Inhibition of JAK-STAT Signaling with Baricitinib Reduces Inflammation and Improves Cellular Homeostasis in Progeria Cells


Abstract
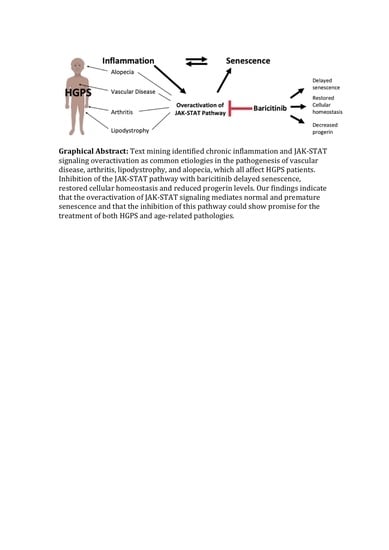
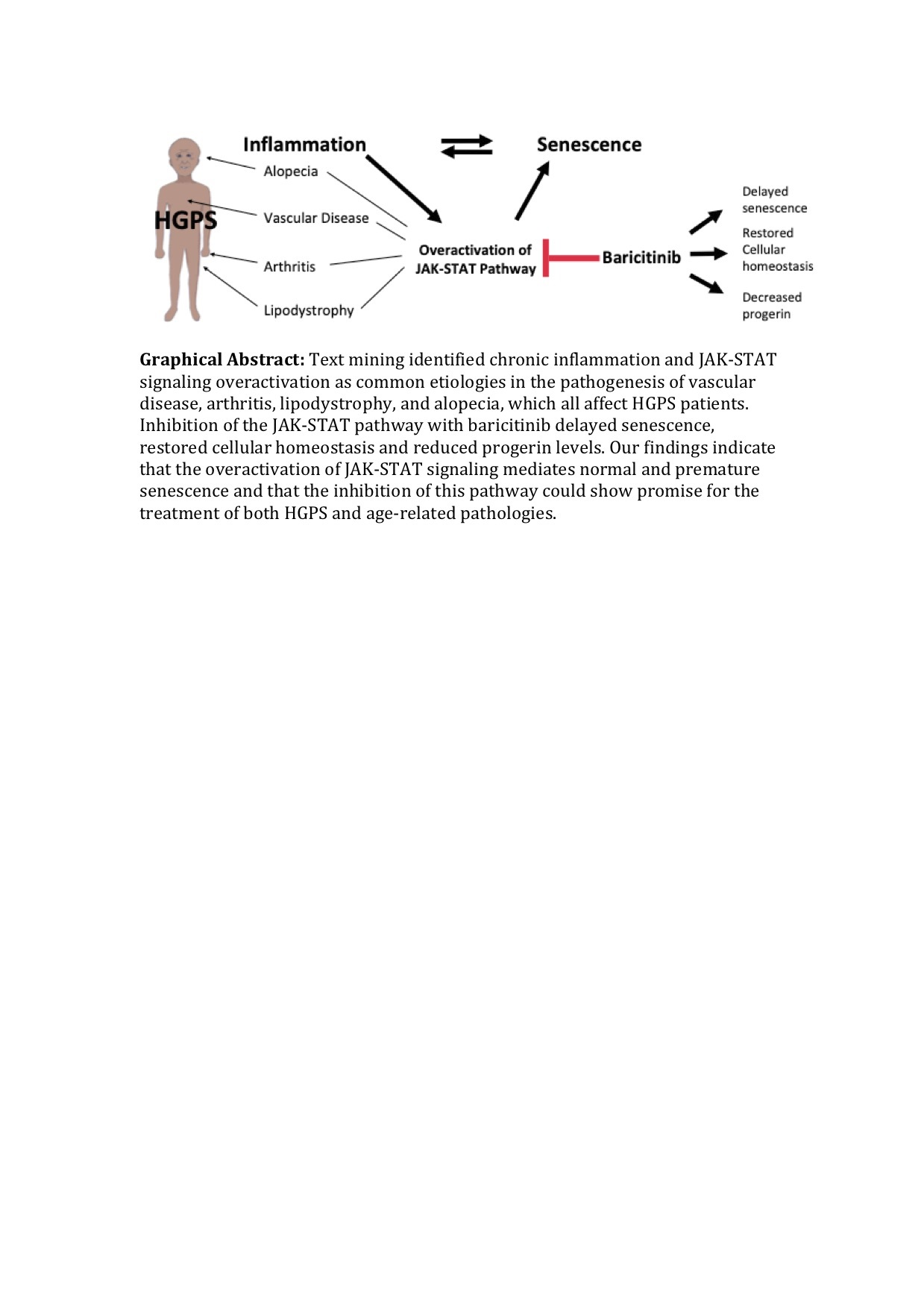
1. Introduction
2. Materials and Methods
2.1. Text Mining Study
2.2. Identification of the Signaling Pathways
2.3. Cell Culture and Drug Treatments
2.4. Determination of Cumulative Population Doubling Determination
2.5. Senescence Associated Beta-Galactosidase Assay
2.6. Cell Cytotoxicity
2.7. Cell Cycle Analysis
2.8. Measurement of Proteasome Activity
2.9. Autophagy Measurement
2.10. Measurement of ROS
2.11. Measurement of Intracellular ATP
2.12. Gene Expression Analysis
2.13. Western Blots Analysis
2.14. Immunocytochemistry
2.15. Statistics
3. Results
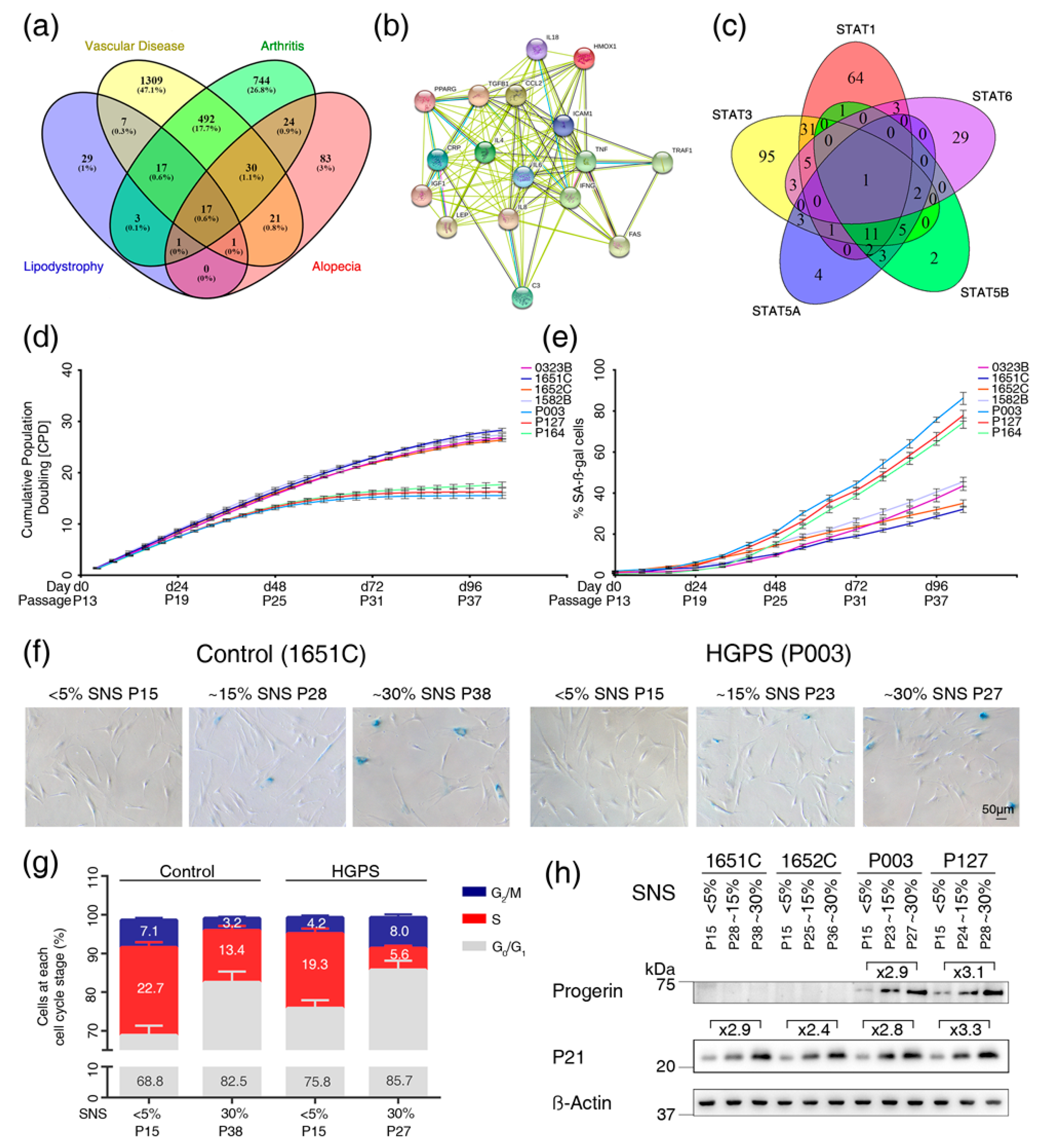
3.1. Text Mining Analysis to Identify Genes Altered in Vascular Disease, Arthritis, Alopecia and Lipodystrophy
3.2. Cell-Based Aging Model to Investigate Normal and Premature Cellular Aging
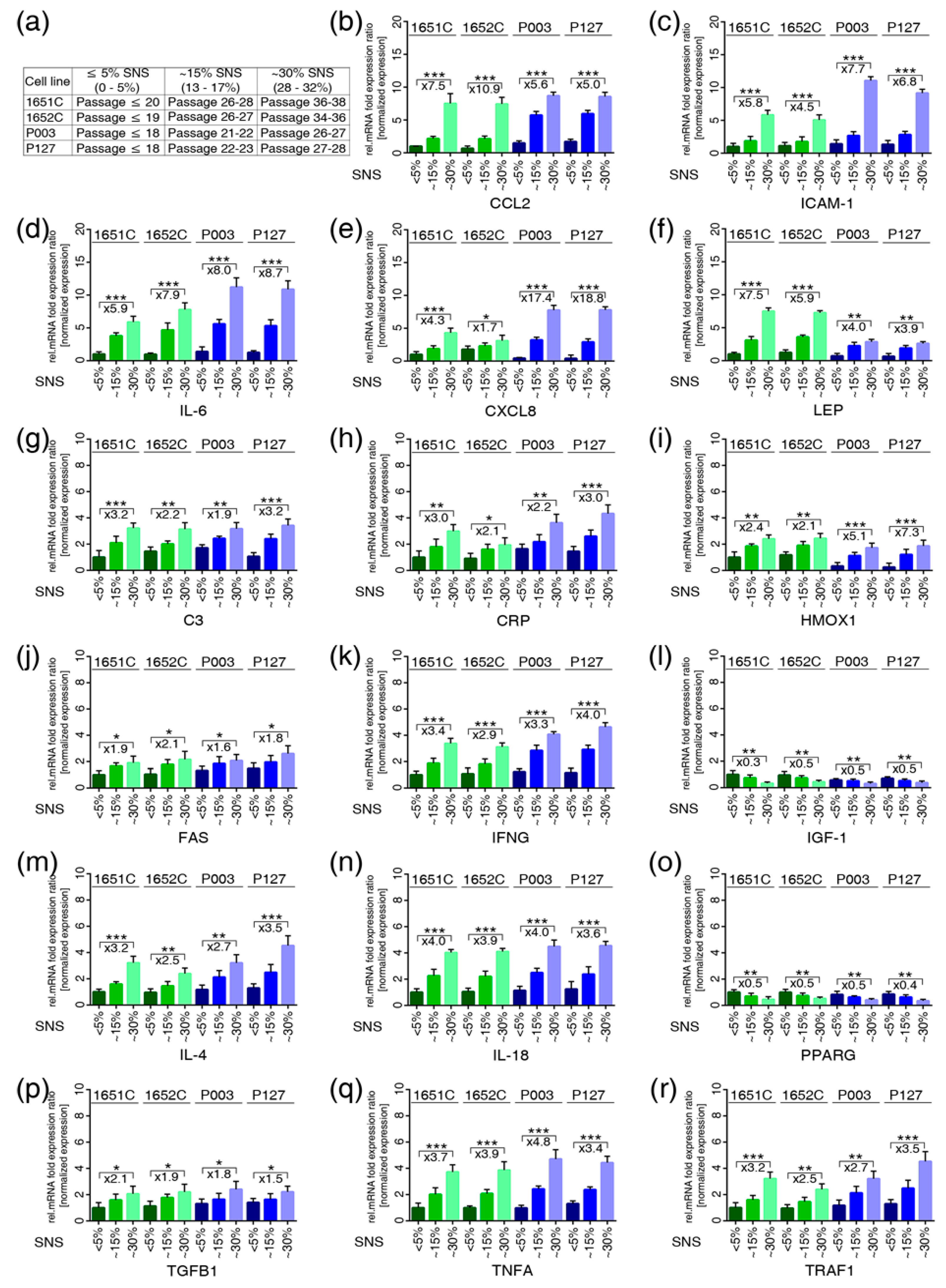
3.3. Profiles of the 17 Genes Associated with Vascular Disease, Arthritis, Lipodystrophy and Alopecia in the Cell-Based Aging Model
3.4. Overactivation of JAK-STAT Signaling During Replicative Senescence in Normal and HGPS Fibroblasts
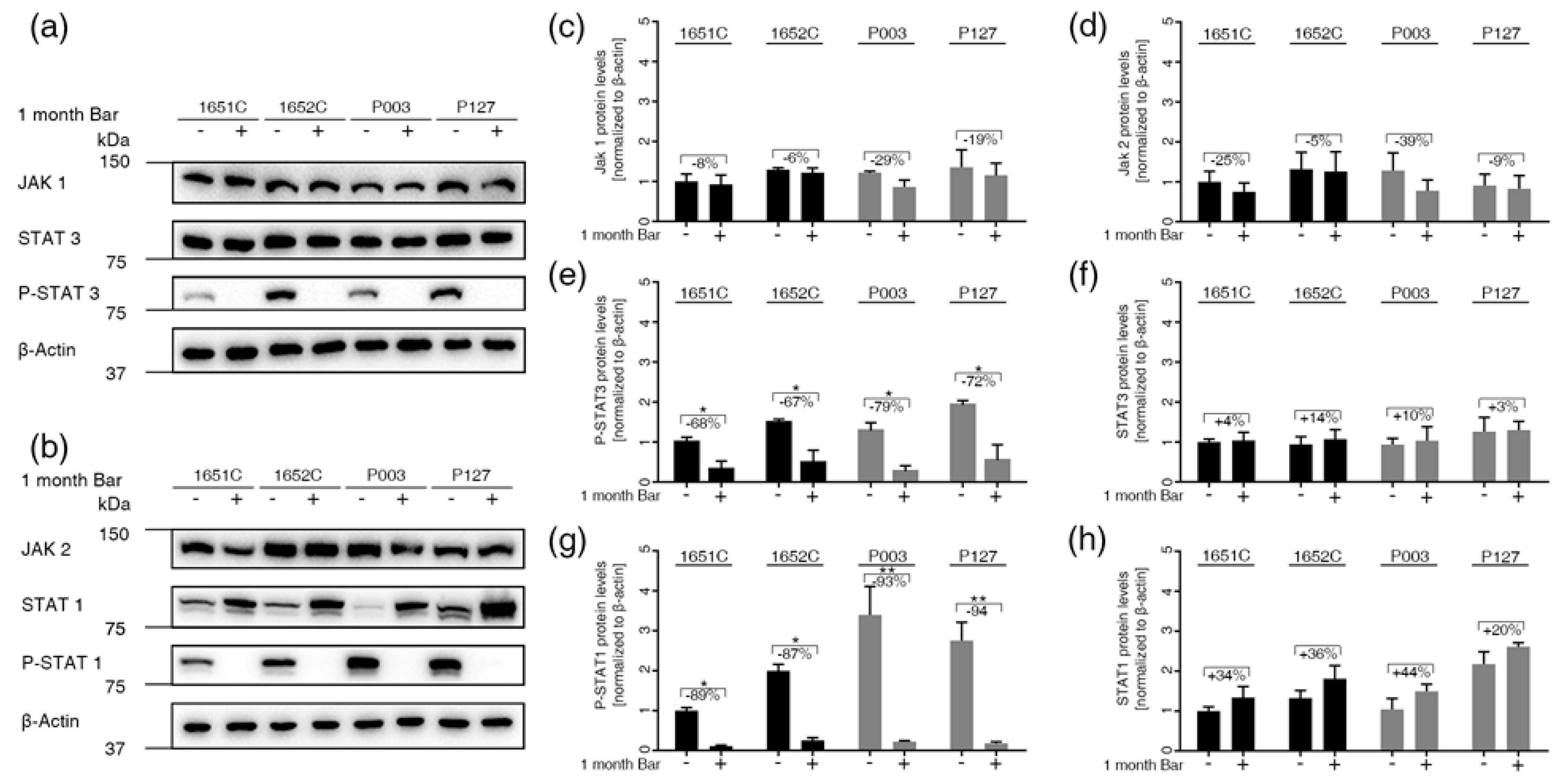
3.5. Baricitinib, a Specific Inhibitor of JAK1 and JAK2, Efficiently Blunts STAT1 and STAT3 Activation
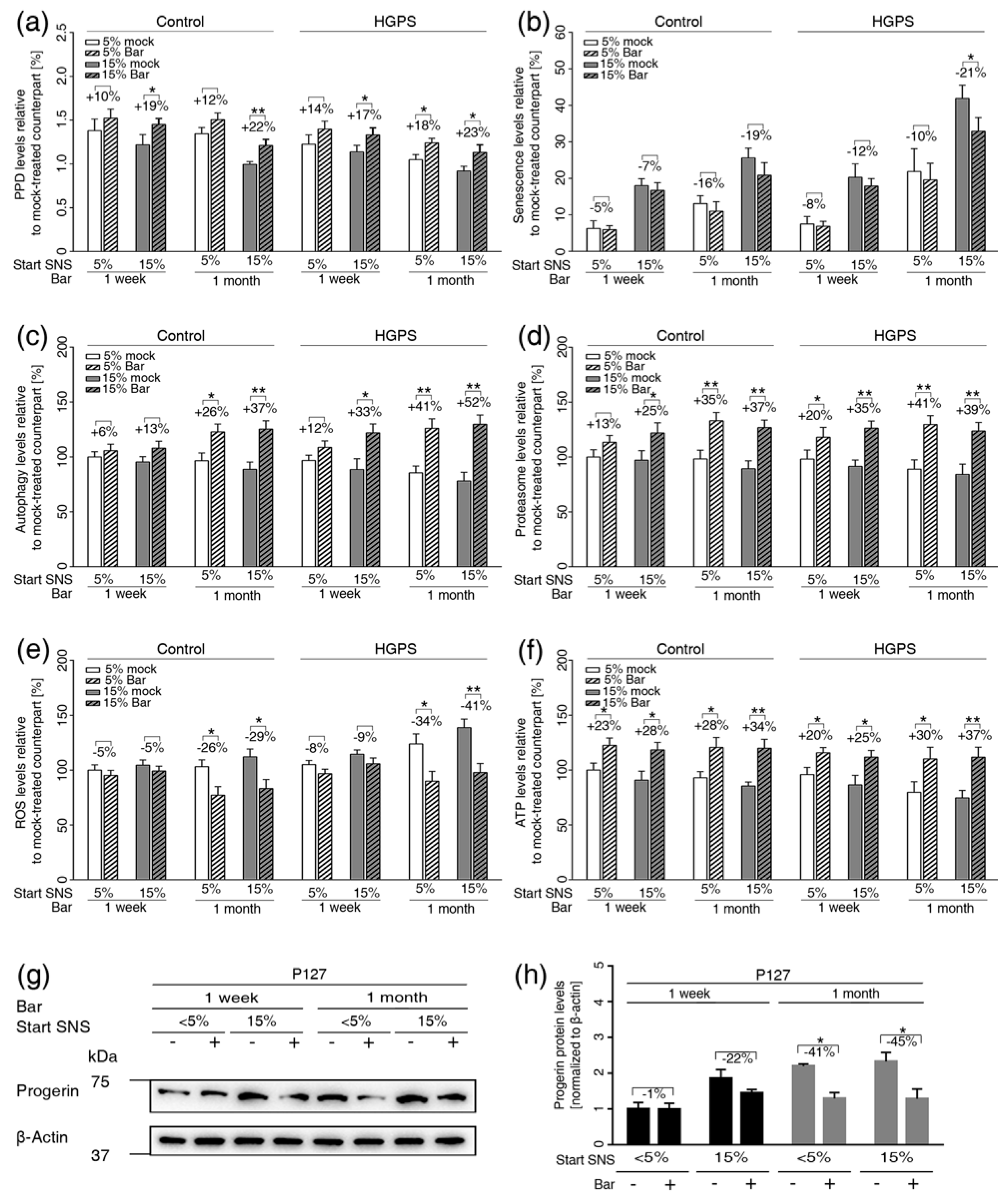
3.6. Inhibition of JAK1 and JAK2 Ameliorates Age-Related Cellular Changes in Normal and HGPS Cells
3.7. JAK-STAT Inhibition Reduces Proinflammatory Factors
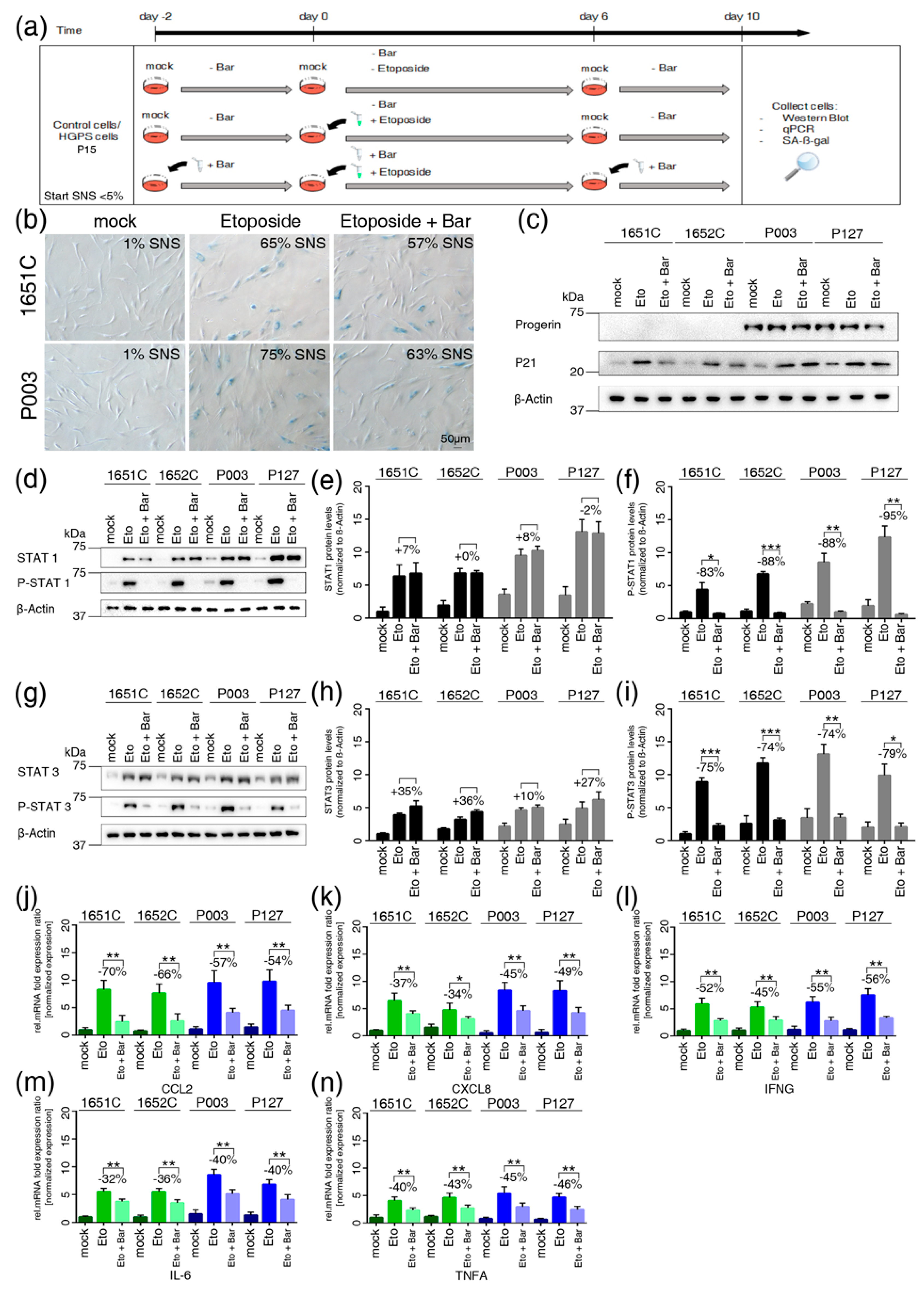
3.8. Etoposide-Induced DNA Damage Overactivates the JAK-STAT Pathway in both Normal and HGPS Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gordon, L.B.; Rothman, F.G.; López-Otín, C.; Misteli, T. Progeria: A paradigm for translational medicine. Cell 2014, 156, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Merideth, M.A.; Gordon, L.B.; Clauss, S.; Sachdev, V.; Smith, A.C.; Perry, M.B.; Brewer, C.C.; Zalewski, C.; Kim, H.J.; Solomon, B.; et al. Phenotype and course of hutchinson-gilford progeria syndrome. N. Engl. J. Med. 2008, 358, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin a cause hutchinson-gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Gordon, L.B.; Massaro, J.; D’Agostino, R.B.S.; Campbell, S.E.; Brazier, J.; Brown, W.T.; Kleinman, M.E.; Kieran, M.W.; Progeria Clinical Trials Collaborative. Impact of farnesylation inhibitors on survival in hutchinson-gilford progeria syndrome. Circulation 2014, 130, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Novelli, G.; Muchir, A.; Sangiuolo, F.; Helbling-Leclerc, A.; D’Apice, M.; Massart, C.; Capon, F.; Sbraccia, P.; Federici, M.; Lauro, R.; et al. Mandibuloacral dysplasia is caused by a mutation in lmna-encoding lamin a/c. Am. J. Hum. Genet. 2002, 71, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Motegi, S.; Yokoyama, Y.; Uchiyama, A.; Ogino, S.; Takeuchi, Y.; Yamada, K.; Hattori, T.; Hashizume, H.; Ishikawa, Y.; Goto, M.; et al. First japanese case of atypical progeroid syndrome/atypical werner syndrome with heterozygous lmna mutation. J. Dermatol. 2014, 41, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Dogramaci, A.C.; Balci, D.D.; Balci, A.; Karazincir, S.; Savas, N.; Topaloglu, C.; Yalcin, F. Is androgenetic alopecia a risk for atherosclerosis? J. Eur. Acad. Dermatol. Venereol. 2009, 23, 673–677. [Google Scholar] [CrossRef]
- Cai, B.; Zeng, J.; Lin, Y.; Lin, Y.; Lin, W.; Lin, W.; Li, Z.; Wang, N. A frameshift mutation in htra1 expands carasil syndrome and peripheral small arterial disease to the chinese population. Neurol. Sci. 2015, 36, 1387–1391. [Google Scholar] [CrossRef]
- Sanon, V.P.; Handelsman, Y.; Pham, S.V.; Chilton, R. Cardiac manifestations of congenital generalized lipodystrophy. Clin. Diabetes 2016, 34, 181–186. [Google Scholar] [CrossRef]
- Loonam, C.R.; Mullen, A. Nutrition and the hiv-associated lipodystrophy syndrome. Nutr. Res. Rev. 2012, 25, 267–287. [Google Scholar] [CrossRef]
- Park, S.; Erdogan, S.; Hwang, D.; Hwang, S.; Han, E.H.; Lim, Y.H. Bee venom promotes hair growth in association with inhibiting 5alpha-reductase expression. Biol. Pharm. Bull. 2016, 39, 1060–1068. [Google Scholar] [CrossRef] [PubMed]
- Cao, K.; Graziotto, J.J.; Blair, C.D.; Mazzulli, J.R.; Erdos, M.R.; Krainc, D.; Collins, F.S. Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in hutchinson-gilford progeria syndrome cells. Sci. Transl. Med. 2011, 3, 89ra58. [Google Scholar] [CrossRef] [PubMed]
- Marji, J.; O’Donoghue, S.I.; McClintock, D.; Satagopam, V.P.; Schneider, R.; Ratner, D.; Worman, H.J.; Gordon, L.B.; Djabali, K. Defective lamin a-rb signaling in hutchinson-gilford progeria syndrome and reversal by farnesyltransferase inhibition. PLoS ONE 2010, 5, e11132. [Google Scholar] [CrossRef] [PubMed]
- Osorio, F.G.; Bárcena, C.; Soria-Valles, C.; Ramsay, A.J.; de Carlos, F.; Cobo, J.; Fueyo, A.; Freije, J.M.; López-Otín, C. Nuclear lamina defects cause atm-dependent nf-κb activation and link accelerated aging to a systemic inflammatory response. Genes Dev. 2012, 26, 2311–2324. [Google Scholar] [CrossRef]
- Gabriel, D.; Roedl, D.; Gordon, L.B.; Djabali, K. Sulforaphane enhances progerin clearance in hutchinson-gilford progeria fibroblasts. Aging Cell 2015, 14, 78–91. [Google Scholar] [CrossRef]
- Kubben, N.; Zhang, W.; Wang, L.; Voss, T.C.; Yang, J.; Qu, J.; Liu, G.H.; Misteli, T. Repression of the antioxidant nrf2 pathway in premature aging. Cell 2016, 165, 1361–1374. [Google Scholar] [CrossRef]
- Fridman, J.S.; Scherle, P.A.; Collins, R.; Burn, T.C.; Li, Y.; Li, J.; Covington, M.B.; Thomas, B.; Collier, P.; Favata, M.F.; et al. Selective inhibition of jak1 and jak2 is efficacious in rodent models of arthritis: Preclinical characterization of incb028050. J. Immunol. 2010, 184, 5298–5307. [Google Scholar] [CrossRef]
- Yates, B.; Braschi, B.; Gray, K.A.; Seal, R.L.; Tweedie, S.; Bruford, E.A. Genenames.Org: The hgnc and vgnc resources in 2017. Nucleic Acids Res. 2017, 45, D619–D625. [Google Scholar] [CrossRef]
- Pasolli, E.; Schiffer, L.; Manghi, P.; Renson, A.; Obenchain, V.; Truong, D.T.; Beghini, F.; Malik, F.; Ramos, M.; Dowd, J.B.; et al. Accessible, curated metagenomic data through experimenthub. Nat. Methods 2017, 14, 1023–1024. [Google Scholar] [CrossRef]
- Rani, J.; Shah, A.B.; Ramachandran, S. Pubmed.Miner: An R package with text-mining algorithms to analyse pubmed abstracts. J. Biosci. 2015, 40, 671–682. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-blast: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time rt–pcr. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Chovatiya, R.; Medzhitov, R. Stress, inflammation, and defense of homeostasi. Mol. Cell. 2014, 54, 281–288. [Google Scholar] [CrossRef]
- Frasca, D.; Blomberg, B.B. Inflammaging decreases adaptive and innate immune responses in mice and humans. Biogerontology 2016, 17, 7–19. [Google Scholar] [CrossRef]
- Han, H.; Cho, J.W.; Lee, S.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E.; et al. Trrust v2: An expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018, 46, D380–D386. [Google Scholar] [CrossRef] [PubMed]
- Essaghir, A.; Toffalini, F.; Knoops, L.; Kallin, A.; van Helden, J.; Demoulin, J.B. Transcription factor regulation can be accurately predicted from the presence of target gene signatures in microarray gene expression data. Nucleic Acids Res. 2010, 38, e120. [Google Scholar] [CrossRef] [PubMed]
- Marbach, D.; Lamparter, D.; Quon, G.; Kellis, M.; Kutalik, Z.; Bergmann, S. Tissue-specific regulatory circuits reveal variable modular perturbations across complex diseases. Nat. Methods 2016, 13, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Osorio, F.G.; Soria-Valles, C.; Santiago-Fernández, O.; Freije, J.M.; López-Otín, C. Nf-κb signaling as a driver of ageing. Int. Rev. Cell. Mol. Biol. 2016, 326, 134–174. [Google Scholar]
- Bridger, J.M.; Kill, I.R.; O’Farrell, M.; Hutchinson, C.J. Internal lamin structures within g1 nuclei of human dermal fibroblasts. J. Cell Sci. 1993, 104, 297–306. [Google Scholar]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef]
- Goldman, R.D.; Shumaker, D.K.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Gordon, L.B.; Gruenbaum, Y.; Khuon, S.; Mendez, M.; Varga, R.; et al. Accumulation of mutant lamin a causes progressive changes in nuclear architecture in hutchinson-gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 8963–8968. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.C.; Levine, R.L. Molecular pathways: Molecular basis for sensitivity and resistance to jak kinase inhibitors. Clin. Cancer Res. 2014, 20, 2051–2059. [Google Scholar] [CrossRef] [PubMed]
- Te Poele, R.H.; Okorokov, A.L.; Jardine, L.; Cummings, J.; Joel, S.P. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Can. Res. 2002, 62, 1876–1883. [Google Scholar]
- Coppe, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Ann. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Alexander, H.D.; Ross, O.A. Age and age-related diseases: Role of inflammation triggers and cytokines. Front. Immunol. 2018, 9, 586. [Google Scholar] [CrossRef] [PubMed]
- Gordon, L.B.; Campbell, S.E.; Massaro, J.M.; D’Agostino, R.B.S.; Kleinman, M.E.; Kieran, M.W.; Moses, M.A. Survey of plasma proteins in children with progeria pre-therapy and on-therapy with lonafarnib. Pediatr. Res. 2018, 83, 982–992. [Google Scholar] [CrossRef]
- Liu, Y.-Z.; Wang, Y.-X.; Jiang, C.-L. Inflammation: The common pathway of stress-related diseases. Front. Hum. Neurosci. 2017, 11, 316. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflammaging: An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Fukushima, Y.; Minato, N.; Hattori, M. The impact of senescence-associated t cells on immunosenescence and age-related disorders. Inflamm. Regen. 2018, 24, 24. [Google Scholar] [CrossRef] [PubMed]
- Turinetto, V.; Vitale, E.; Giachino, C. Senescence in human mesenchymal stem cells: Functional changes and implications in stem cell-based therapy. Int. J. Mol. Sci. 2016, 17, 1164. [Google Scholar] [CrossRef] [PubMed]
- Erusalimsky, J.D.; Kurz, D.J. Cellular senescence in vivo: Its relevance in ageing and cardiovascular disease. Exp. Gerontol. 2005, 40, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Stout, M.B.; Justice, J.N.; Nicklas, B.J.; Kirkland, J.L. Physiological aging: Links among adipose tissue dysfunction, diabetes, and frailty. Physiology 2017, 32, 9–19. [Google Scholar] [CrossRef]
- McCulloch, K.; Litherland, G.J.; Rai, T.S. Reviewcellular senescence in osteoarthritis pathology. Aging Cell 2017, 16, 210–218. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | NF-κB1 | Rela | STAT3 | STAT1 | STAT5A | STAT5B | STAT6 |
|---|---|---|---|---|---|---|---|
| C3 | |||||||
| CCL2 | √ | √ | √ | √ | |||
| CRP | √ | √ | √ | ||||
| CXCL8 | √ | √ | √ | √ | √ | ||
| FAS | √ | √ | √ | √ | |||
| HMOX1 | √ | √ | √ | √ | |||
| ICAM1 | √ | √ | √ | √ | |||
| IGF1 | √ | √ | √ | ||||
| IL6 | √ | √ | √ | √ | √ | ||
| IL18 | √ | √ | |||||
| PPARG | √ | √ | √ | ||||
| TGFB1 | √ | √ | |||||
| TNF | √ | √ | √ | √ | √ | ||
| TRAF1 | √ | √ | |||||
| LEP | √ | ||||||
| IL4 | √ | √ | √ | ||||
| IFN-G | √ | √ | √ | √ | √ | √ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, C.; Arnold, R.; Henriques, G.; Djabali, K. Inhibition of JAK-STAT Signaling with Baricitinib Reduces Inflammation and Improves Cellular Homeostasis in Progeria Cells. Cells 2019, 8, 1276. https://doi.org/10.3390/cells8101276
Liu C, Arnold R, Henriques G, Djabali K. Inhibition of JAK-STAT Signaling with Baricitinib Reduces Inflammation and Improves Cellular Homeostasis in Progeria Cells. Cells. 2019; 8(10):1276. https://doi.org/10.3390/cells8101276
Chicago/Turabian StyleLiu, Chang, Rouven Arnold, Gonçalo Henriques, and Karima Djabali. 2019. "Inhibition of JAK-STAT Signaling with Baricitinib Reduces Inflammation and Improves Cellular Homeostasis in Progeria Cells" Cells 8, no. 10: 1276. https://doi.org/10.3390/cells8101276
APA StyleLiu, C., Arnold, R., Henriques, G., & Djabali, K. (2019). Inhibition of JAK-STAT Signaling with Baricitinib Reduces Inflammation and Improves Cellular Homeostasis in Progeria Cells. Cells, 8(10), 1276. https://doi.org/10.3390/cells8101276