From OPC to Oligodendrocyte: An Epigenetic Journey


 and
and {kind=link}
Abstract
:1. Introduction
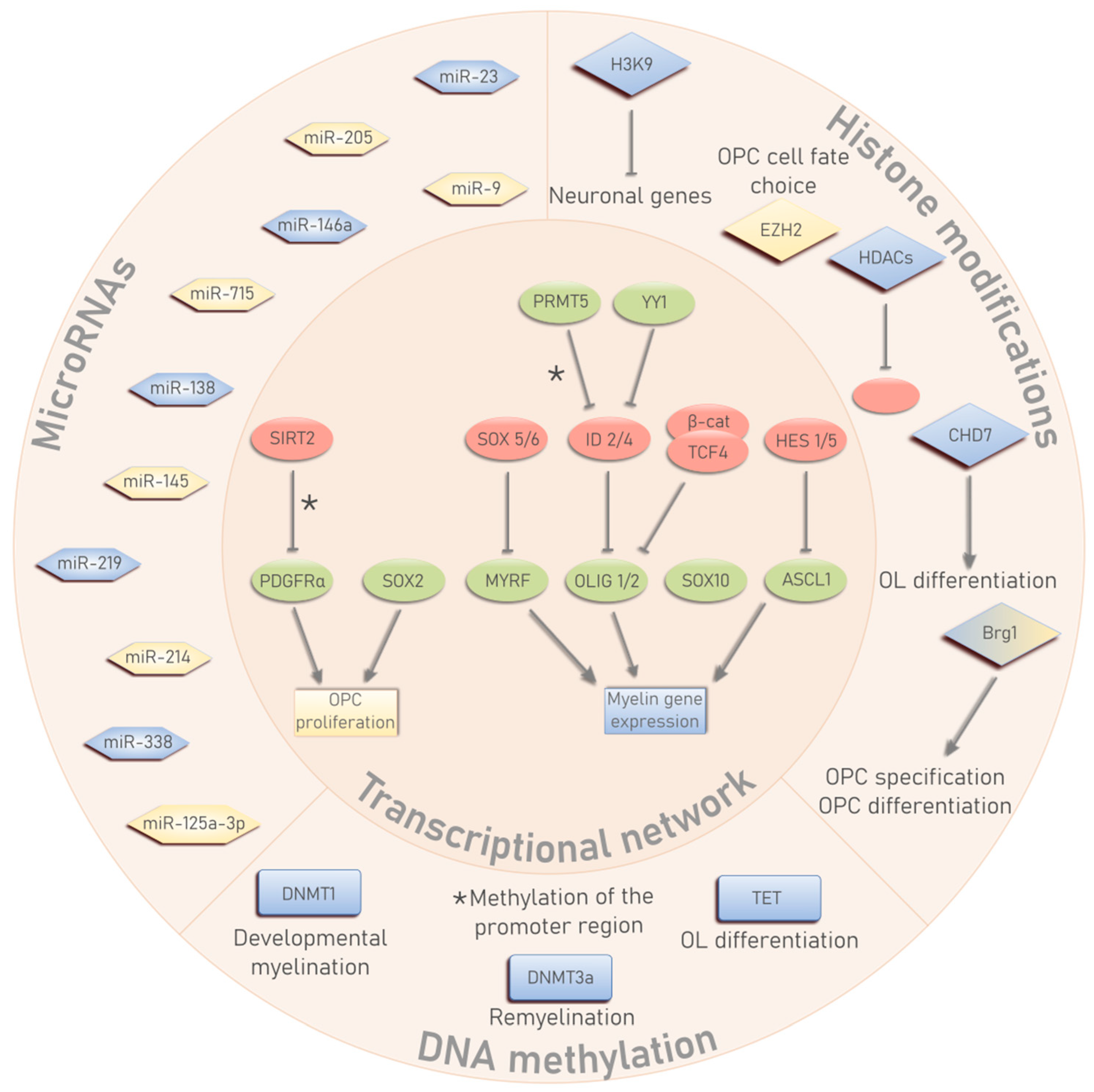
2. OL Differentiation and the Transcriptional Network
3. The Epigenetic Triumvirate in OL Development
3.1. DNA Methylation
3.2. Histone Modifications
3.3. MicroRNAs
4. Implications in Ageing and CNS Myelin Disorders
4.1. Ageing
4.2. Multiple Sclerosis
4.3. Other Diseases with Myelopathy
5. Therapeutic Perspectives: From Pharmaceuticals to (epi) Gene Therapy to IPSCs
6. Concluding Remarks
Funding
Conflicts of Interest
References
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and pathology. Acta. Neuropathol. 2010, 119, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.R.; Polito, A.; Levine, J.M.; Reynolds, R. NG2-expressing glial progenitor cells: An abundant and widespread population of cycling cells in the adult rat CNS. Mol. Cell. Neurosci. 2003, 24, 476–488. [Google Scholar] [CrossRef]
- Fernandez, C.A.; Gaultier, A. Adult oligodendrocyte progenitor cells—Multifaceted regulators of the CNS in health and disease. Brain Behav. Immun. 2016, 57, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bergles, D.E.; Richardson, W.D. Oligodendrocyte Development and Plasticity. Cold Spring Harb. Perspect. Biol. 2015, 8, 020453. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, N.A.; Fuss, B. Extracellular cues influencing oligodendrocyte differentiation and (re)myelination. Exp. Neurol. 2016, 283, 512–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, M.W.; Metz, L.M.; Kovalchuk, O. Epigenetic changes in patients with multiple sclerosis. Nat. Rev. Neurol. 2013, 9, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Moyon, S.; Hernandez, M.; Casaccia, P. Epigenetic control of oligodendrocyte development: Adding new players to old keepers. Curr. Opin. Neurobiol. 2016, 39, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, O.; Alvarez-Buylla, A. Oligodendrogenesis in the subventricular zone and the role of epidermal growth factor. Brain Res. Rev. 2011, 67, 147–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbaz, B.; Popko, B. Molecular Control of Oligodendrocyte Development. Trends Neurosci. 2019, 42, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Armada-Moreira, A.; Ribeiro, F.F.; Sebastião, A.M.; Xapelli, S. Neuroinflammatory modulators of oligodendrogenesis. Neuroimmunol. Neuroinflammation 2015, 2, 263–273. [Google Scholar] [Green Version]
- Sock, E.; Wegner, M. Transcriptional control of myelination and remyelination. Trends cell boil. 2011, 21, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; He, Y.; Richardson, W.D.; Casaccia, P. Two-tier transcriptional control of oligodendrocyte differentiation. Curr. Opin. Neurobiol. 2009, 19, 479–485. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Zuo, H.; Maher, B.J.; Serwanski, D.R.; LoTurco, J.J.; Lu, Q.R.; Nishiyama, A. Olig2-dependent developmental fate switch of NG2 cells. Dev. Camb. Engl. 2012, 139, 2299–2307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegener, A.; Deboux, C.; Bachelin, C.; Frah, M.; Kerninon, C.; Seilhean, D.; Weider, M.; Wegner, M.; Nait-Oumesmar, B. Gain of Olig2 function in oligodendrocyte progenitors promotes remyelination. Brain A J. Neurol. 2015, 138, 120–135. [Google Scholar] [CrossRef] [PubMed]
- Maire, C.L.; Wegener, A.; Kerninon, C.; Nait Oumesmar, B. Gain-of-Function of Olig Transcription Factors Enhances Oligodendrogenesis and Myelination. Stem Cells 2010, 28, 1611–1622. [Google Scholar] [CrossRef]
- Arnett, H.A.; Fancy, S.P.; Alberta, J.A.; Zhao, C.; Plant, S.R.; Kaing, S.; Raine, C.S.; Rowitch, D.H.; Franklin, R.J.; Stiles, C.D. bHLH transcription factor Olig1 is required to repair demyelinated lesions in the CNS. Science 2004, 306, 2111–2115. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Bercury, K.K.; Ahrendsen, J.T.; Macklin, W.B. Olig1 function is required for oligodendrocyte differentiation in the mouse brain. J. Neurosci. 2015, 35, 4386–4402. [Google Scholar] [CrossRef]
- Sugimori, M.; Nagao, M.; Parras, C.M.; Nakatani, H.; Lebel, M.; Guillemot, F.; Nakafuku, M. Ascl1 is required for oligodendrocyte development in the spinal cord. Dev. Camb. Engl. 2008, 135, 1271–1281. [Google Scholar] [CrossRef] [Green Version]
- Nakatani, H.; Martin, E.; Hassani, H.; Clavairoly, A.; Maire, C.L.; Viadieu, A.; Kerninon, C.; Delmasure, A.; Frah, M.; Weber, M.; et al. Ascl1/Mash1 Promotes Brain Oligodendrogenesis during Myelination and Remyelination. J. Neurosci. 2013, 33, 9752–9768. [Google Scholar] [CrossRef] [Green Version]
- Samanta, J.; Kessler, J.A. Interactions between ID and OLIG proteins mediate the inhibitory effects of BMP4 on oligodendroglial differentiation. Dev. Camb. Engl. 2004, 131, 4131–4142. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Sdrulla, A.; Johnson, J.E.; Yokota, Y.; Barres, B.A. A role for the helix-loop-helix protein Id2 in the control of oligodendrocyte development. Neuron 2001, 29, 603–614. [Google Scholar] [CrossRef]
- Turnescu, T.; Arter, J.; Reiprich, S.; Tamm, E.R.; Waisman, A.; Wegner, M. Sox8 and Sox10 jointly maintain myelin gene expression in oligodendrocytes. Glia 2018, 66, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Hornig, J.; Frob, F.; Vogl, M.R.; Hermans-Borgmeyer, I.; Tamm, E.R.; Wegner, M. The transcription factors Sox10 and Myrf define an essential regulatory network module in differentiating oligodendrocytes. PLoS Genet. 2013, 9, 1003907. [Google Scholar] [CrossRef] [PubMed]
- Stolt, C.C.; Rehberg, S.; Ader, M.; Lommes, P.; Riethmacher, D.; Schachner, M.; Bartsch, U.; Wegner, M. Terminal differentiation of myelin-forming oligodendrocytes depends on the transcription factor Sox10. Genes Dev. 2002, 16, 165–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stolt, C.C.; Schlierf, A.; Lommes, P.; Hillgartner, S.; Werner, T.; Kosian, T.; Sock, E.; Kessaris, N.; Richardson, W.D.; Lefebvre, V.; et al. SoxD proteins influence multiple stages of oligodendrocyte development and modulate SoxE protein function. Dev. Cell 2006, 11, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Ma, D.; Zawadzka, M.; Fancy, S.P.; Elis-Williams, L.; Bouvier, G.; Stockley, J.H.; de Castro, G.M.; Wang, B.; Jacobs, S.; et al. Sox2 Sustains Recruitment of Oligodendrocyte Progenitor Cells following CNS Demyelination and Primes Them for Differentiation during Remyelination. J. Neurosci. Neurosci. 2015, 35, 11482–11499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Zhu, X.; Gui, X.; Croteau, C.; Song, L.; Xu, J.; Wang, A.; Bannerman, P.; Guo, F. Sox2 Is Essential for Oligodendroglial Proliferation and Differentiation during Postnatal Brain Myelination and CNS Remyelination. J. Neurosci. 2018, 38, 1802–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, F.; Chen, Y.; Hoang, T.; Montgomery, R.L.; Zhao, X.H.; Bu, H.; Hu, T.; Taketo, M.M.; van Es, J.H.; Clevers, H.; et al. HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta-catenin-TCF interaction. Nat. Neurosci. 2009, 12, 829–838. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Dupree, J.; Wang, J.; Sandoval, J.; Li, J.; Liu, H.; Shi, Y.; Nave, K.A.; Casaccia-Bonnefil, P. The transcription factor Yin Yang 1 is essential for oligodendrocyte progenitor differentiation. Neuron 2007, 55, 217–230. [Google Scholar] [CrossRef]
- Howng, S.Y.; Avila, R.L.; Emery, B.; Traka, M.; Lin, W.; Watkins, T.; Cook, S.; Bronson, R.; Davisson, M.; Barres, B.A.; et al. ZFP191 is required by oligodendrocytes for CNS myelination. Genes Dev. 2010, 24, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.; Chung, S.H.; Jiang, P.; Dehghan, S.; Deng, W. Development of glial restricted human neural stem cells for oligodendrocyte differentiation in vitro and in vivo. Sci. Rep. 2019, 9, 9013. [Google Scholar] [CrossRef] [PubMed]
- Soundarapandian, M.M.; Selvaraj, V.; Lo, U.G.; Golub, M.S.; Feldman, D.H.; Pleasure, D.E.; Deng, W. Zfp488 promotes oligodendrocyte differentiation of neural progenitor cells in adult mice after demyelination. Sci. Rep. 2011, 1, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, Q.; Chen, Y.; Wang, H.; Xu, X.; Yang, B.; He, Q.; Shou, W.; Chen, Y.; Higashi, Y.; van den Berghe, V.; et al. Dual-mode modulation of Smad signaling by Smad-interacting protein Sip1 is required for myelination in the central nervous system. Neuron 2012, 73, 713–728. [Google Scholar] [CrossRef] [PubMed]
- Emery, B.; Agalliu, D.; Cahoy, J.D.; Watkins, T.A.; Dugas, J.C.; Mulinyawe, S.B.; Ibrahim, A.; Ligon, K.L.; Rowitch, D.H.; Barres, B.A. Myelin gene regulatory factor is a critical transcriptional regulator required for CNS myelination. Cell 2009, 138, 172–185. [Google Scholar] [CrossRef]
- Fulton, D.; Paez, P.M.; Campagnoni, A.T. The Multiple Roles of Myelin Protein Genes During the Development of the Oligodendrocyte. Asn. Neuro. 2010, 2, AN20090051. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, S.; Yurlova, L.; Simons, M. Central nervous system myelin: Structure, synthesis and assembly. Trends Cell Biol. 2011, 21, 585–593. [Google Scholar] [CrossRef]
- Emery, B.; Lu, Q.R. Transcriptional and Epigenetic Regulation of Oligodendrocyte Development and Myelination in the Central Nervous System. Cold Spring Harb. Perspect. Biol. 2015, 7, a020461. [Google Scholar] [CrossRef] [Green Version]
- Dulac, C. Brain function and chromatin plasticity. Nature 2010, 465, 728–735. [Google Scholar] [CrossRef] [Green Version]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487. [Google Scholar] [CrossRef]
- Copray, S.; Huynh, J.L.; Sher, F.; Casaccia-Bonnefil, P.; Boddeke, E. Epigenetic mechanisms facilitating oligodendrocyte development, maturation and aging. Glia 2009, 57, 1579–1587. [Google Scholar] [CrossRef]
- Illingworth, R.S.; Gruenewald-Schneider, U.; Webb, S.; Kerr, A.R.W.; James, K.D.; Turner, D.J.; Smith, C.; Harrison, D.J.; Andrews, R.; Bird, A.P. Orphan CpG Islands Identify Numerous Conserved Promoters in the Mammalian Genome. PLoS Genet. 2010, 6, e1001134. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.L.; Veenstra, G.J.; Wade, P.A.; Vermaak, D.; Kass, S.U.; Landsberger, N.; Strouboulis, J.; Wolffe, A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998, 19, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Fuks, F.; Hurd, P.J.; Wolf, D.; Nan, X.; Bird, A.P.; Kouzarides, T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J. Biol. Chem. 2003, 278, 4035–4040. [Google Scholar] [CrossRef] [PubMed]
- Day, J.J.; Kennedy, A.J.; Sweatt, J.D. DNA Methylation and Its Implications and Accessibility for Neuropsychiatric Therapeutics. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 591–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.X.; Riggs, A.D. DNA methylation and demethylation in mammals. J. Biol. Chem. 2011, 286, 18347–18353. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Lu, J.; Cheng, J.; Rao, Q.; Li, Z.; Hou, H.; Lou, Z.; Zhang, L.; Li, W.; Gong, W.; et al. Structural insight into substrate preference for TET-mediated oxidation. Nature 2015, 527, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-C.; Wang, K.-Y.; Shen, C.-K.J. The mammalian de novo DNA methyltransferases DNMT3A and DNMT3B are also DNA 5-hydroxymethylcytosine dehydroxymethylases. J. Biol. Chem. 2012, 287, 33116–33121. [Google Scholar] [CrossRef]
- Chen, C.C.; Wang, K.Y.; Shen, C.K. DNA 5-methylcytosine demethylation activities of the mammalian DNA methyltransferases. J. Biol. Chem. 2013, 288, 9084–9091. [Google Scholar] [CrossRef] [PubMed]
- Globisch, D.; Munzel, M.; Muller, M.; Michalakis, S.; Wagner, M.; Koch, S.; Bruckl, T.; Biel, M.; Carell, T. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS ONE 2010, 5, 15367. [Google Scholar] [CrossRef]
- Roubroeks, J.A.Y.; Smith, R.G.; van den Hove, D.L.A.; Lunnon, K. Epigenetics and DNA methylomic profiling in Alzheimer’s disease and other neurodegenerative diseases. J. Neurochem. 2017, 143, 158–170. [Google Scholar] [CrossRef]
- van den Hove, D.L.; Chouliaras, L.; Rutten, B.P. The role of 5-hydroxymethylcytosine in aging and Alzheimer’s disease: Current status and prospects for future studies. Curr. Alzheimer Res. 2012, 9, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Ransom, B.R.; Yamate, C.L.; Black, J.A.; Waxman, S.G. Rat optic nerve: Disruption of gliogenesis with 5-azacytidine during early postnatal development. Brain Res. 1985, 337, 41–49. [Google Scholar] [CrossRef]
- Moyon, S.; Huynh, J.L.; Dutta, D.; Zhang, F.; Ma, D.; Yoo, S.; Lawrence, R.; Wegner, M.; John, G.R.; Emery, B.; et al. Functional Characterization of DNA Methylation in the Oligodendrocyte Lineage. Cell Rep. 2016, 15, 748–760. [Google Scholar] [CrossRef] [Green Version]
- Moyon, S.; Ma, D.; Huynh, J.L.; Coutts, D.J.C.; Zhao, C.; Casaccia, P.; Franklin, R.J.M. Efficient Remyelination Requires DNA Methylation. eNeuro 2017, 4, ENEURO.0336–0316.2017. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Dai, J.; Ma, Y.; Mi, Y.; Cui, D.; Ju, G.; Macklin, W.B.; Jin, W. Dynamics of ten-eleven translocation hydroxylase family proteins and 5-hydroxymethylcytosine in oligodendrocyte differentiation. Glia 2014, 62, 914–926. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Vogel, G.; Yu, Z.; Almazan, G.; Richard, S. Type II arginine methyltransferase PRMT5 regulates gene expression of inhibitors of differentiation/DNA binding Id2 and Id4 during glial cell differentiation. J. Biol. Chem. 2011, 286, 44424–44432. [Google Scholar] [CrossRef] [PubMed]
- Fang, N.; Cheng, J.; Zhang, C.; Chen, K.; Zhang, C.; Hu, Z.; Bi, R.; Furber, K.L.; Thangaraj, M.; Nazarali, A.J.; et al. Sirt2 epigenetically down-regulates PDGFRalpha expression and promotes CG4 cell differentiation. Cell Cycle 2019, 18, 1095–1109. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381. [Google Scholar] [CrossRef]
- He, H.; Hu, Z.; Xiao, H.; Zhou, F.; Yang, B. The tale of histone modifications and its role in multiple sclerosis. Hum. Genom. 2018, 12, 31. [Google Scholar] [CrossRef]
- Marin-Husstege, M.; Muggironi, M.; Liu, A.; Casaccia-Bonnefil, P. Histone deacetylase activity is necessary for oligodendrocyte lineage progression. J. Neurosci. 2002, 22, 10333–10345. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Li, J.; Casaccia-Bonnefil, P. Histone modifications affect timing of oligodendrocyte progenitor differentiation in the developing rat brain. J. Cell Biol. 2005, 169, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Swiss, V.A.; Nguyen, T.; Dugas, J.; Ibrahim, A.; Barres, B.; Androulakis, I.P.; Casaccia, P. Identification of a gene regulatory network necessary for the initiation of oligodendrocyte differentiation. PLoS ONE 2011, 6, e18088. [Google Scholar] [CrossRef] [PubMed]
- Conway, G.D.; O’Bara, M.A.; Vedia, B.H.; Pol, S.U.; Sim, F.J. Histone deacetylase activity is required for human oligodendrocyte progenitor differentiation. Glia 2012, 60, 1944–1953. [Google Scholar] [CrossRef] [PubMed]
- Egawa, N.; Shindo, A.; Hikawa, R.; Kinoshita, H.; Liang, A.C.; Itoh, K.; Lok, J.; Maki, T.; Takahashi, R.; Lo, E.H.; et al. Differential roles of epigenetic regulators in the survival and differentiation of oligodendrocyte precursor cells. Glia 2019, 67, 718–728. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Sandoval, J.; Casaccia-Bonnefil, P. Events at the transition between cell cycle exit and oligodendrocyte progenitor differentiation: The role of HDAC and YY1. Neuron Glia Biol. 2007, 3, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Bercury, K.K.; Jin, W.; Macklin, W.B. Olig1 Acetylation and Nuclear Export Mediate Oligodendrocyte Development. J. Neurosci. 2015, 35, 15875–15893. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Magri, L.; Zhang, F.; Marsh, N.O.; Albrecht, S.; Huynh, J.L.; Kaur, J.; Kuhlmann, T.; Zhang, W.; Slesinger, P.A.; et al. Chromatin landscape defined by repressive histone methylation during oligodendrocyte differentiation. J. Neurosci. 2015, 35, 352–365. [Google Scholar] [CrossRef]
- Sher, F.; Rößler, R.; Brouwer, N.; Balasubramaniyan, V.; Boddeke, E.; Copray, S. Differentiation of Neural Stem Cells into Oligodendrocytes: Involvement of the Polycomb Group Protein Ezh2. Stem Cells 2008, 26, 2875–2883. [Google Scholar] [CrossRef]
- Koreman, E.; Sun, X.; Lu, Q.R. Chromatin remodeling and epigenetic regulation of oligodendrocyte myelination and myelin repair. Mol. Cell. Neurosci. 2018, 87, 18–26. [Google Scholar] [CrossRef]
- Scaglione, A.; Patzig, J.; Liang, J.; Frawley, R.; Bok, J.; Mela, A.; Yattah, C.; Zhang, J.; Teo, S.X.; Zhou, T.; et al. PRMT5-mediated regulation of developmental myelination. Nat. Commun. 2018, 9, 2840. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Murata, K.; Ishida, J.; Kanou, A.; Kasuya, Y.; Fukamizu, A. Severe Hypomyelination and Developmental Defects Are Caused in Mice Lacking Protein Arginine Methyltransferase 1 (PRMT1) in the Central Nervous System. J. Biol. Chem. 2016, 291, 2237–2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregath, A.; Lu, Q.R. Epigenetic modifications-insight into oligodendrocyte lineage progression, regeneration, and disease. FEBS Lett. 2018, 592, 1063–1078. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Banine, F.; Feistel, K.; Foster, S.; Xing, R.; Struve, J.; Sherman, L.S. Brg1 directly regulates Olig2 transcription and is required for oligodendrocyte progenitor cell specification. Dev. Biol. 2016, 413, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Chen, Y.; Kim, B.; Wang, H.; Zhao, C.; He, X.; Liu, L.; Liu, W.; Wu, L.M.; Mao, M.; et al. Olig2 targets chromatin remodelers to enhancers to initiate oligodendrocyte differentiation. Cell 2013, 152, 248–261. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Marie, C.; Zhao, C.; Kim, B.; Wang, J.; Deng, Y.; Clavairoly, A.; Frah, M.; Wang, H.; He, X.; et al. Chd7 cooperates with Sox10 and regulates the onset of CNS myelination and remyelination. Nat. Neurosci 2016, 19, 678–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabian, M.R.; Sundermeier, T.R.; Sonenberg, N. Understanding how miRNAs post-transcriptionally regulate gene expression. Prog. Mol. Subcell. Biol. 2010, 50, 1–20. [Google Scholar]
- Rodriguez, A.; Griffiths-Jones, S.; Ashurst, J.L.; Bradley, A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004, 14, 1902–1910. [Google Scholar] [CrossRef]
- Lewis, B.P.; Shih, I.H.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of mammalian microRNA targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef]
- Eulalio, A.; Huntzinger, E.; Nishihara, T.; Rehwinkel, J.; Fauser, M.; Izaurralde, E. Deadenylation is a widespread effect of miRNA regulation. RNA 2009, 15, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Sayed, D.; Abdellatif, M. MicroRNAs in development and disease. Physiol. Rev. 2011, 91, 827–887. [Google Scholar] [CrossRef] [PubMed]
- Barca-Mayo, O.; Lu, Q.R. Fine-Tuning Oligodendrocyte Development by microRNAs. Front. Neurosci. 2012, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzpatrick, J.M.; Anderson, R.C.; McDermott, K.W. MicroRNA: Key regulators of oligodendrocyte development and pathobiology. Int. J. Biochem. Cell Biol. 2015, 65, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Dugas, J.C.; Cuellar, T.L.; Scholze, A.; Ason, B.; Ibrahim, A.; Emery, B.; Zamanian, J.L.; Foo, L.C.; McManus, M.T.; Barres, B.A. Dicer1 and miR-219 Are required for normal oligodendrocyte differentiation and myelination. Neuron 2010, 65, 597–611. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Shin, J.Y.; McManus, M.T.; Ptacek, L.J.; Fu, Y.H. Dicer ablation in oligodendrocytes provokes neuronal impairment in mice. Ann. Neurol. 2009, 66, 843–857. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ren, C.; Qu, X.; Wu, X.; Dong, F.; Chand, Y.K.; Fan, H.; Yao, R.; Geng, D. miR-219 attenuates demyelination in cuprizone-induced demyelinated mice by regulating monocarboxylate transporter 1. Eur. J. Neurosci. 2017, 45, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Moyano, A.L.; Ma, Z.; Deng, Y.; Lin, Y.; Zhao, C.; Zhang, L.; Jiang, M.; He, X.; Ma, Z.; et al. miR-219 Cooperates with miR-338 in Myelination and Promotes Myelin Repair in the CNS. Dev. Cell 2017, 40, 566–582. [Google Scholar] [CrossRef] [PubMed]
- Dugas, J.C.; Notterpek, L. MicroRNAs in oligodendrocyte and Schwann cell differentiation. Dev. Neurosci. 2011, 33, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; He, X.; Han, X.; Yu, Y.; Ye, F.; Chen, Y.; Hoang, T.; Xu, X.; Mi, Q.S.; Xin, M.; et al. MicroRNA-mediated control of oligodendrocyte differentiation. Neuron 2010, 65, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Barough, S.; Massumi, M.; Kouchesfahani, H.M.; Ai, J. Derivation of pre-oligodendrocytes from human endometrial stromal cells by using overexpression of microRNA 338. J. Mol. Neurosci. Mn 2013, 51, 337–343. [Google Scholar] [CrossRef]
- Smirnova, L.; Grafe, A.; Seiler, A.; Schumacher, S.; Nitsch, R.; Wulczyn, F.G. Regulation of miRNA expression during neural cell specification. Eur. J. Neurosci. 2005, 21, 1469–1477. [Google Scholar] [CrossRef]
- Lau, P.; Verrier, J.D.; Nielsen, J.A.; Johnson, K.R.; Notterpek, L.; Hudson, L.D. Identification of dynamically regulated microRNA and mRNA networks in developing oligodendrocytes. J. Neurosci. 2008, 28, 11720–11730. [Google Scholar] [CrossRef]
- Buller, B.; Chopp, M.; Ueno, Y.; Zhang, L.; Zhang, R.L.; Morris, D.; Zhang, Y.; Zhang, Z.G. Regulation of serum response factor by miRNA-200 and miRNA-9 modulates oligodendrocyte progenitor cell differentiation. Glia 2012, 60, 1906–1914. [Google Scholar] [CrossRef] [Green Version]
- Lecca, D.; Marangon, D.; Coppolino, G.T.; Mendez, A.M.; Finardi, A.; Costa, G.D.; Martinelli, V.; Furlan, R.; Abbracchio, M.P. MiR-125a-3p timely inhibits oligodendroglial maturation and is pathologically up-regulated in human multiple sclerosis. Sci. Rep. 2016, 6, 34503. [Google Scholar] [CrossRef]
- Huang, B.; Luo, W.; Sun, L.; Zhang, Q.; Jiang, L.; Chang, J.; Qiu, X.; Wang, E. MiRNA-125a-3p is a negative regulator of the RhoA-actomyosin pathway in A549 cells. Int. J. Oncol. 2013, 42, 1734–1742. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Li, P.; Ni, Y.; Zhao, J.; Liu, Z. Decreased microRNA-125a-3p contributes to upregulation of p38 MAPK in rat trigeminal ganglions with orofacial inflammatory pain. PLoS ONe 2014, 9, e111594. [Google Scholar] [CrossRef]
- Yin, F.; Zhang, J.N.; Wang, S.W.; Zhou, C.H.; Zhao, M.M.; Fan, W.H.; Fan, M.; Liu, S. MiR-125a-3p regulates glioma apoptosis and invasion by regulating Nrg1. PLoS ONE 2015, 10, e0116759. [Google Scholar] [CrossRef]
- Liu, X.S.; Chopp, M.; Pan, W.L.; Wang, X.L.; Fan, B.Y.; Zhang, Y.; Kassis, H.; Zhang, R.L.; Zhang, X.M.; Zhang, Z.G. MicroRNA-146a Promotes Oligodendrogenesis in Stroke. Mol. Neurobiol. 2017, 54, 227–237. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Z.G.; Lu, M.; Wang, X.; Shang, X.; Elias, S.B.; Chopp, M. MiR-146a promotes remyelination in a cuprizone model of demyelinating injury. Neuroscience 2017, 348, 252–263. [Google Scholar] [CrossRef]
- Lin, S.T.; Huang, Y.; Zhang, L.; Heng, M.Y.; Ptacek, L.J.; Fu, Y.H. MicroRNA-23a promotes myelination in the central nervous system. Proc. Natl. Acad Sci. USA 2013, 110, 17468–17473. [Google Scholar] [CrossRef] [Green Version]
- Bronstein, J.M.; Tiwari-Woodruff, S.; Buznikov, A.G.; Stevens, D.B. Involvement of OSP/claudin-11 in oligodendrocyte membrane interactions: Role in biology and disease. J. Neurosci. Res. 2000, 59, 706–711. [Google Scholar] [CrossRef]
- Letzen, B.S.; Liu, C.; Thakor, N.V.; Gearhart, J.D.; All, A.H.; Kerr, C.L. MicroRNA expression profiling of oligodendrocyte differentiation from human embryonic stem cells. PLoS ONE 2010, 5, e10480. [Google Scholar] [CrossRef]
- Hoffmann, S.A.; Hos, D.; Kuspert, M.; Lang, R.A.; Lovell-Badge, R.; Wegner, M.; Reiprich, S. Stem cell factor Sox2 and its close relative Sox3 have differentiation functions in oligodendrocytes. Dev. Camb. Engl. 2014, 141, 39–50. [Google Scholar] [CrossRef] [Green Version]
- Budde, H.; Schmitt, S.; Fitzner, D.; Opitz, L.; Salinas-Riester, G.; Simons, M. Control of oligodendroglial cell number by the miR-17-92 cluster. Dev. Camb. Engl. 2010, 137, 2127–2132. [Google Scholar] [CrossRef] [Green Version]
- Shields, S.A.; Gilson, J.M.; Blakemore, W.F.; Franklin, R.J. Remyelination occurs as extensively but more slowly in old rats compared to young rats following gliotoxin-induced CNS demyelination. Glia 1999, 28, 77–83. [Google Scholar] [CrossRef]
- Sim, F.J.; Zhao, C.; Penderis, J.; Franklin, R.J. The age-related decrease in CNS remyelination efficiency is attributable to an impairment of both oligodendrocyte progenitor recruitment and differentiation. J. Neurosci. 2002, 22, 2451–2459. [Google Scholar] [CrossRef]
- Nicaise, A.M.; Wagstaff, L.J.; Willis, C.M.; Paisie, C.; Chandok, H.; Robson, P.; Fossati, V.; Williams, A.; Crocker, S.J. Cellular senescence in progenitor cells contributes to diminished remyelination potential in progressive multiple sclerosis. Proc. Natl. Acad. Sci. USA 2019, 116, 9030–9039. [Google Scholar] [CrossRef] [Green Version]
- Ryan, J.M.; Cristofalo, V.J. Histone acetylation during aging of human cells in culture. Biochem. Biophys. Res. Commun. 1972, 48, 735–742. [Google Scholar] [CrossRef]
- Chouliaras, L.; Lardenoije, R.; Kenis, G.; Mastroeni, D.; Hof, P.R.; van Os, J.; Steinbusch, H.W.M.; van Leeuwen, F.W.; Rutten, B.P.F.; van den Hove, D.L.A. Age-related Disturbances in DNA (hydroxy)methylation in APP/PS1 Mice. Transl. Neurosci. 2018, 9, 190–202. [Google Scholar] [CrossRef]
- Calvanese, V.; Lara, E.; Kahn, A.; Fraga, M.F. The role of epigenetics in aging and age-related diseases. Ageing Res. Rev. 2009, 8, 268–276. [Google Scholar] [CrossRef]
- Zhou, J.; Wu, Y.C.; Xiao, B.J.; Guo, X.D.; Zheng, Q.X.; Wu, B. Age-related Changes in the Global DNA Methylation Profile of Oligodendrocyte Progenitor Cells Derived from Rat Spinal Cords. Curr. Med Sci. 2019, 39, 67–74. [Google Scholar] [CrossRef]
- Shen, S.; Liu, A.; Li, J.; Wolubah, C.; Casaccia-Bonnefil, P. Epigenetic memory loss in aging oligodendrocytes in the corpus callosum. Neurobiol. Aging 2008, 29, 452–463. [Google Scholar] [CrossRef] [Green Version]
- Shen, S.; Sandoval, J.; Swiss, V.A.; Li, J.; Dupree, J.; Franklin, R.J.; Casaccia-Bonnefil, P. Age-dependent epigenetic control of differentiation inhibitors is critical for remyelination efficiency. Nat. Neurosci 2008, 11, 1024–1034. [Google Scholar] [CrossRef] [Green Version]
- Pusic, A.D.; Kraig, R.P. Youth and environmental enrichment generate serum exosomes containing miR-219 that promote CNS myelination. Glia 2014, 62, 284–299. [Google Scholar] [CrossRef]
- Fares, R.P.; Belmeguenai, A.; Sanchez, P.E.; Kouchi, H.Y.; Bodennec, J.; Morales, A.; Georges, B.; Bonnet, C.; Bouvard, S.; Sloviter, R.S.; et al. Standardized environmental enrichment supports enhanced brain plasticity in healthy rats and prevents cognitive impairment in epileptic rats. PLoS ONE 2013, 8, e53888. [Google Scholar] [CrossRef]
- Loma, I.; Heyman, R. Multiple Sclerosis: Pathogenesis and Treatment. Curr. Neuropharmacol. 2011, 9, 409–416. [Google Scholar] [CrossRef]
- Zurawski, J.; Stankiewicz, J. Multiple Sclerosis Re-Examined: Essential and Emerging Clinical Concepts. Am. J. Med. 2017, 10, 1016. [Google Scholar]
- Ebers, G.C.; Bulman, D.E.; Sadovnick, A.D.; Paty, D.W.; Warren, S.; Hader, W.; Murray, T.J.; Seland, T.P.; Duquette, P.; Grey, T.; et al. A population-based study of multiple sclerosis in twins. New Engl. J. Med. 1986, 315, 1638–1642. [Google Scholar] [CrossRef]
- Ascherio, A.; Munger, K.L.; Lennette, E.T.; Spiegelman, D.; Hernan, M.A.; Olek, M.J.; Hankinson, S.E.; Hunter, D.J. Epstein-Barr virus antibodies and risk of multiple sclerosis: A prospective study. JAMA 2001, 286, 3083–3088. [Google Scholar] [CrossRef]
- Tsai, C.N.; Tsai, C.L.; Tse, K.P.; Chang, H.Y.; Chang, Y.S. The Epstein-Barr virus oncogene product, latent membrane protein 1, induces the downregulation of E-cadherin gene expression via activation of DNA methyltransferases. Proc. Natl. Acad Sci. USA 2002, 99, 10084–10089. [Google Scholar] [CrossRef]
- Baranzini, S.E.; Mudge, J.; van Velkinburgh, J.C.; Khankhanian, P.; Khrebtukova, I.; Miller, N.A.; Zhang, L.; Farmer, A.D.; Bell, C.J.; Kim, R.W.; et al. Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature 2010, 464, 1351–1356. [Google Scholar] [CrossRef]
- Graves, M.C.; Benton, M.; Lea, R.A.; Boyle, M.; Tajouri, L.; Macartney-Coxson, D.; Scott, R.J.; Lechner-Scott, J. Methylation differences at the HLA-DRB1 locus in CD4+ T-Cells are associated with multiple sclerosis. Mult. Scler. J. 2013, 20, 1033–1041. [Google Scholar] [CrossRef]
- Liggett, T.; Melnikov, A.; Tilwalli, S.; Yi, Q.; Chen, H.; Replogle, C.; Feng, X.; Reder, A.; Stefoski, D.; Balabanov, R.; et al. Methylation patterns of cell-free plasma DNA in relapsing-remitting multiple sclerosis. J. Neurol. Sci. 2010, 290, 16. [Google Scholar] [CrossRef]
- Guan, H.; Nagarkatti, P.S.; Nagarkatti, M. CD44 Reciprocally Regulates the Differentiation of Encephalitogenic Th1/Th17 and Th2/Regulatory T Cells through Epigenetic Modulation Involving DNA Methylation of Cytokine Gene Promoters, Thereby Controlling the Development of Experimental Autoimmune Encephalomyelitis. J. Immunol. 2011, 186, 6955–6964. [Google Scholar] [Green Version]
- Franklin, R.J.; Ffrench-Constant, C. Remyelination in the CNS: From biology to therapy. Nat. Rev. Neurosci. 2008, 9, 839–855. [Google Scholar] [CrossRef]
- Kuhlmann, T.; Miron, V.; Cui, Q.; Wegner, C.; Antel, J.; Bruck, W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain: J. Neurol. 2008, 131, 1749–1758. [Google Scholar] [CrossRef] [Green Version]
- Cadavid, D.; Mellion, M.; Hupperts, R.; Edwards, K.R.; Calabresi, P.A.; Drulović, J.; Giovannoni, G.; Hartung, H.-P.; Arnold, D.L.; Fisher, E.; et al. Safety and efficacy of opicinumab in patients with relapsing multiple sclerosis (SYNERGY): A randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2019, 18, 845–856. [Google Scholar] [CrossRef]
- Moscarello, M.A.; Wood, D.D.; Ackerley, C.; Boulias, C. Myelin in multiple sclerosis is developmentally immature. J. Clin. Investig. 1994, 94, 146–154. [Google Scholar] [CrossRef]
- Mastronardi, F.G.; Noor, A.; Wood, D.D.; Paton, T.; Moscarello, M.A. Peptidyl argininedeiminase 2 CpG island in multiple sclerosis white matter is hypomethylated. J. Neurosci. Res. 2007, 85, 2006–2016. [Google Scholar] [CrossRef]
- Calabrese, R.; Zampieri, M.; Mechelli, R.; Annibali, V.; Guastafierro, T.; Ciccarone, F.; Coarelli, G.; Umeton, R.; Salvetti, M.; Caiafa, P. Methylation-dependent PAD2 upregulation in multiple sclerosis peripheral blood. Mult. Scler. 2012, 18, 299–304. [Google Scholar] [CrossRef]
- Olsen, J.A.; Kenna, L.A.; Tipon, R.C.; Spelios, M.G.; Stecker, M.M.; Akirav, E.M. A Minimally-invasive Blood-derived Biomarker of Oligodendrocyte Cell-loss in Multiple Sclerosis. EBioMedicine 2016, 10, 227–235. [Google Scholar] [CrossRef] [Green Version]
- Al-Mahdawi, S.; Anjomani Virmouni, S.; Pook, M.A. DNA Methylation in Neurodegenerative Diseases A2. In Epigenetic Biomarkers and Diagnostics; Giménez, G., Luis, J., Eds.; Academic Press: Boston, ME, USA, 2016; pp. 401–415. [Google Scholar]
- Jakubowski, J.L.; Labrie, V. Epigenetic Biomarkers for Parkinson’s Disease: From Diagnostics to Therapeutics. J. Parkinson’s Dis. 2016, 7, 1–12. [Google Scholar] [CrossRef]
- Pihlstrom, L.; Berge, V.; Rengmark, A.; Toft, M. Parkinson’s disease correlates with promoter methylation in the alpha-synuclein gene. Mov. Disord. 2015, 30, 577–580. [Google Scholar] [CrossRef]
- Huynh, J.L.; Garg, P.; Thin, T.H.; Yoo, S.; Dutta, R.; Trapp, B.D.; Haroutunian, V.; Zhu, J.; Donovan, M.J.; Sharp, A.J.; et al. Epigenome-wide differences in pathology-free regions of multiple sclerosis-affected brains. Nat. Neurosci 2014, 17, 121–130. [Google Scholar] [CrossRef]
- Pedre, X.; Mastronardi, F.; Bruck, W.; Lopez-Rodas, G.; Kuhlmann, T.; Casaccia, P. Changed histone acetylation patterns in normal-appearing white matter and early multiple sclerosis lesions. J. Neurosci. 2011, 31, 3435–3445. [Google Scholar] [CrossRef]
- Noorbakhsh, F.; Ellestad, K.K.; Maingat, F.; Warren, K.G.; Han, M.H.; Steinman, L.; Baker, G.B.; Power, C. Impaired neurosteroid synthesis in multiple sclerosis. Brain J. Neurol. 2011, 134, 2703–2721. [Google Scholar] [CrossRef] [Green Version]
- Junker, A.; Krumbholz, M.; Eisele, S.; Mohan, H.; Augstein, F.; Bittner, R.; Lassmann, H.; Wekerle, H.; Hohlfeld, R.; Meinl, E. MicroRNA profiling of multiple sclerosis lesions identifies modulators of the regulatory protein CD47. Brain J. Neurol. 2009, 132, 3342–3352. [Google Scholar] [CrossRef] [Green Version]
- Bruinsma, I.B.; van Dijk, M.; Bridel, C.; van de Lisdonk, T.; Haverkort, S.Q.; Runia, T.F.; Steinman, L.; Hintzen, R.Q.; Killestein, J.; Verbeek, M.M.; et al. Regulator of oligodendrocyte maturation, miR-219, a potential biomarker for MS. J. Neuroinflamm. 2017, 14, 235. [Google Scholar] [CrossRef]
- Mendizabal, I.; Berto, S.; Usui, N.; Toriumi, K.; Chatterjee, P.; Douglas, C.; Huh, I.; Jeong, H.; Layman, T.; Tamminga, C.A.; et al. Cell type-specific epigenetic links to schizophrenia risk in the brain. Genome Biol. 2019, 20, 135. [Google Scholar] [CrossRef] [Green Version]
- Kozlenkov, A.; Roussos, P.; Timashpolsky, A.; Barbu, M.; Rudchenko, S.; Bibikova, M.; Klotzle, B.; Byne, W.; Lyddon, R.; Di Narzo, A.F.; et al. Differences in DNA methylation between human neuronal and glial cells are concentrated in enhancers and non-CpG sites. Nucleic Acids Res. 2014, 42, 109–127. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.; Zou, Z.; Tian, H.; Zhang, Y.; Zhou, H.; Liu, L. Stem Cell-Based Therapies for Ischemic Stroke. Biomed Res. Int. 2014, 2014, 17. [Google Scholar] [CrossRef] [PubMed]
- Kassis, H.; Chopp, M.; Liu, X.S.; Shehadah, A.; Roberts, C.; Zhang, Z.G. Histone deacetylase expression in white matter oligodendrocytes after stroke. Neurochem. Int. 2014, 77, 17–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felling, R.J.; Song, H. Epigenetic mechanisms of neuroplasticity and the implications for stroke recovery. Exp. Neurol. 2015, 268, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Chopp, M.; Kassis, H.; Jia, L.F.; Hozeska-Solgot, A.; Zhang, R.L.; Chen, C.; Cui, Y.S.; Zhang, Z.G. Valproic acid increases white matter repair and neurogenesis after stroke. Neuroscience 2012, 220, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Chuang, D.M. HDAC inhibitors mitigate ischemia-induced oligodendrocyte damage: Potential roles of oligodendrogenesis, VEGF, and anti-inflammation. Am. J. Transl. Res. 2014, 6, 206–223. [Google Scholar] [PubMed]
- Ziemka-Nalecz, M.; Jaworska, J.; Sypecka, J.; Polowy, R.; Filipkowski, R.K.; Zalewska, T. Sodium Butyrate, a Histone Deacetylase Inhibitor, Exhibits Neuroprotective/Neurogenic Effects in a Rat Model of Neonatal Hypoxia-Ischemia. Mol. Neurobiol. 2017, 54, 5300–5318. [Google Scholar] [CrossRef] [PubMed]
- Dincman, T.A.; Beare, J.E.; Ohri, S.S.; Gallo, V.; Hetman, M.; Whittemore, S.R. Histone deacetylase inhibition is cytotoxic to oligodendrocyte precursor cells in vitro and in vivo. Int. J. Dev. Neurosci. 2016, 54, 53–61. [Google Scholar] [CrossRef]
- Tan, K.S.; Armugam, A.; Sepramaniam, S.; Lim, K.Y.; Setyowati, K.D.; Wang, C.W.; Jeyaseelan, K. Expression profile of MicroRNAs in young stroke patients. PLoS ONE 2009, 4, 7689. [Google Scholar] [CrossRef]
- Delaloy, C.; Liu, L.; Lee, J.A.; Su, H.; Shen, F.; Yang, G.Y.; Young, W.L.; Ivey, K.N.; Gao, F.B. MicroRNA-9 coordinates proliferation and migration of human embryonic stem cell-derived neural progenitors. Cell Stem Cell 2010, 6, 323–335. [Google Scholar] [CrossRef]
- Birch, D.; Britt, B.C.; Dukes, S.C.; Kessler, J.A.; Dizon, M.L. MicroRNAs participate in the murine oligodendroglial response to perinatal hypoxia-ischemia. Pediatric Res. 2014, 76, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Li, J.S.; Yao, Z.X. MicroRNA patents in demyelinating diseases: A new diagnostic and therapeutic perspective. Recent Pat. Dna Gene Seq. 2012, 6, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Dharap, A.; Vemuganti, R. Ischemic pre-conditioning alters cerebral microRNAs that are upstream to neuroprotective signaling pathways. J. Neurochem. 2010, 113, 1685–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.H.; Su, S.Y.; Liu, J.L. Differential Regulation of microRNAs in Patients with Ischemic Stroke. Curr. Neurovascular Res. 2015, 12, 214–221. [Google Scholar] [CrossRef]
- Engelen, M.; Kemp, S.; Poll-The, B.T. X-linked adrenoleukodystrophy: Pathogenesis and treatment. Curr. Neurol. Neurosci. Rep. 2014, 14, 486. [Google Scholar] [CrossRef] [PubMed]
- Schluter, A.; Sandoval, J.; Fourcade, S.; Diaz-Lagares, A.; Ruiz, M.; Casaccia, P.; Esteller, M.; Pujol, A. Epigenomic signature of adrenoleukodystrophy predicts compromised oligodendrocyte differentiation. Brain Pathol. 2018, 10, 12595. [Google Scholar] [CrossRef] [PubMed]
- Engelen, M.; Kemp, S.; de Visser, M.; van Geel, B.M.; Wanders, R.J.A.; Aubourg, P.; Poll-The, B.T. X-linked adrenoleukodystrophy (X-ALD): Clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J. Rare Dis. 2012, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Khan, M.; Pujol, A.; Baarine, M.; Singh, I. Histone deacetylase inhibitor upregulates peroxisomal fatty acid oxidation and inhibits apoptotic cell death in abcd1-deficient glial cells. PLoS ONE 2013, 8, e70712. [Google Scholar] [CrossRef]
- Lin, S.-T.; Ptácek, L.J.; Fu, Y.-H. Adult-onset autosomal dominant leukodystrophy: Linking nuclear envelope to myelin. J. Neurosci. 2011, 31, 1163–1166. [Google Scholar] [CrossRef]
- Lin, S.T.; Fu, Y.H. miR-23 regulation of lamin B1 is crucial for oligodendrocyte development and myelination. Dis. Models Mech. 2009, 2, 178–188. [Google Scholar] [CrossRef]
- Iwamoto, K.; Bundo, M.; Yamada, K.; Takao, H.; Iwayama-Shigeno, Y.; Yoshikawa, T.; Kato, T. DNA methylation status of SOX10 correlates with its downregulation and oligodendrocyte dysfunction in schizophrenia. J. Neurosci. 2005, 25, 5376–5381. [Google Scholar] [CrossRef] [PubMed]
- Fabianowska-Majewska, K.; Wyczechowska, D.; Czyz, M. Inhibition of dna methylation by 5-aza-2’-deoxycytidine correlates with induction of K562 cells differentiation. Adv. Exp. Med. Biol. 2000, 486, 343–347. [Google Scholar] [PubMed]
- Raj, K.; Mufti, G.J. Azacytidine (Vidaza®) in the treatment of myelodysplastic syndromes. Ther. Clin. Risk Manag. 2006, 2, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Gnyszka, A.; Jastrzebski, Z.; Flis, S. DNA methyltransferase inhibitors and their emerging role in epigenetic therapy of cancer. Anticancer Res. 2013, 33, 2989–2996. [Google Scholar] [PubMed]
- Stresemann, C.; Lyko, F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer 2008, 123, 8–13. [Google Scholar] [CrossRef]
- Waryah, C.B.; Moses, C.; Arooj, M.; Blancafort, P. Zinc Fingers, TALEs, and CRISPR Systems: A Comparison of Tools for Epigenome Editing. Methods Mol. Biol. 2018, 1767, 19–63. [Google Scholar]
- Thakore, P.I.; Black, J.B.; Hilton, I.B.; Gersbach, C.A. Editing the Epigenome: Technologies for Programmable Transcriptional Modulation and Epigenetic Regulation. Nat. Methods 2016, 13, 127–137. [Google Scholar] [CrossRef]
- Laity, J.H.; Lee, B.M.; Wright, P.E. Zinc finger proteins: New insights into structural and functional diversity. Curr. Opin. Struct. Biol. 2001, 11, 39–46. [Google Scholar] [CrossRef]
- Rots, M.G.; Jeltsch, A. Editing the Epigenome: Overview, Open Questions, and Directions of Future Development. Methods Mol. Biol. 2018, 1767, 3–18. [Google Scholar]
- Dai, W.J.; Zhu, L.Y.; Yan, Z.Y.; Xu, Y.; Wang, Q.L.; Lu, X.J. CRISPR-Cas9 for in vivo Gene Therapy: Promise and Hurdles. Mol. Ther. Nucleic Acids 2016, 5, 349. [Google Scholar] [CrossRef]
- Wang, S.; Bates, J.; Li, X.; Schanz, S.; Chandler-Militello, D.; Levine, C.; Maherali, N.; Studer, L.; Hochedlinger, K.; Windrem, M.; et al. Human iPSC-derived oligodendrocyte progenitor cells can myelinate and rescue a mouse model of congenital hypomyelination. Cell Stem Cell 2013, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Douvaras, P.; Rusielewicz, T.; Kim, K.H.; Haines, J.D.; Casaccia, P.; Fossati, V. Epigenetic Modulation of Human Induced Pluripotent Stem Cell Differentiation to Oligodendrocytes. Int. J. Mol. Sci. 2016, 17, 614. [Google Scholar] [CrossRef] [PubMed]
- Douvaras, P.; Wang, J.; Zimmer, M.; Hanchuk, S.; O’Bara, M.A.; Sadiq, S.; Sim, F.J.; Goldman, J.; Fossati, V. Efficient Generation of Myelinating Oligodendrocytes from Primary Progressive Multiple Sclerosis Patients by Induced Pluripotent Stem Cells. Stem Cell Rep. 2014, 3, 250–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tiane, A.; Schepers, M.; Rombaut, B.; Hupperts, R.; Prickaerts, J.; Hellings, N.; van den Hove, D.; Vanmierlo, T. From OPC to Oligodendrocyte: An Epigenetic Journey. Cells 2019, 8, 1236. https://doi.org/10.3390/cells8101236
Tiane A, Schepers M, Rombaut B, Hupperts R, Prickaerts J, Hellings N, van den Hove D, Vanmierlo T. From OPC to Oligodendrocyte: An Epigenetic Journey. Cells. 2019; 8(10):1236. https://doi.org/10.3390/cells8101236
Chicago/Turabian StyleTiane, Assia, Melissa Schepers, Ben Rombaut, Raymond Hupperts, Jos Prickaerts, Niels Hellings, Daniel van den Hove, and Tim Vanmierlo. 2019. "From OPC to Oligodendrocyte: An Epigenetic Journey" Cells 8, no. 10: 1236. https://doi.org/10.3390/cells8101236