Intracellular Redox-Balance Involvement in Temozolomide Resistance-Related Molecular Mechanisms in Glioblastoma

 , ,
, ,
Abstract
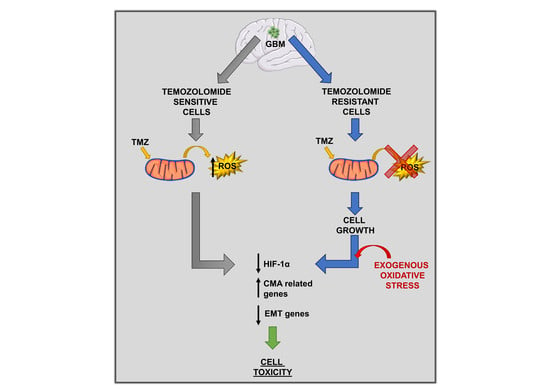
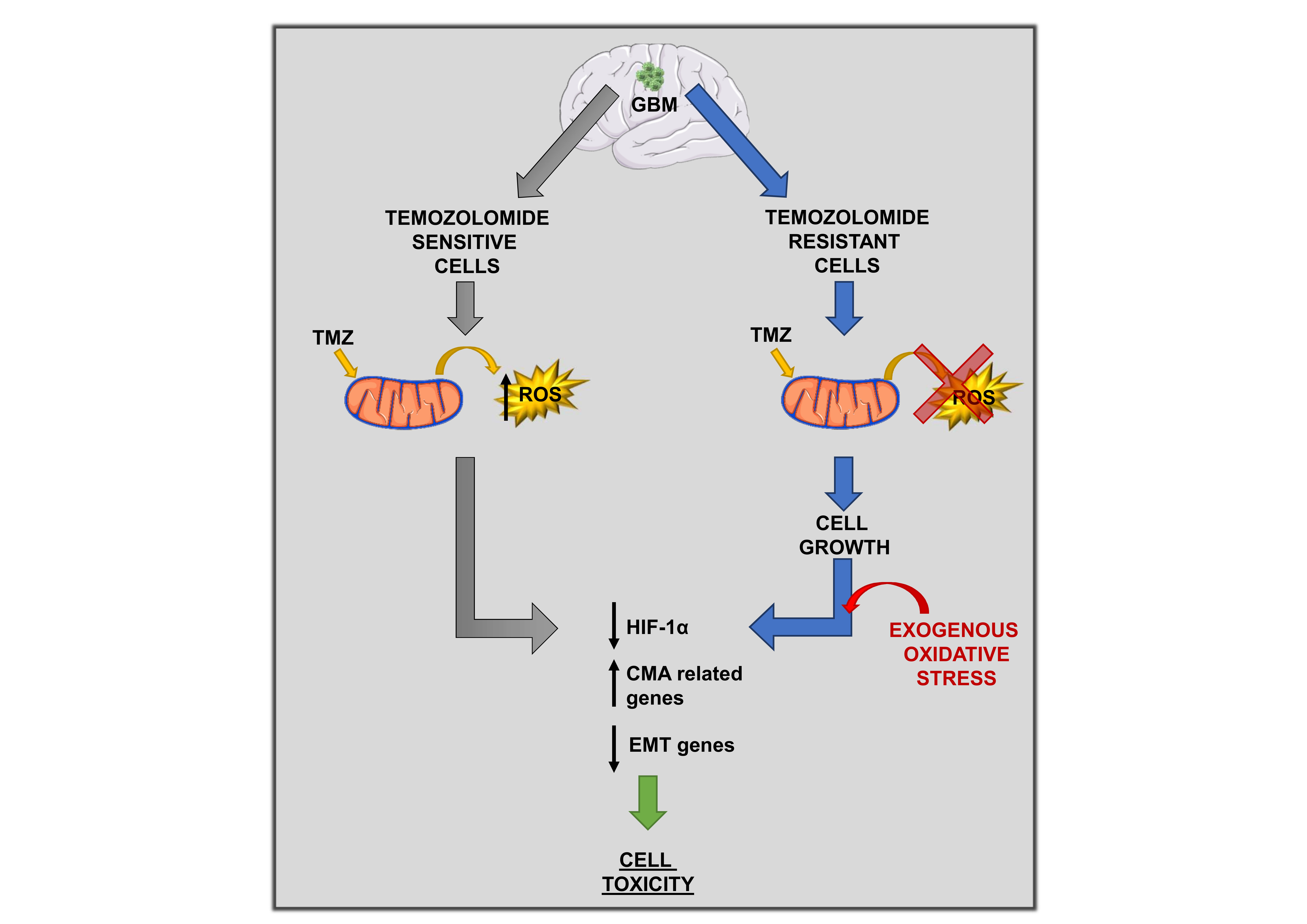
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. Biochemical Assays
2.3. RNA Extraction and Real-Time PCR
2.4. Wounding Assay
2.5. Protein Studies
2.6. Mitochondrial Complex Activity Studies
2.7. Statistical Analyses
3. Results
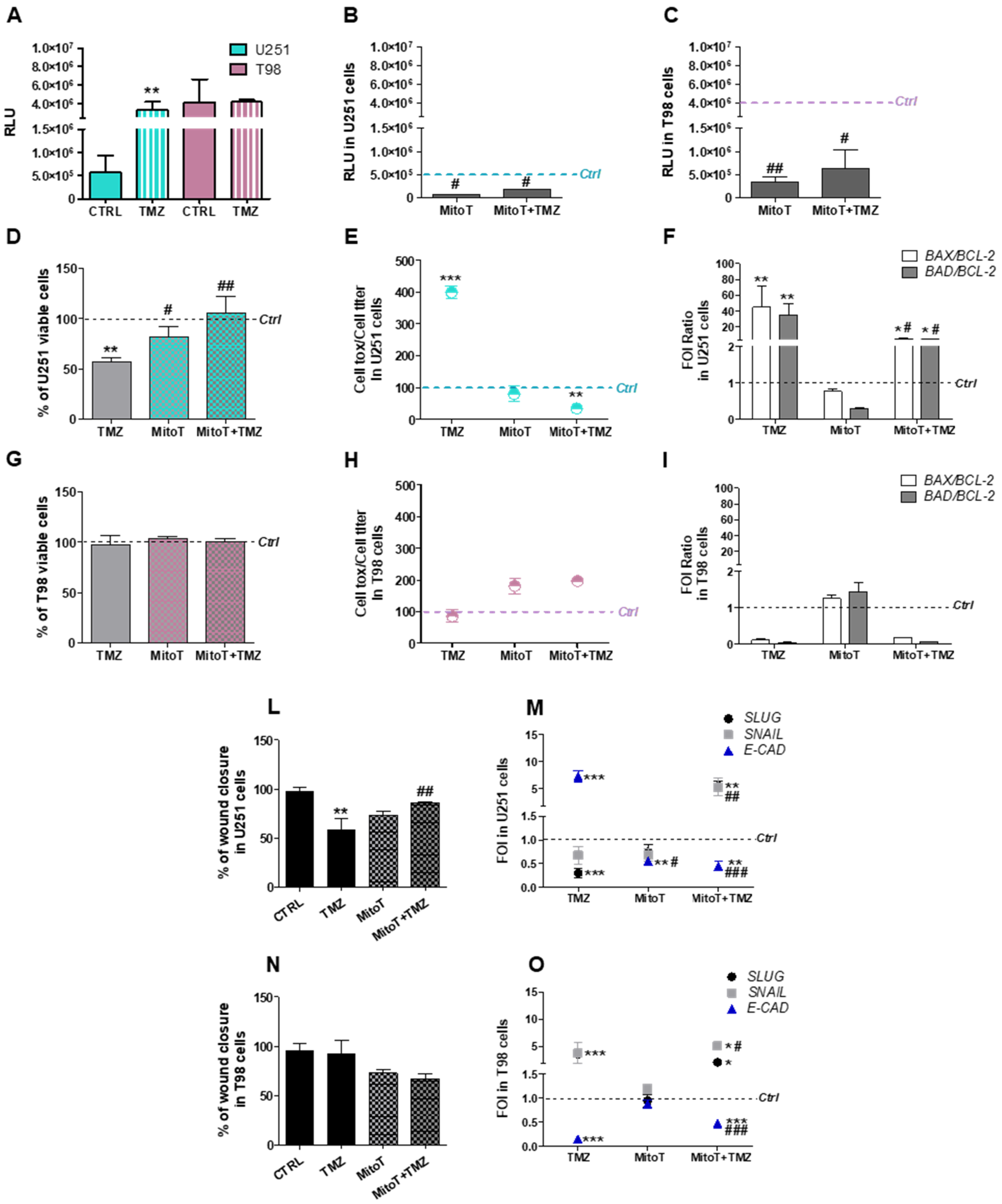
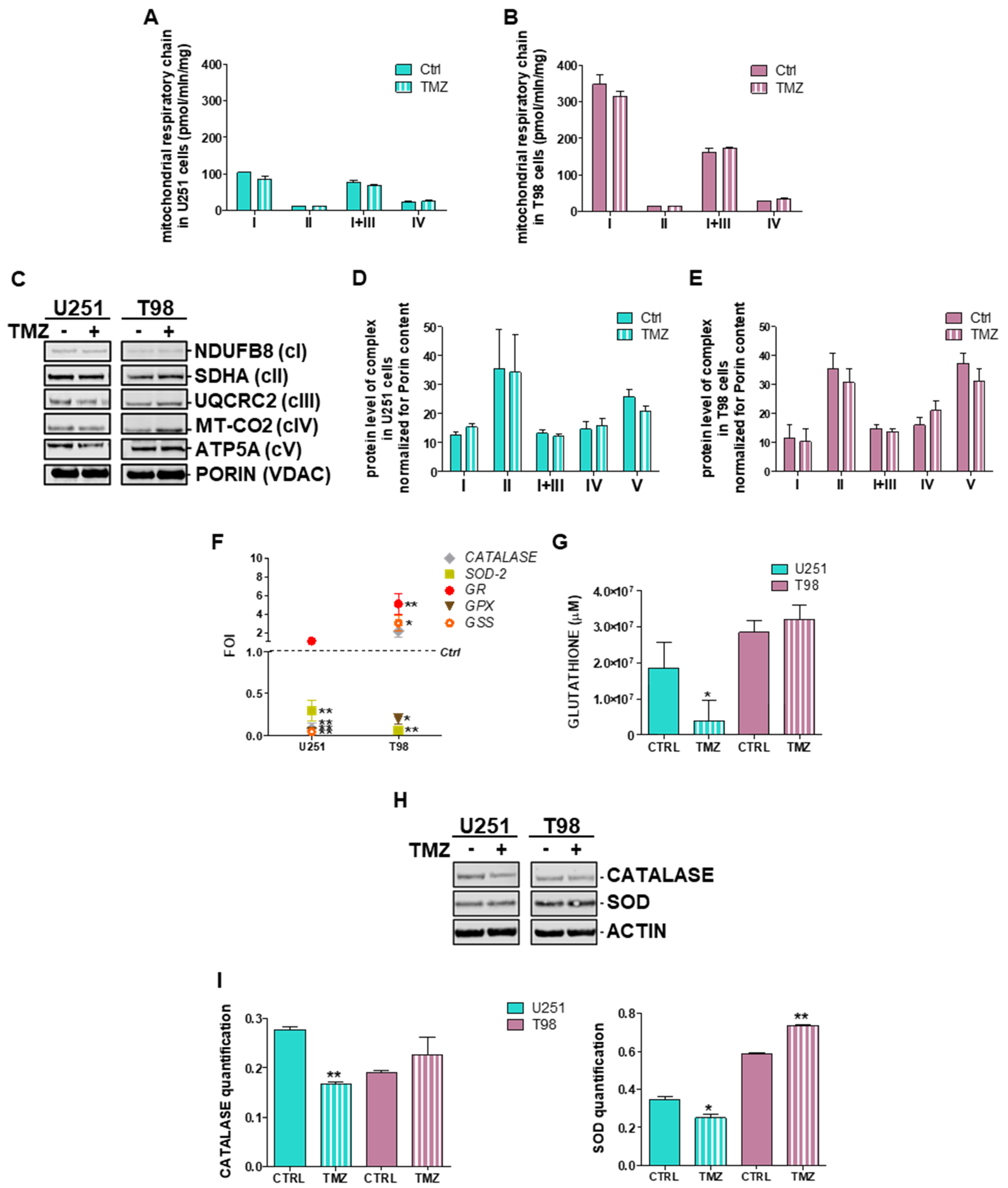
3.1. Mitochondrial ROS are Crucial for TMZ Responsiveness in U251 Cells
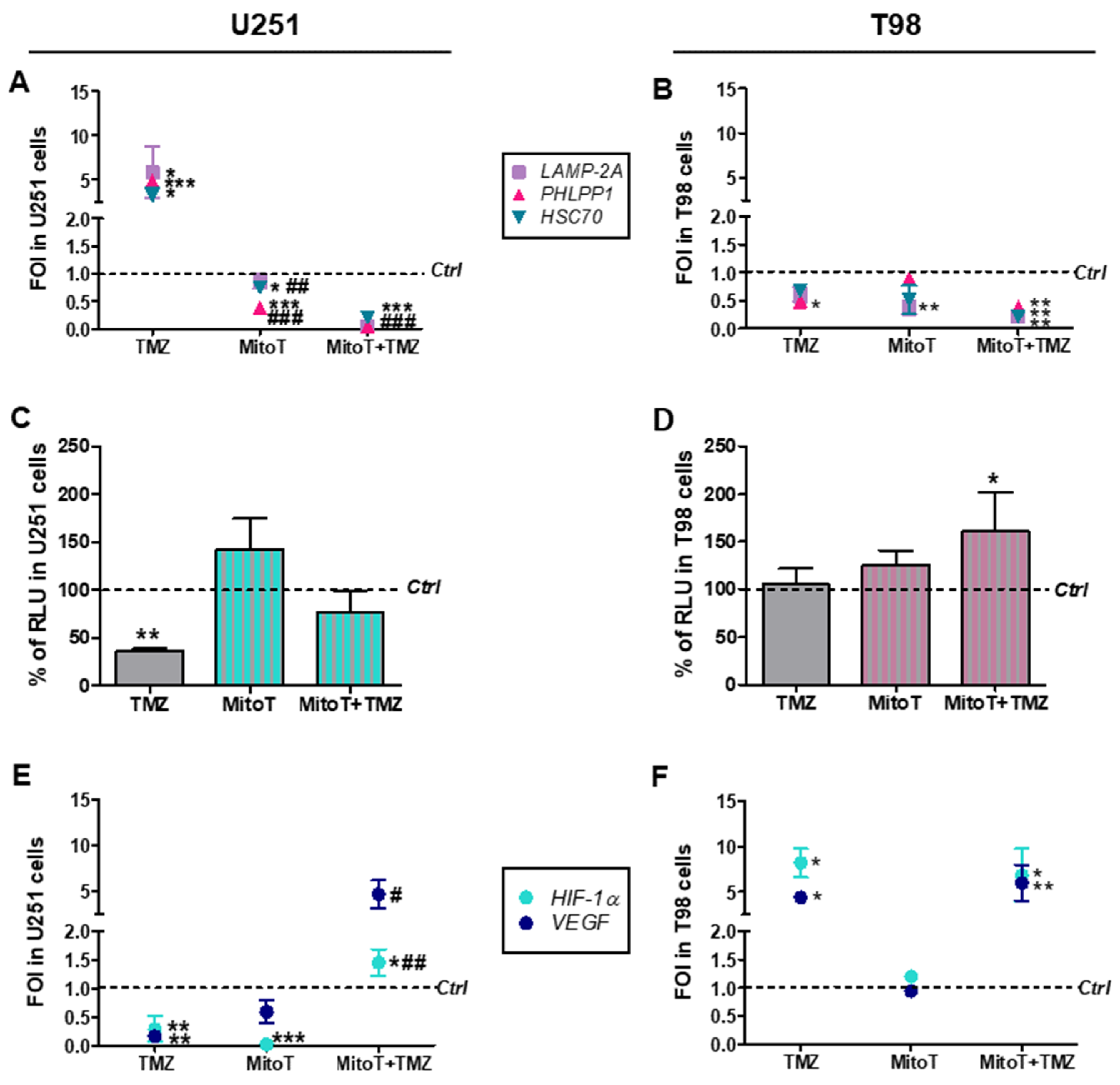
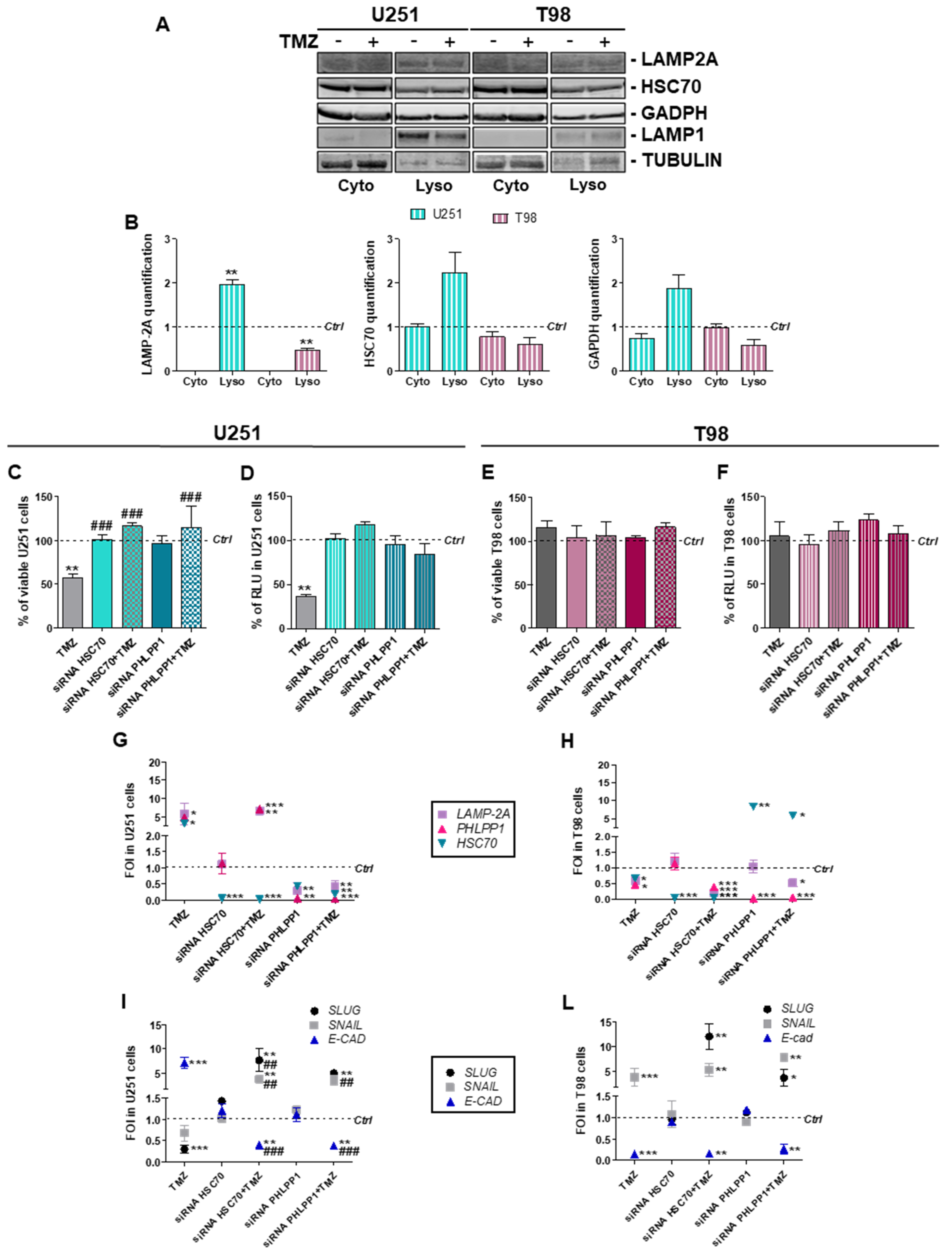
3.2. Involvement of Chaperone Mediated Autophagy in GBM Responsiveness to TMZ
3.3. Chaperone Mediated Autophagy Activity is Dependent Upon Different Proteins
3.4. Deregulation of Redox-Homeostasis is Involved in GBM Responsiveness to TMZ
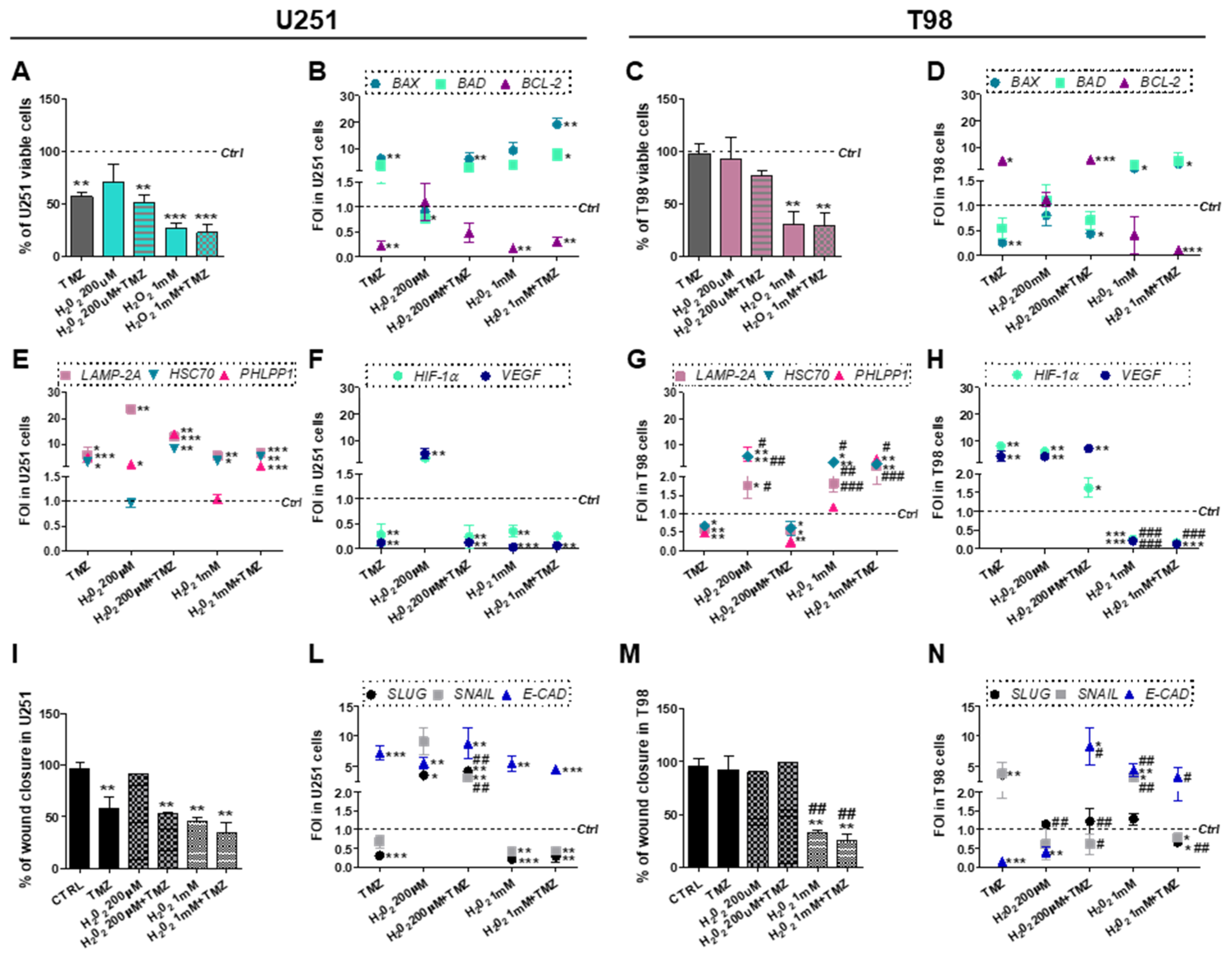
3.5. Chemical-Induced Oxidative Stress Weakens the Resistance of T98 Cells to TMZ
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. New Engl. J. Nucl. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Valtorta, S.; Dico, A.L.; Raccagni, I.; Gaglio, D.; Belloli, S.; Politi, L.S.; Martelli, C.; Diceglie, C.; Bonanomi, M.; Ercoli, G.; et al. Metformin and temozolomide, a synergic option to overcome resistance in glioblastoma multiforme models. Oncotarget 2017, 8, 113090–113104. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, Z.; Shu, F.; Jin, Y.; Liu, H.; Wang, Q.; Yang, Y. Activation of AMP-activated protein kinase by temozolomide contributes to apoptosis in glioblastoma cells via p53 activation and mTORC1 inhibition. J. Biol. Chem. 2010, 285, 40461–40471. [Google Scholar] [CrossRef]
- Yan, Y.; Xu, Z.; Dai, S.; Qian, L.; Sun, L.; Gong, Z. Targeting autophagy to sensitive glioma to temozolomide treatment. J. Exp. Clin. Cancer Res. 2016, 35, 23. [Google Scholar] [CrossRef]
- Roos, W.P.; Batista, L.F.Z.; Naumann, S.C.; Wick, W.; Weller, M.; Menck, C.F.M.; Kaina, B. Apoptosis in malignant glioma cells triggered by the temozolomide-induced DNA lesion O6-methylguanine. Oncogene 2007, 26, 186–197. [Google Scholar] [CrossRef]
- Lo Dico, A.; Martelli, C.; Valtorta, S.; Raccagni, I.; Diceglie, C.; Belloli, S.; Gianelli, U.; Vaira, V.; Politi, L.S.; Bosari, S.; et al. Identification of imaging biomarkers for the assessment of tumour response to different treatments in a preclinical glioma model. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 1093–1105. [Google Scholar] [CrossRef]
- Lo Dico, A.; Martelli, C.; Diceglie, C.; Lucignani, G.; Ottobrini, L. Hypoxia-Inducible Factor-1α Activity as a Switch for Glioblastoma Responsiveness to Temozolomide. Front. Oncol. 2018, 8, 249–259. [Google Scholar] [CrossRef]
- Lo Dico, A.; Valtorta, S.; Ottobrini, L.; Moresco, R.M. Role of Metformin and AKT Axis Modulation in the Reversion of Hypoxia Induced TMZ-Resistance in Glioma Cells. Front. Oncol. 2019, 9, 463. [Google Scholar] [CrossRef]
- Kahlert, U.D.; Nikkhah, G.; Maciaczyk, J. Epithelial-to-mesenchymal(-like) transition as a relevant molecular event in malignant gliomas. Cancer Lett. 2013, 331, 131–138. [Google Scholar] [CrossRef]
- Das, C.K.; Mandal, M.; Kögel, D. Pro-survival autophagy and cancer cell resistance to therapy. Cancer Metastasis Rev. 2018, 37, 749–766. [Google Scholar] [CrossRef]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef]
- Colella, B.; Faienza, F.; Di Bartolomeo, S.; Colella, B.; Faienza, F.; Di Bartolomeo, S. EMT Regulation by Autophagy: A New Perspective in Glioblastoma Biology. Cancers (Basel) 2019, 11, 312. [Google Scholar] [CrossRef] [PubMed]
- Trejo-Solís, C.; Serrano-Garcia, N.; Escamilla-Ramírez, Á.; Castillo-Rodríguez, R.; Jimenez-Farfan, D.; Palencia, G.; Calvillo, M.; Alvarez-Lemus, M.; Flores-Nájera, A.; Cruz-Salgado, A.; et al. Autophagic and Apoptotic Pathways as Targets for Chemotherapy in Glioblastoma. Int. J. Mol. Sci. 2018, 19, 3773. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Buccarelli, M.; Marconi, M.; Pacioni, S.; De Pascalis, I.; Giorgio D’alessandris, Q.; Martini, M.; Ascione, B.; Malorni, W.; Larocca, L.M.; Pallini, R.; et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 2018, 9, 841–857. [Google Scholar] [CrossRef]
- Kriel, J.; Müller-Nedebock, K.; Maarman, G.; Mbizana, S.; Ojuka, E.; Klumperman, B.; Loos, B. Coordinated autophagy modulation overcomes glioblastoma chemoresistance through disruption of mitochondrial bioenergetics. Scientific Reports 2018, 8, 10348. [Google Scholar] [CrossRef]
- Ding, Z.-B.; Fu, X.-T.; Shi, Y.-H.; Zhou, J.; Peng, Y.-F.; Liu, W.-R.; Shi, G.-M.; Gao, Q.; Wang, X.-Y.; Song, K.; et al. Lamp2a is required for tumor growth and promotes tumor recurrence of hepatocellular carcinoma. Int. J. Oncol. 2016, 49, 2367–2376. [Google Scholar] [CrossRef]
- Saha, T. LAMP2A overexpression in breast tumors promotes cancer cell survival via chaperone-mediated autophagy. Autophagy 2012, 8, 1643–1656. [Google Scholar] [CrossRef]
- Cabello, C.M.; Bair, W.B.; Wondrak, G.T. Experimental therapeutics: Targeting the redox Achilles heel of cancer. Curr. Opin. Investig. Drugs 2007, 8, 1022–1037. [Google Scholar]
- Chiu, W.-T.; Shen, S.-C.; Chow, J.-M.; Lin, C.-W.; Shia, L.-T.; Chen, Y.-C. Contribution of reactive oxygen species to migration/invasion of human glioblastoma cells U87 via ERK-dependent COX-2/PGE2 activation. Neurobiol. Dis. 2010, 37, 118–129. [Google Scholar] [CrossRef]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Glutathione and its role in cellular functions. Free Radic. Biol. Med. 1999, 27, 916–921. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Landriscina, M.; Maddalena, F.; Laudiero, G.; Esposito, F. Adaptation to Oxidative Stress, Chemoresistance, and Cell Survival. Antioxid. Redox Signal. 2009, 11, 2701–2716. [Google Scholar] [CrossRef]
- Laurent, A.; Nicco, C.; Chéreau, C.; Goulvestre, C.; Alexandre, J.; Alves, A.; Lévy, E.; Goldwasser, F.; Panis, Y.; Soubrane, O.; et al. Controlling tumor growth by modulating endogenous production of reactive oxygen species. Cancer Res. 2005, 65, 948–956. [Google Scholar]
- Lo Dico, A.; Valtorta, S.; Martelli, C.; Belloli, S.; Gianelli, U.; Tosi, D.; Bosari, S.; Degrassi, A.; Russo, M.; Raccagni, I.; et al. Validation of an engineered cell model for in vitro and in vivo hif-1α evaluation by different imaging modalities. Mol. Imaging Biol. 2014, 16, 210–223. [Google Scholar] [CrossRef]
- Monzio Compagnoni, G.; Kleiner, G.; Bordoni, A.; Fortunato, F.; Ronchi, D.; Salani, S.; Guida, M.; Corti, C.; Pichler, I.; Bergamini, C.; et al. Mitochondrial dysfunction in fibroblasts of Multiple System Atrophy. Biochim. Biophys. Acta-Mol. Basis Dis. 2018, 1864, 3588–3597. [Google Scholar] [CrossRef]
- Kiffin, R.; Christian, C.; Knecht, E.; Cuervo, A.M. Activation of chaperone-mediated autophagy during oxidative stress. Mol. Biol. Cell 2004, 15, 4829–4840. [Google Scholar] [CrossRef]
- Baas, A.S.; Berk, B.C. Differential Activation of Mitogen-Activated Protein Kinases by H2O2 and O2− in Vascular Smooth Muscle Cells. Circ. Res. 1995, 77, 29–36. [Google Scholar] [CrossRef]
- Salazar-Ramiro, A.; Ramírez-Ortega, D.; Pérez de la Cruz, V.; Hérnandez-Pedro, N.Y.; González-Esquivel, D.F.; Sotelo, J.; Pineda, B. Role of Redox Status in Development of Glioblastoma. Front. Immunol. 2016, 7, 156. [Google Scholar] [CrossRef]
- Chen, H.; Wang, J.; Liu, Z.; Yang, H.; Zhu, Y.; Zhao, M.; Liu, Y.; Yan, M. Mitochondrial DNA depletion causes decreased ROS production and resistance to apoptosis. Int. J. Mol. Med. 2016, 38, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Mei, Z.; He, Y.; Feng, J.; Shi, J.; Du, Y.; Qian, L.; Huang, Q.; Jie, Z. MicroRNA-141 promotes the proliferation of non-small cell lung cancer cells by regulating expression of PHLPP1 and PHLPP2. FEBS Lett. 2014, 588, 3055–3061. [Google Scholar] [CrossRef] [PubMed]
- Teng, D.-C.; Sun, J.; An, Y.-Q.; Hu, Z.-H.; Liu, P.; Ma, Y.-C.; Han, B.; Shi, Y. Role of PHLPP1 in inflammation response: Its loss contributes to gliomas development and progression. Int. Immunopharmacol. 2016, 34, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Arias, E.; Koga, H.; Diaz, A.; Mocholi, E.; Patel, B.; Cuervo, A.M. Lysosomal mTORC2/PHLPP1/Akt Regulate Chaperone-Mediated Autophagy. Mol. Cell 2015, 59, 270–284. [Google Scholar] [CrossRef]
- Zhu, Z.; Du, S.; Du, Y.; Ren, J.; Ying, G.; Yan, Z. Glutathione reductase mediates drug resistance in glioblastoma cells by regulating redox homeostasis. J. Neurochem. 2018, 144, 93–104. [Google Scholar] [CrossRef]
- Smith, P.S.; Zhao, W.; Robbins, M. Knocking down catalase leads to increased ROS production and proliferation in glioma cells. Cancer Res. 2006, 66, 1037. [Google Scholar]
- Chang, K.-Y.; Hsu, T.-I.; Hsu, C.-C.; Tsai, S.-Y.; Liu, J.-J.; Chou, S.-W.; Liu, M.-S.; Liou, J.-P.; Ko, C.-Y.; Chen, K.-Y.; et al. Specificity protein 1-modulated superoxide dismutase 2 enhances temozolomide resistance in glioblastoma, which is independent of O6-methylguanine-DNA methyltransferase. Redox Biol. 2017, 13, 655–664. [Google Scholar] [CrossRef]
- Iida, M.; Sunaga, S.; Hirota, N.; Kuribayashi, N.; Sakagami, H.; Takeda, M.; Matsumoto, K. Effect of glutathione-modulating compounds on hydrogen-peroxide-induced cytotoxicity in human glioblastoma and glioma cell lines. J. Cancer Res. Clin. Oncol. 1997, 123, 619–622. [Google Scholar] [CrossRef]
- Rocha, C.R.R.; Garcia, C.C.M.; Vieira, D.B.; Quinet, A.; de Andrade-Lima, L.C.; Munford, V.; Belizário, J.E.; Menck, C.F.M. Glutathione depletion sensitizes cisplatin- and temozolomide-resistant glioma cells in vitro and in vivo. Cell Death Dis. 2014, 5, e1505. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| BAX | ATGGACGGGTCCGGGGAG | ATCCAGCCCAACAGCCGC |
| BAD | CCCAGAGTTTGAGCCGAGTG | CCCATCCCTTCGTCGTCCT |
| BCL-2 | GATTGTGGCCTTCTTTGAG | CAAACTGAGCAGAGTCTTC |
| PHLPP1 | CCTACCTTCTCCAGTGCACT | CCAGCAGTTCCAAGTTTCCT |
| LAMP2A | TGCTGGCTACCATGGGGCTG | GCAGCTGCCTGTGGAGTGAGT |
| HSC70 | ATTGATCTTGGCACCACCTA | GGGTGCAGGAGGTATGCCTGTGA |
| HIF-1α | TGATTGCATCTCCATCTCCTAC | GACTCAAAGCGACAGATAACACG |
| VEGF | CGAGGGCCTGGAGTGTGT | CGCATAATCTGCATGGTGATG |
| SNAIL | GCGAGCTGCAGGACTCTAAT | CCCGCAATGGTCCACAAAAC |
| SLUG | CATGCCTGTCATACCACAAC | GGTGTCAGATGGAGGAGG |
| E-CAD | GATCAAGTCAAGCGTGAGTCG | AGCCTCT CAATGGCGAACAC |
| SOD-2 | TTAACGCGCAGATCATGCA | GGTGGCGTTGAGATTGTTCA |
| CATALASE | TAAGACTGACCAGGGCA | CAAACCTTGGTGAGATCGAA |
| GR | AACATCCCAACTGTGGTCTTCAGC | TTGGTAACTGCGTGATACATCGGG |
| GPX | CGCAACGATGTTGCCTGGAACTTT | AGGCTCGATGTCAATGGTCTGGAA |
| GSS | ATGCTGTGCAGATGGACTTCAACC | TGGATGTCAAACAGACGAGCGGTA |
| β-ACTIN | TCAAGATCATTGCTCCTCCTG | CCAGAGGCGTACAGGGATAG |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lo Dico, A.; Salvatore, D.; Martelli, C.; Ronchi, D.; Diceglie, C.; Lucignani, G.; Ottobrini, L. Intracellular Redox-Balance Involvement in Temozolomide Resistance-Related Molecular Mechanisms in Glioblastoma. Cells 2019, 8, 1315. https://doi.org/10.3390/cells8111315
Lo Dico A, Salvatore D, Martelli C, Ronchi D, Diceglie C, Lucignani G, Ottobrini L. Intracellular Redox-Balance Involvement in Temozolomide Resistance-Related Molecular Mechanisms in Glioblastoma. Cells. 2019; 8(11):1315. https://doi.org/10.3390/cells8111315
Chicago/Turabian StyleLo Dico, Alessia, Daniela Salvatore, Cristina Martelli, Dario Ronchi, Cecilia Diceglie, Giovanni Lucignani, and Luisa Ottobrini. 2019. "Intracellular Redox-Balance Involvement in Temozolomide Resistance-Related Molecular Mechanisms in Glioblastoma" Cells 8, no. 11: 1315. https://doi.org/10.3390/cells8111315
APA StyleLo Dico, A., Salvatore, D., Martelli, C., Ronchi, D., Diceglie, C., Lucignani, G., & Ottobrini, L. (2019). Intracellular Redox-Balance Involvement in Temozolomide Resistance-Related Molecular Mechanisms in Glioblastoma. Cells, 8(11), 1315. https://doi.org/10.3390/cells8111315