2 times | ||||
| TFAM (Decrement by 30–70%) | Human lung A549 and H460 carcinomas | Glycolytic flux | Down 30–70% | [54] | |
| STAT3 | Human liver HepG2 and Hep3B carcinomas; human HCV virus-related hepatocarcinoma | GLUT1, HKII | mRNA content | Up ~1.4 times | [55,56,57] |
| Protein content | Up ~1.3 times | ||||
| Glucose consumption | Up ~1 time | ||||
| Lactate production | Up ~1.6 times | ||||
| E2F | Rat rhabdomyosarcoma | Fetal-type PFK-2/F2,6BPase | mRNA content | Not reported | [58] |
| AR | Human prostate LNCaP and LAPC4 carcinomas | HKII, PFK-P, ENO, PGK | mRNA content | Up ~1–3 times | [59,60] |
| ECAR | Up ~2.5–5 times | ||||
| ChREBP | Human hepatocarcinoma HepG2 | PYK-LR | mRNA content | Up ~2 times | [61] |
| ChREBP (SUPRESSION) | Human colon HCT116 carcinoma | Glucose uptake | Down ~50–60% Down ~60–70% | [62] | |
| Lactate production | |||||
| p53-Induced Phosphatase | |||||
| TIGAR (Normoxia) | Human bone U20S osteosarcoma | Fru-2,6-BP2 | Metabolite content | Down ~70–80% | [31] |
| TIGAR (Hypoxia) | Human ovarian A2780 and SW48 carcinomas | HKII | Activity | Up ~1.4 times | [63] |
| Oncogene | |||||
| c-MYC | Human Burkitt’s P493 lymphoma; mouse Eµ-Myc lymphoma | HKII, PFK-1, GAPDH, TPI, ENO, LDH-A, MCT1 | mRNA content | Up ~1–17 times | [64,65,66,67] |
| Protein content | Up ~2–3 times | ||||
| Glycolysis flux | Up ~1.5–3 times | ||||
| KRAS | Mutant KRASG13D human colon HTC116, DLD1 carcinomas | GLUT1, HKII | mRNA content | Up ~1.7–5 times | [68,69] |
| Protein content | Up ~3–5 times | ||||
| Glycolysis flux | Up ~2 times | ||||
| Tumor Suppressors | |||||
| Inactive RB | Human retinoblastoma biopsies | ALDO, LDH | Activity | Up ~1.2–10 times | [70] |
| HKII, HPI, TPI, GAPDH, ENO, PGK, PYK | Activity | Down ~20–80% | [71] | ||
| PTEN (Suppression) | Human prostate DU-145, 22Rv1 carcinomas | HKII | Protein content | Up ~1.5 times | [72] |
| Glucose consumption | Up ~1.2–1.4 times | ||||
| Lactate production | Up ~1.2 times | ||||
| PTEN (Overexpression) | Human ovarian carcinoma cells A2780 and SKOV-3 | Glucose consumption | Down 40–70% | [73] | |
| Protein Kinases | |||||
| JNK/PARP14 (Suppression) | Human liver Hep3B and Huh7 carcinomas | PYKM2 | Protein content | Up ~1–1.4 times | [74] |
| Activity | Up ~1.6 times | ||||
| mTOR | Human cervix HeLa carcinoma; human myeloid MOLM-14 leukemia | GLUT1, PFK1, Glc6PDH, R5PE | mRNA content | Up ~1–3 times | [75,76] |
| Glucose consumption | Up ~2 times | ||||
| Transcription Regulator (Protein) | Cancer Cell | Target | Measured Parameter | Variation | Refs. |
|---|---|---|---|---|---|
| Transcription Factors | |||||
| HIF-1α (Hypoxia) | Human Burkitt’s P493-6 lymphoma | PDK | mRNA content | Up 4times | [80] |
| Kidney RCC4, cervix HeLa, liver Hep3B, lung A495, colon HCT116 carcinomas | COX4-2 | mRNA content | Up 2 times | [81] | |
| Human Burkitt’s P493-6 lymphoma, renal clear cell RCCA, kidney RCC4, cervix HeLa, liver Hep3B, lung A495, kidney RCC4, colon HCT116, breast T47D, MDA-MB-468 and MDA-MB-231 carcinomas, fibrosarcoma HT1080 | BNIP3 | mRNA content | Up 1–5 times | [81,82,83] | |
| BNIP3 | Protein content | Up 0.5–3 times | |||
| COX4-1 | Protein content | Down 80% | |||
| PDH | Activity | Down 50% | |||
| Total oxygen consumption | Down 80% | ||||
| p53 (Normoxia) | Nonmutant p53 human breast MCF-7, human colon HCT116 carcinomas | PDK2 | Protein content | Down 75% | [84] |
| Nonmutant p53 human liver HepG2; human colon HTC116 and H460, human cervix HeLa, human nonsmall-cell lung H1299, human large-cell lung H460 carcinomas | GA, SOC2c, ND1, COX4, 2OGDH, ATPS, AIF, Parkin | mRNA content | Up 3–12 times | [50,85,86,87,88] | |
| Protein content | Up two times | ||||
| Total oxygen consumption | Up 0.5-fold | ||||
| Δψm and OxPhos | Up 3 times | ||||
| Glutaminolysis | Up 6 times | ||||
| p53 (Hypoxia) | Nonmutant cervix human carcinoma (HeLa) | COX4, 2OGDH and ATPS | Protein content | Down 40–90% | [52] |
| Δψm and OxPhos flux | Down 75–85% | ||||
| p53 (Normoxia/hypoxia) | Mutant p53 R248Q human cervix HeLa carcinoma | 2OGDH, GA, ND1, COX4, ATPS | Protein content | Down 10–50% | [52] |
| Total oxygen consumption, Δψm and OxPhos flux | Down 50% | ||||
| PGC1-α | Human prostate PC3 carcinoma overexpressing PGC1α | SDH, IDH3, AAT | mRNA content | Up 5 times | [89] |
| OAA, fum, mal, ATP | Metabolite content | Up 1–2 times | |||
| β-oxidation and OxPhos fluxes | Up 0.5–2.2-fold | ||||
| NRF-1 | Human cervix HeLa cancer | COX, ND1; SDH; bc-1 complex; ATPS | Gene promoter activity * | Up 2–10 times | [90,91] |
| Human breast MDA-MB-231 knockdown NRF-1 carcinoma | ATP | Metabolite content Total oxygen consumption | Down 40% 20% | [19] | |
| TFAM (Knockdown) | Human liver Hep-2, human lung A549, human laryngeal HNE2 carcinomas | mtDNA content | Down 60% | [54,92,93] | |
| Total oxygen consumption | Down 40% | ||||
| STAT3 | Human bladder T24 carcinoma | ND1, SDH | Activity OxPhos flux | Up 40–80% Up 70% | [94,95] |
| FOXO3a | Human colon DLD-1 carcinoma | Cyt c, ND1, FH | mRNA content Total oxygen consumption | Down 40% Down 75% | [96] |
| Human colon DLD-1 carcinoma | PDK4 | mRNA content | Up 1.5 times | ||
| E2F | Human sarcoma Saos2 | ND1, ACO, FH | mRNA content | Up 4 times | [97] |
| AR | Human prostate LNCaP and LAPC4 carcinoma | 2OGDH, FH, ND1, ATPS | mRNA content OxPhos | Up > 2 times Up 2 times | [59,60] |
| ER | Human breast MCF7; human lung H1793 carcinoma; ERα-transformed MDA-MB-231 | TNRF, COX | mRNA content | Up 0.8–5 times | [98,99,100] |
| mtDNA content | Up 0.6-fold | ||||
| HACoADH activity | Activity | Up 0.5 times | |||
| Total oxygen consumption | Up 0.8-fold | ||||
| PPARs | Human cervix HeLa; human osteosarcoma 143B; human breast MDA-MB-231 exposed to PPAR pan-agonist | CS, CPT-I and CPTII, COX | Activity | Up 0.1–1-fold | [101] |
| Cellular ATP, Δψm | Up 10–40% | ||||
| p53-Induced Phosphatase | |||||
| TIGAR | Human glioma T98G and LNT-299 overexpressing TIGAR | Total oxygen consumption and OxPhos | Up 10–50% | [102] | |
| Oncogenes | |||||
| c-MYC | Human Burkitt’s P493 lymphoma, glioma SF188, human-transformed CRL-2091 fibroblasts; rat TGR1-transformed fibroblasts | Mitochondrial biogenesis proteins; mitochondrial stability proteins; COX4, PDH, FH, ATPS, GA, glutamine transporters SLC38A5 and SLC1A5 | mRNA content | Up 2–5 times | [103,104,105,106] |
| GA | Activity | Up 2.5-fold | |||
| Acetyl-CoA | Metabolite content | Up 1.6-fold | |||
| HRAS | No mutant HRAS-transformed mouse 3T3 fibroblasts | SDH, COX4 | Protein content | Up 0.2–0.4-fold | [107,108,109] |
| OxPhos flux | Up 0.5-fold | ||||
| Mutant HRAS G12V and Q61L mouse 3T3 fibroblasts | OxPhos flux Oxygen consumption | Down 30–50% | |||
| Mutant HRAS G12V-transformed human bronchial epithelial NHBE cells | OxPhos flux | Up 0.5-fold | [110] | ||
| KRAS | Nonmutant KRAS-transformed mouse 3T3 fibroblasts | ND1 | Activity | Down 20% | [111] |
| Oxygen consumption | Down 30% | ||||
| Mutant KRAS G12V-transformed human embryonic 239 kidney | ND1 | Protein content | Down 25% | [112] | |
| Oxygen consumption | Down 60% | ||||
| Tumor Suppressor | |||||
| RB inactivation | Human breast MDA-MB-231 carcinoma | ATP | Metabolite content | Up 25 % | [113] |
| RB | Human osteosarcoma Saos2 and U2OS | BNIP3 | Protein content | Down 50–70% | [114] |
| PTEN | Human glioma SF767, A172, and U87MG | ND1, SDH, bc1 complex, COX, ATPS | Protein content OxPhos flux | Up 30–80% Up 40% | [115,116] |
| Protein Kinase | |||||
| JNK | Human breast BT-549, MDA-MB-231 carcinoma | GA | mRNA content | Up 2 times | [117,118] |
| Human cervix Hela carcinoma | ND1 | Activity Oxygen consumption and OxPhos | Down 30% Down 20% | ||
| mTOR | Human breast MCF7 carcinoma | ND1, IDH3, COX, ATPS | mRNA content | Up 0.6–2 times | [119,120] |
| Human kidney HEK-293 carcinoma | ATG13 | Protein content | Down 70% | [121] | |
| Plasma Membrane Receptors | |||||
| Notch1 blocking | Human breast MDA-MB-231 carcinoma | Total oxygen consumption | Down 50% | [122] | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Enríquez, S.; Marín-Hernández, Á.; Gallardo-Pérez, J.C.; Pacheco-Velázquez, S.C.; Belmont-Díaz, J.A.; Robledo-Cadena, D.X.; Vargas-Navarro, J.L.; Corona de la Peña, N.A.; Saavedra, E.; Moreno-Sánchez, R. Transcriptional Regulation of Energy Metabolism in Cancer Cells. Cells 2019, 8, 1225. https://doi.org/10.3390/cells8101225
Rodríguez-Enríquez S, Marín-Hernández Á, Gallardo-Pérez JC, Pacheco-Velázquez SC, Belmont-Díaz JA, Robledo-Cadena DX, Vargas-Navarro JL, Corona de la Peña NA, Saavedra E, Moreno-Sánchez R. Transcriptional Regulation of Energy Metabolism in Cancer Cells. Cells. 2019; 8(10):1225. https://doi.org/10.3390/cells8101225
Chicago/Turabian StyleRodríguez-Enríquez, Sara, Álvaro Marín-Hernández, Juan Carlos Gallardo-Pérez, Silvia Cecilia Pacheco-Velázquez, Javier Alejandro Belmont-Díaz, Diana Xochiquetzal Robledo-Cadena, Jorge Luis Vargas-Navarro, Norma Angélica Corona de la Peña, Emma Saavedra, and Rafael Moreno-Sánchez. 2019. "Transcriptional Regulation of Energy Metabolism in Cancer Cells" Cells 8, no. 10: 1225. https://doi.org/10.3390/cells8101225