The Charcot–Marie Tooth Disease Mutation R94Q in MFN2 Decreases ATP Production but Increases Mitochondrial Respiration under Conditions of Mild Oxidative Stress

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Immunoblotting
2.3. Measurement of Mitochondrial Oxygen Consumption
2.4. Cell Proliferation Assay
2.5. Quantification of ATP Levels
2.6. Measurement of Total Cellular GSH
2.7. Lactate Measurement
2.8. RNA Isolation and PCR
2.9. Microscopy and Image Analysis
2.10. Statistical Analyses
3. Results
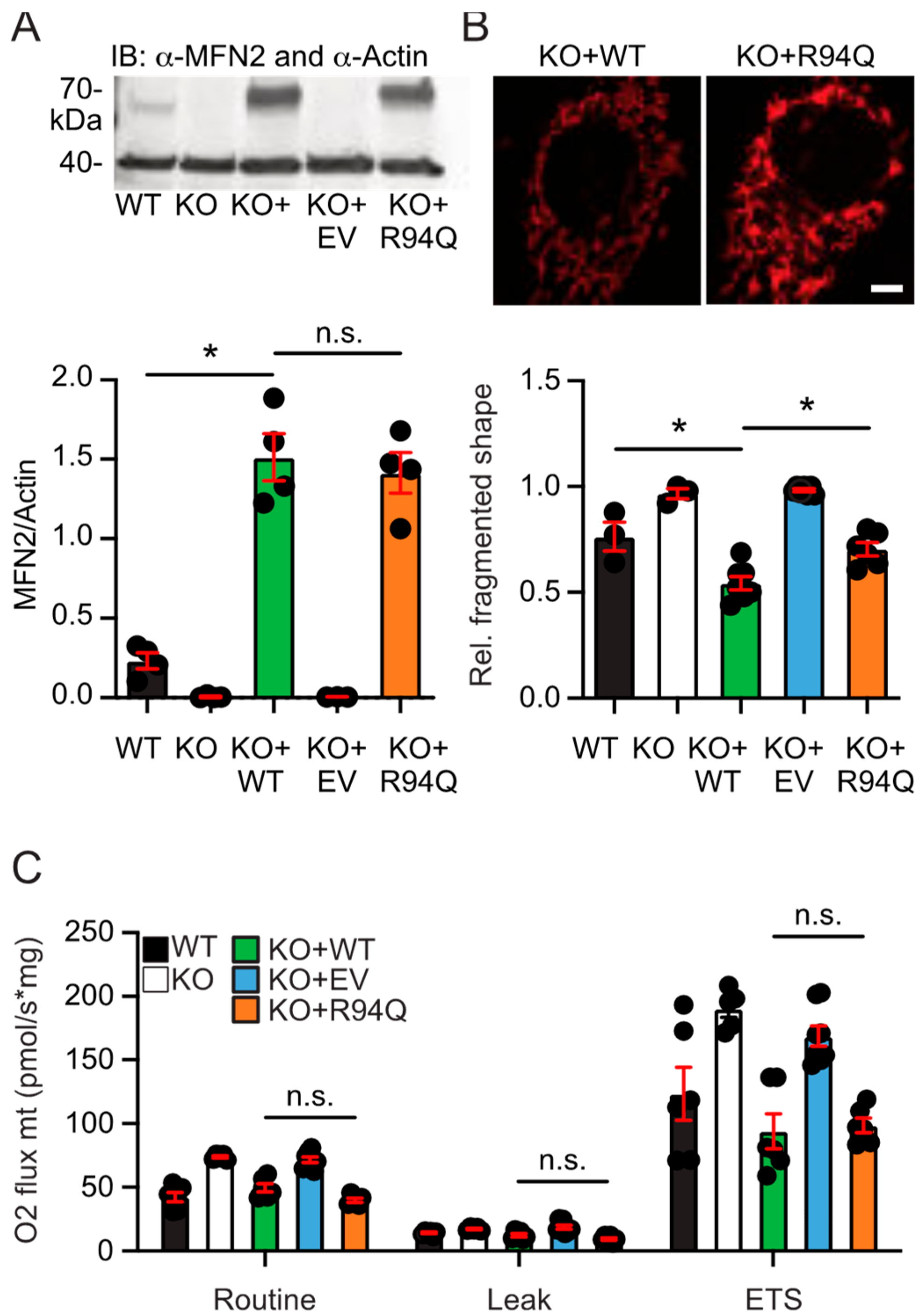
3.1. More Fragmented Mitochondria but Similar Basal Respiration in Cells Expressing Wildtype and Disease-Causing R94Q MFN2
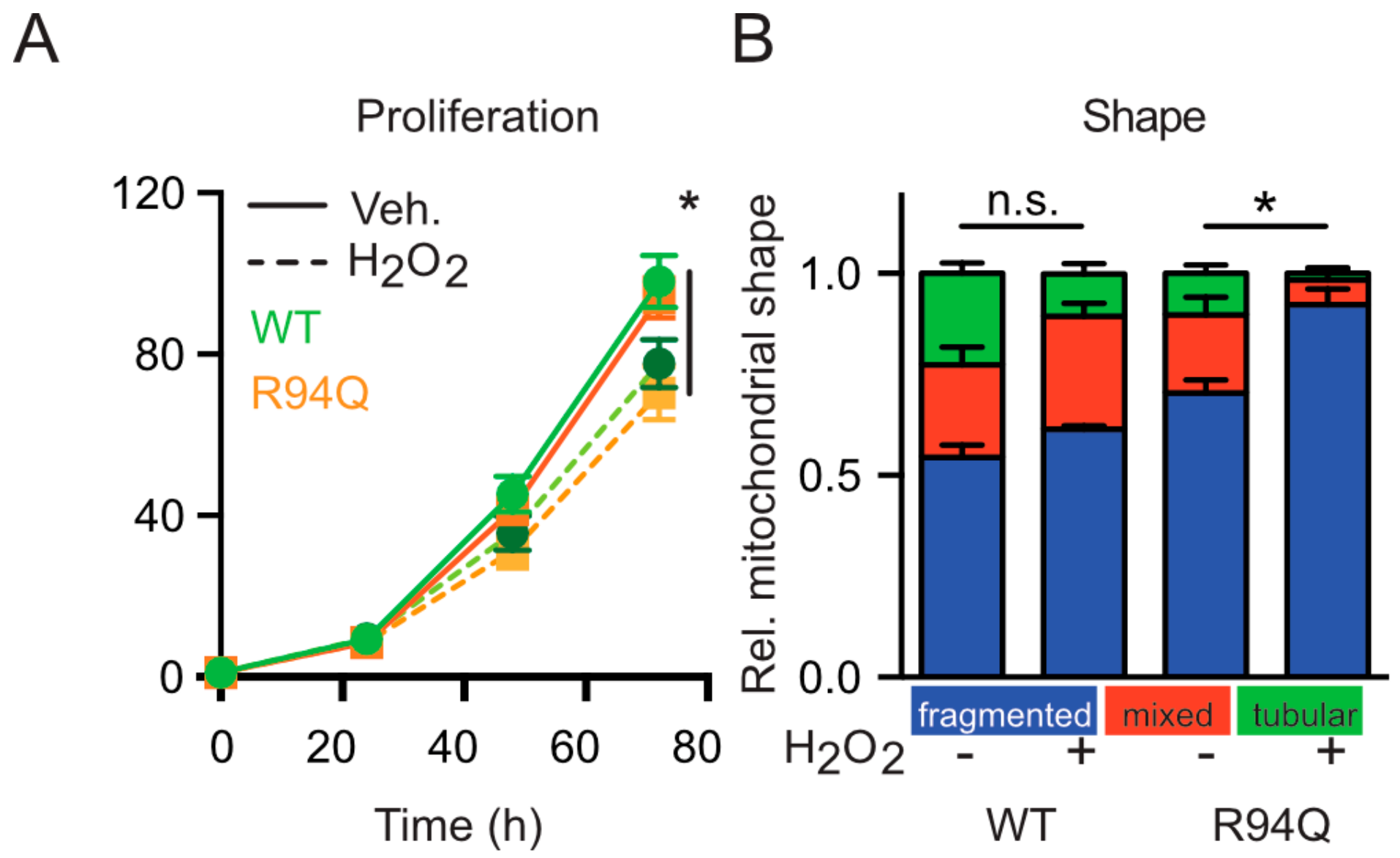
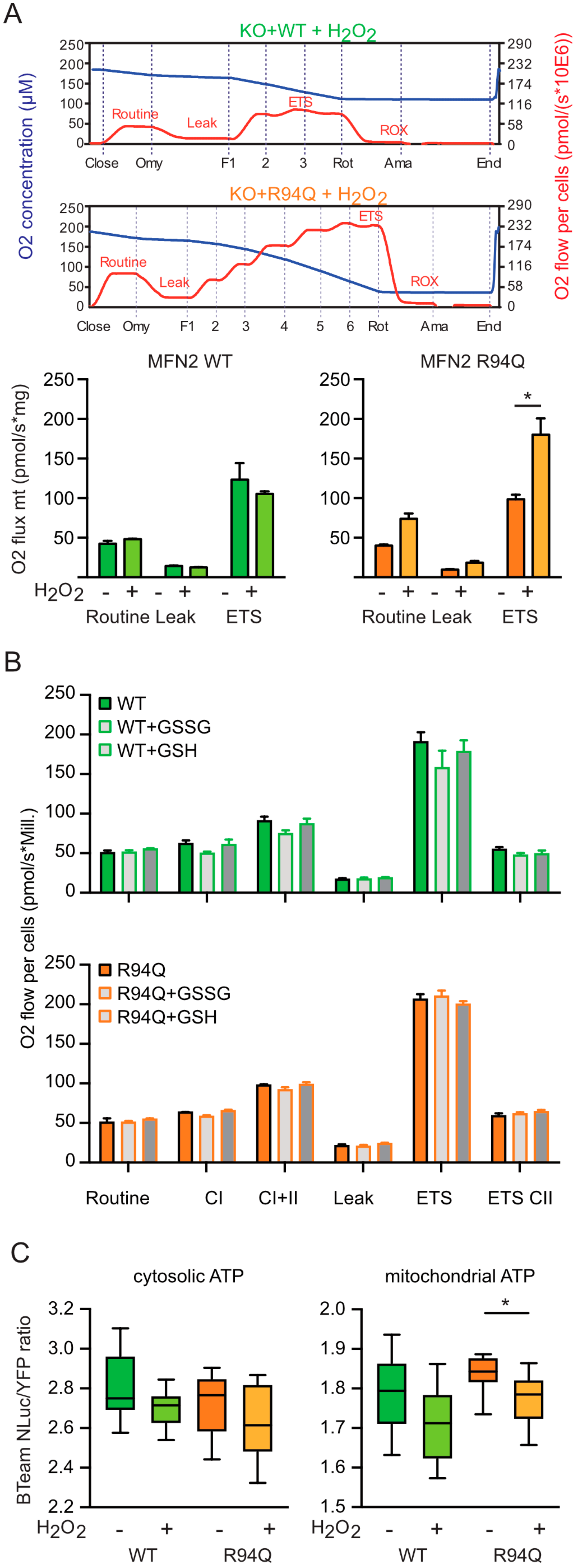
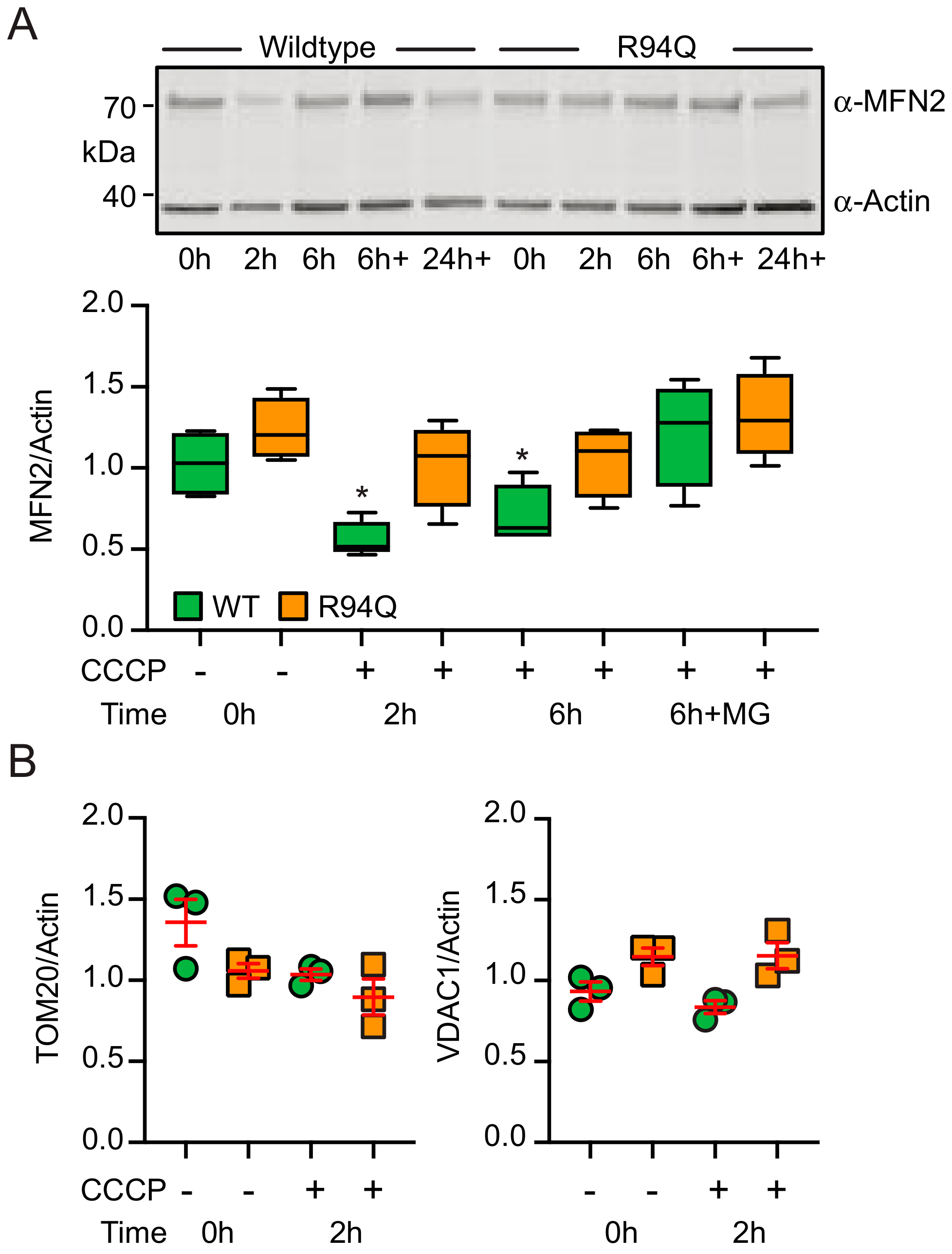
3.2. Mild Oxidative Stress Causes Increased Mitochondrial Fragmentation and Uncoupling in Cells Expressing R94Q MFN2
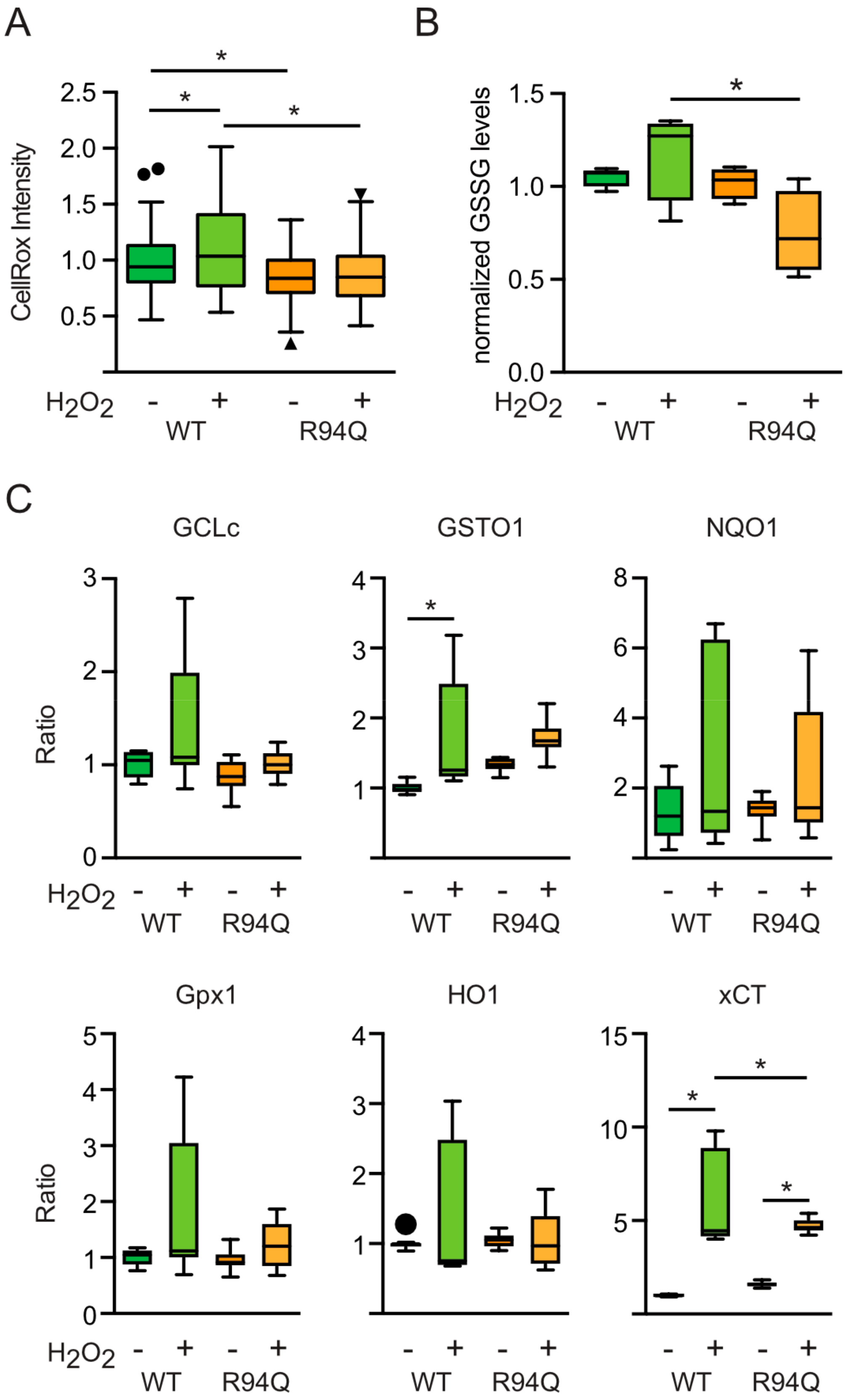
3.3. Rather Reduced and Not Increased Oxidative Stress in Cells Expressing R94Q MFN2
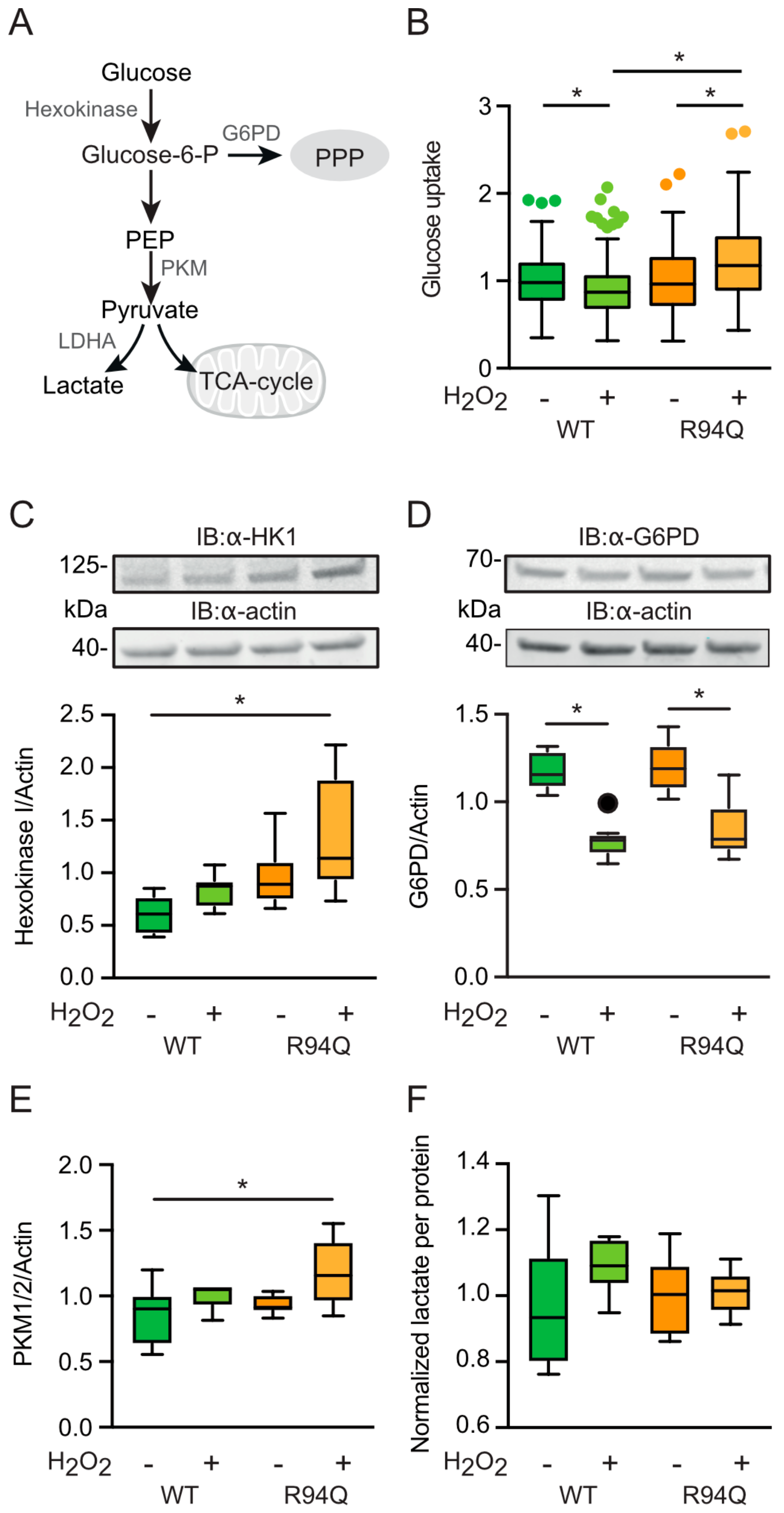
3.4. Increased Glucose Uptake and Fueling of Mitochondria with Pyruvate in R94Q Cells
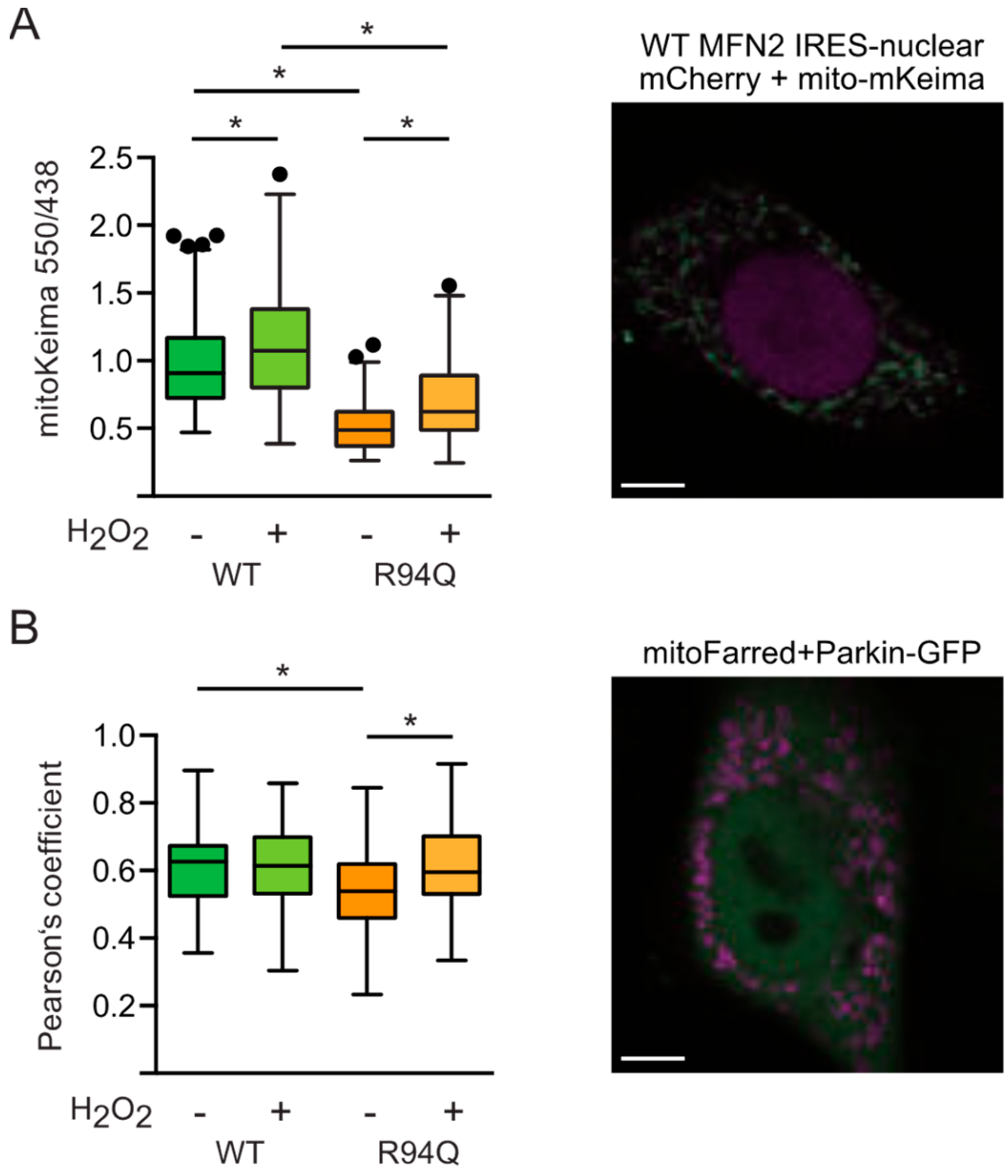
3.5. Reduced Mitophagy in R94Q Cells
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vallat, J.-M. Dominantly inherited peripheral neuropathies. J. Neuropathol. Exp. Neurol. 2003, 62, 699–714. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Calvo, J.; Funalot, B.; Ouvrier, R.A.; Lazaro, L.; Toutain, A.; De Mas, P.; Bouche, P.; Gilbert-Dussardier, B.; Arne-Bes, M.-C.; Carrière, J.-P.; et al. Genotype-phenotype correlations in Charcot-Marie-Tooth disease type 2 caused by mitofusin 2 mutations. Arch. Neurol. 2009, 66, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Santel, A.; Fuller, M.T. Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 2001, 114, 867–874. [Google Scholar] [PubMed]
- Bach, D.; Pich, S.; Soriano, F.X.; Vega, N.; Baumgartner, B.; Oriola, J.; Daugaard, J.R.; Lloberas, J.; Camps, M.; Zierath, J.R.; et al. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J. Biol. Chem. 2003, 278, 17190–17197. [Google Scholar] [CrossRef]
- Pich, S.; Bach, D.; Briones, P.; Liesa, M.; Camps, M.; Testar, X.; Palacín, M.; Zorzano, A. The Charcot-Marie-Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum. Mol. Genet. 2005, 14, 1405–1415. [Google Scholar] [CrossRef]
- Kawalec, M.; Boratyńska-Jasińska, A.; Beręsewicz, M.; Dymkowska, D.; Zabłocki, K.; Zabłocka, B. Mitofusin 2 Deficiency Affects Energy Metabolism and Mitochondrial Biogenesis in MEF Cells. PLoS ONE 2015, 10, e0134162. [Google Scholar] [CrossRef]
- Thaher, O.; Wolf, C.; Dey, P.N.; Pouya, A.; Wüllner, V.; Tenzer, S.; Methner, A. The thiol switch C684 in Mitofusin-2 mediates redox-induced alterations of mitochondrial shape and respiration. Neurochem. Int. 2017. [Google Scholar] [CrossRef]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Cosson, P.; Marchetti, A.; Ravazzola, M.; Orci, L. Mitofusin-2 Independent Juxtaposition of Endoplasmic Reticulum and Mitochondria: An Ultrastructural Study. PLoS ONE 2012, 7, e46293. [Google Scholar] [CrossRef]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc. Natl. Acad. Sci. USA 2015, 112, E2174–E2181. [Google Scholar] [CrossRef] [PubMed]
- Naon, D.; Zaninello, M.; Giacomello, M.; Varanita, T.; Grespi, F.; Lakshminaranayan, S.; Serafini, A.; Semenzato, M.; Herkenne, S.; Hernández-Alvarez, M.I.; et al. Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc. Natl. Acad. Sci. USA 2016, 113, 11249–11254. [Google Scholar] [CrossRef] [PubMed]
- Züchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Cartoni, R.; Arnaud, E.; Médard, J.-J.; Poirot, O.; Courvoisier, D.S.; Chrast, R.; Martinou, J.-C. Expression of mitofusin 2R94Q in a transgenic mouse leads to Charcot–Marie–Tooth neuropathy type 2A. Brain 2010, 133, 1460–1469. [Google Scholar] [CrossRef]
- Guillet, V.; Gueguen, N.; Cartoni, R.; Chevrollier, A.; Desquiret, V.; Angebault, C.; Amati-Bonneau, P.; Procaccio, V.; Bonneau, D.; Martinou, J.-C.; et al. Bioenergetic defect associated with mKATP channel opening in a mouse model carrying a mitofusin 2 mutation. FASEB J. 2011, 25, 1618–1627. [Google Scholar] [CrossRef]
- Van Hameren, G.; Campbell, G.; Deck, M.; Berthelot, J.; Gautier, B.; Quintana, P.; Chrast, R.; Tricaud, N. In vivo real-time dynamics of ATP and ROS production in axonal mitochondria show decoupling in mouse models of peripheral neuropathies. Acta Neuropathol. Commun. 2019, 7, 86. [Google Scholar] [CrossRef]
- Bernard-Marissal, N.; van Hameren, G.; Juneja, M.; Pellegrino, C.; Louhivuori, L.; Bartesaghi, L.; Rochat, C.; El Mansour, O.; Médard, J.-J.; Croisier, M.; et al. Altered interplay between endoplasmic reticulum and mitochondria in Charcot-Marie-Tooth type 2A neuropathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2328–2337. [Google Scholar] [CrossRef]
- Nickel, N.; Cleven, A.; Enders, V.; Lisak, D.; Schneider, L.; Methner, A. Androgen-inducible gene 1 increases the ER Ca(2+) content and cell death susceptibility against oxidative stress. Gene 2016, 586, 62–68. [Google Scholar] [CrossRef]
- Pfeiffer, A.; Schneider, J.; Bueno, D.; Dolga, A.; Voss, T.-D.; Lewerenz, J.; Wüllner, V.; Methner, A. Bcl-xL knockout attenuates mitochondrial respiration and causes oxidative stress that is compensated by pentose phosphate pathway activity. Free Radic. Biol. Med. 2017, 112, 350–359. [Google Scholar] [CrossRef]
- Yoshida, T.; Kakizuka, A.; Imamura, H. BTeam, a Novel BRET-based Biosensor for the Accurate Quantification of ATP Concentration within Living Cells. Sci. Rep. 2016, 6, 39618. [Google Scholar] [CrossRef]
- Pesta, D.; Gnaiger, E. High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol. Biol. 2012, 810, 25–58. [Google Scholar] [PubMed]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Shutt, T.; Geoffrion, M.; Milne, R.; McBride, H.M. The intracellular redox state is a core determinant of mitochondrial fusion. EMBO Rep. 2012, 13, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Baxter, P.S.; Hardingham, G.E. Adaptive regulation of the brain’s antioxidant defences by neurons and astrocytes. Free Radic. Biol. Med. 2016, 100, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef]
- Pfeiffer, A.; Jaeckel, M.; Lewerenz, J.; Noack, R.; Pouya, A.; Schacht, T.; Hoffmann, C.; Winter, J.; Schweiger, S.; Schäfer, M.K.E.; et al. Mitochondrial function and energy metabolism in neuronal HT22 cells resistant to oxidative stress. Br. J. Pharmacol. 2014, 171, 2147–2158. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef]
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006, 441, 1162–1166. [Google Scholar] [CrossRef]
- Katayama, H.; Kogure, T.; Mizushima, N.; Yoshimori, T.; Miyawaki, A. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem. Biol. 2011, 18, 1042–1052. [Google Scholar] [CrossRef]
- Leichert, L.I.; Dick, T.P. Incidence and physiological relevance of protein thiol switches. Biol. Chem. 2015, 396, 389–399. [Google Scholar] [CrossRef]
- Groitl, B.; Jakob, U. Thiol-based redox switches. Biochim. Biophys. Acta 2014, 1844, 1335–1343. [Google Scholar] [CrossRef] [PubMed]
- Milkovic, L.; Cipak Gasparovic, A.; Zarkovic, N. Overview on major lipid peroxidation bioactive factor 4-hydroxynonenal as pluripotent growth-regulating factor. Free Radic. Res. 2015, 49, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dorn, G.W. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Choubey, V.; Cagalinec, M.; Liiv, J.; Safiulina, D.; Hickey, M.A.; Kuum, M.; Liiv, M.; Anwar, T.; Eskelinen, E.-L.; Kaasik, A. BECN1 is involved in the initiation of mitophagy: It facilitates PARK2 translocation to mitochondria. Autophagy 2014, 10, 1105–1119. [Google Scholar] [CrossRef] [PubMed]
- Gong, G.; Song, M.; Csordás, G.; Kelly, D.P.; Matkovich, S.J.; Dorn, G.W., II. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 2015, 350, aad2459. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, F.; Ronchi, D.; Salani, S.; Nizzardo, M.; Fortunato, F.; Bordoni, A.; Stuppia, G.; Del Bo, R.; Piga, D.; Fato, R.; et al. Selective mitochondrial depletion, apoptosis resistance, and increased mitophagy in human Charcot-Marie-Tooth 2A motor neurons. Hum. Mol. Genet. 2016, 25, 4266–4281. [Google Scholar] [CrossRef] [PubMed]
- Sebastián, D.; Sorianello, E.; Segalés, J.; Irazoki, A.; Ruiz Bonilla, V.; Sala, D.; Planet, E.; Berenguer Llergo, A.; Muñoz, J.P.; Sánchez Feutrie, M.; et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 2016, 35, 1677–1693. [Google Scholar] [CrossRef]
- Zhou, Y.; Carmona, S.; Muhammad, A.K.M.G.; Bell, S.; Landeros, J.; Vazquez, M.; Ho, R.; Franco, A.; Lu, B.; Dorn, G.W.; et al. Restoring mitofusin balance prevents axonal degeneration in a Charcot-Marie-Tooth type 2A model. J. Clin. Investig. 2019, 130, 1756–1771. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wolf, C.; Zimmermann, R.; Thaher, O.; Bueno, D.; Wüllner, V.; Schäfer, M.K.E.; Albrecht, P.; Methner, A. The Charcot–Marie Tooth Disease Mutation R94Q in MFN2 Decreases ATP Production but Increases Mitochondrial Respiration under Conditions of Mild Oxidative Stress. Cells 2019, 8, 1289. https://doi.org/10.3390/cells8101289
Wolf C, Zimmermann R, Thaher O, Bueno D, Wüllner V, Schäfer MKE, Albrecht P, Methner A. The Charcot–Marie Tooth Disease Mutation R94Q in MFN2 Decreases ATP Production but Increases Mitochondrial Respiration under Conditions of Mild Oxidative Stress. Cells. 2019; 8(10):1289. https://doi.org/10.3390/cells8101289
Chicago/Turabian StyleWolf, Christina, Rahel Zimmermann, Osamah Thaher, Diones Bueno, Verena Wüllner, Michael K.E. Schäfer, Philipp Albrecht, and Axel Methner. 2019. "The Charcot–Marie Tooth Disease Mutation R94Q in MFN2 Decreases ATP Production but Increases Mitochondrial Respiration under Conditions of Mild Oxidative Stress" Cells 8, no. 10: 1289. https://doi.org/10.3390/cells8101289
APA StyleWolf, C., Zimmermann, R., Thaher, O., Bueno, D., Wüllner, V., Schäfer, M. K. E., Albrecht, P., & Methner, A. (2019). The Charcot–Marie Tooth Disease Mutation R94Q in MFN2 Decreases ATP Production but Increases Mitochondrial Respiration under Conditions of Mild Oxidative Stress. Cells, 8(10), 1289. https://doi.org/10.3390/cells8101289