Abstract
Intestinal dysbiosis has recently become known as an important driver of gastrointestinal and liver disease. It remains poorly understood, however, how gastrointestinal microbes bypass the intestinal mucosa and enter systemic circulation to enact an inflammatory immune response. In the context of chronic liver disease (CLD), insults that drive hepatic inflammation and fibrogenesis (alcohol, fat) can drastically increase intestinal permeability, hence flooding the liver with gut-derived microbiota. Consequently, this may result in exacerbated liver inflammation and fibrosis through activation of liver-resident Kupffer and stellate cells by bacterial, viral, and fungal antigens transported to the liver via the portal vein. This review summarizes the current understanding of microbial translocation in CLD, the cell-specific hepatic response to intestinal antigens, and how this drives the development and progression of hepatic inflammation and fibrosis. Further, we reviewed current and future therapies targeting intestinal permeability and the associated, potentially harmful anti-microbial immune response with respect to their potential in terms of limiting the development and progression of liver fibrosis and end-stage cirrhosis.
1. Introduction
The progressive accumulation of extracellular matrix (ECM) in the liver, termed fibrosis, is the result of chronic liver damage due to a variety of insults: viral hepatitis (hepatitis B virus (HBV) and hepatitis C virus (HCV)), alcohol or fatty liver disease, drug-induced liver damage, or autoimmunity. While the prevalence of HCV- and HBV-mediated fibrosis has been on the decline since the advent of direct acting antivirals (DAAs) for HCV and improved vaccination/education strategies for HBV, other etiologies are on the rise [1]. The prevalence of non-alcoholic fatty liver disease (NAFLD) and its inflammatory form, non-alcoholic steatohepatitis (NASH), have increased in step with the obesity epidemic and are significant contributors to fibrosis, particularly in Western countries [1]. Indeed, while the prevalence of viral hepatitis dropped between 2000 and 2017, alcoholic cirrhosis increased by 16% and NASH cirrhosis by 33% [1].
The term gut–liver axis defines a bidirectional interaction between the gastrointestinal tract and the liver [2]. While the liver contributes bile acids, IgA, and antimicrobial peptides to the gut via the biliary tract, the portal vein transports gastrointestinal metabolites from the gut into the liver [2]. In the absence of disease, the mucosal barriers within the intestinal tract remain intact, preventing the transport of luminal microbes into the liver. In chronic liver disease (CLD) however, the intestinal barrier is impaired as a result of lifestyle choices (e.g., alcohol or obesity) or portal hypertension secondary to advanced fibrosis/cirrhosis, resulting in the translocation of microbes and their products into the blood.
Translocation of gut-derived antigens into the portal circulation enacts a potent inflammatory response in the liver, which has been well described in alcoholic and fatty liver disease, as well as liver cirrhosis [2]. These antigens not only drive hepatic inflammation and progressive fibrosis, but also contribute to mortality in end-stage liver disease due to their role in secondary infections such as spontaneous bacterial peritonitis (SBP) and hepatic encephalopathy [3]. While intestinal permeability is not the primary driver of liver inflammation and fibrosis, it has become evident that the inflammatory response to microbial antigens as a result of increased intestinal permeability strongly influences the progression of disease.
This review will focus on the multi-systemic nature of the gut–liver axis in health and disease. We have described (1) the intestinal barriers and mechanisms by which they become impaired in chronic liver disease, (2) the contribution of microbial antigens to liver inflammation and fibrosis, and (3), current therapies used to prevent either intestinal permeability or the hepatic inflammatory and fibrotic response.
2. Gut Microbiota and Bacterial Translocation in Chronic Liver Diseases
2.1. Gut Microbiota in Chronic Liver Diseases
The human gastrointestinal (GI) tract is estimated to contain more than 1014 microorganisms which collectively form the gut microbiota [4]. High motility and acidity within the esophagus and stomach limit colonization, however microbial numbers steadily increase throughout the small bowel, reaching the highest density in the colon where 1012 bacteria can be found in every gram of dry feces [5].
The intestinal microbiota is composed primarily of bacteria (60% of dry fecal mass), but is also abundant in archaea, eukarya, and viral species [6]. Sequencing of 16S ribosomal DNA from mucosal and stool samples has shown that Firmicutes and Bacteroidetes are the two most abundant phyla in human feces, followed by Proteobacteria, Actinobacteria, Fusobacteria, and Verrucomicrobia species [7]. Mucosal and fecal microbiota harbor distinct microbial profiles, characterized by an abundance of Bacteroidetes in stool, while human colonic crypts are colonized mainly with Firmicutes and Proteobacteria [8].
The non-bacterial intestinal microbiota contributes significantly to health and disease, but has been largely unappreciated to date. Eukaryotes account for less than 0.03% of fecal microbes and are primarily composed of 200–300 fungal species [9]. The study of intestinal viromes has been limited due to challenges in isolation and culture; however, recent metagenomic analyses have revealed that bacteriophages comprise ~90% of the gut virome and contribute significantly to bacterial dynamics [10].
The gut microbiota is essential for proper digestion and, furthermore, plays an important role in facilitating gut immune responses against potential pathogens. Indeed, commensal Bacteroides and Lactobacillus spp. can stimulate the release and activation of antimicrobial peptides such as C-type lectins and pro-defensins from intestinal Paneth cells [11,12], activate intestinal B cells to express secretory IgA [13,14], and stimulate the production of protective mucus from colonic goblet cells [15]—all mechanisms that prevent bacterial translocation across the mucosa [16]. Bacteriophage adherence to mucus layers has also been hypothesized to protect against bacterial colonization and infiltration [17].
Disturbances within the gut microbiota, termed “dysbiosis”, are linked to numerous diseases, many of which are hepatic in nature [18]. This is likely due to the bidirectional nature of the gut–liver axis: nutrient rich portal vein blood entering the liver originates from the gut, while hepatic bile from gallbladder travels into the intestines to facilitate digestion [19]. Consequently, the insults that drive CLD, including caloric excess (NAFLD/NASH), alcoholism (ALD), and biliary damage (primary sclerosing cholangitis, primary biliary cirrhosis), can have significant effects on the gut microbiota, leading to intestinal permeability and exacerbation of inflammation and fibrosis.
Many studies have suggested an association between gut microbiome alteration and chronic liver diseases. Both Mouzaki et al. and Silva et al. demonstrated a reduction in Bacteroidetes and Firmicutes spp. in both NAFLD and NASH patients compared to adult healthy controls [20,21]. In pediatric studies, Zhu et al. measured a decrease in Firmicutes and increased Bacteroidetes in children with obesity or NASH [22]. A more recent, larger cohort study challenged these findings, finding a decrease of total Bacteroidetes in both NASH and NALFD pediatric patients, in agreement with adult studies [23].
In ALD, a reduction of Lactobacillus spp. has been recorded in both alcohol-consuming patients and mouse models [24,25]. Lactobacilli are beneficial bacteria commonly used in probiotics that have been shown to inhibit pathogen colonization [26]. Patients with ALD have also been found to have lower abundance of Bifidobacterium and Enterobacterium, and increased Proteobacteria, Fusobacteria, and Actinobacteria [27,28].
Changes in patient gut microbiota have also been measured in the context of worsening disease state. Indeed, significant differences in gut microbiota have been observed in NALFD subjects who had progressed to steatohepatitis or moderate fibrosis (F ≥ 2) when compared to patients with earlier stages of the disease. Boursier et al. found that NASH patients possessed a significantly larger abundance of Bacteroides and a reduction in Prevotella compared to NAFLD patients [29]. Recently, Bastian et al. also confirmed a significantly higher proportion of Bacteroides in fibrotic (F2–F4) patients compared to patients with minimal fibrosis (F0–F1). Two large studies by Loomba et al. and Caussy et al. also found a reduction in Firmicutes spp. and an enrichment of Proteobacteria spp. in patients with cirrhosis compared to those with minimal fibrosis [30]. In addition, Bajaj et al. recently demonstrated that periodontal therapy improves gut dysbiosis and systemic inflammation in cirrhotic patients [31]. Cirrhotic patients treated with scaling and root planning followed by oral hygiene showed a reduction in Enterobacteriaceae and Streptococcaceae, and a decrease of inflammatory markers interleukin (IL)-1β and IL-6 [31]. Together, these findings suggest that certain bacteria, likely Bacteriodes and Proteobacteria spp., and other factors such as oral health may play important roles in liver fibrosis progression.
2.2. Physical and Chemical Barriers of the Intestinal Mucosa
To maintain a healthy coexistence with commensal microbes and prevent bacterial dissemination, the gastrointestinal tract is lined by a cellular epithelium. This physical barrier is composed primarily of epithelial cells, with the addition of specialized cell types that differ between the small and large intestine. While all epithelial cells arise from intestinal epithelial stem cells (IESC) at the base of crypts, they differentiate into a variety of cells, including enterocytes (colonocytes in the large intestine), goblet cells, Paneth cells, tuft cells, and Microfold cells (M cells) [32]. Apart from hormone-secreting enteroendocrine cells and nutrient-absorbing enterocytes, the remaining epithelial cells are largely responsible for defending against microbial invasion (Figure 1).
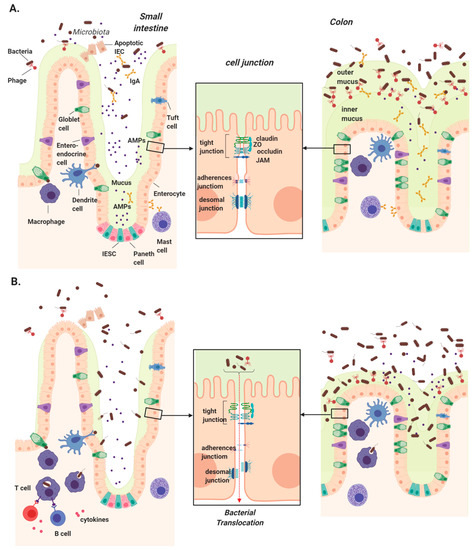
Figure 1.
Intestinal mucosal barriers in health and chronic liver disease. (A) Several physical and chemical defenses make up the intestinal mucosa, which serves to protect us from luminal microbes. Intestinal epithelial stem cells (IESCs) located at the base of crypts give rise to all epithelial cells. Goblet cells secrete mucins to form a thin mucus layer in the small intestine and two thick layers in colon, the innermost of which is devoid of bacteria. Enterocytes/colonocytes and Paneth cells secrete antimicrobial peptides (AMPs) primarily in the small intestine, while mast cells secrete IgA, which travels through the epithelium and is concentrated in the colon. Underlying the epithelium, dendritic cells and macrophages continuously surveil luminal contents using trans-epithelial dendrites. (B) Disruption of these physical barriers can lead to intestinal permeability and increased microbial translocation in chronic liver disease. These include the reduction of secreted mucus, AMPs, and IgA, permitting microbial access to the epithelial layer. Downregulation, altered localization, or rearrangement of tight junction components can also significantly impact intestinal permeability, allowing microbial translocation into the portal circulation where they are transported into the liver.
Goblet cells secret mucin proteins to form a highly glycosylated mucus layer over the vast epithelial surface, and this layer is significantly thinner in the small intestine compared to the colon due to a lower goblet cell density and bacterial colonization [33]. IgA secreted across the intestinal epithelium also comprises a significant component of the chemical defense. IgA is secreted by plasma cells in lymphoid follicles of the lamina propria, and transported via polymeric immunoglobulin receptors (pIgR) on the basolateral surface of epithelial cells into the lumen [34].
Epithelial integrity is maintained by junctions between intestinal epithelial cells (IECs) that provide selective nutrient permeability while preventing microbial translocation. There are three major types of cell junction that typically form near the apical end of the cells’ side walls: tight junctions (TJs), gap junctions, and adherens junctions [35]. Among them, TJs form the most rigid and impenetrable seal, hence their name. This junction is a complex of more than 50 proteins, most of which are transmembrane proteins such as occludin, claudin, and the junctional adhesion molecule (JAM) family proteins such as zonula occludens (ZO)-1, which connect with the cytoskeleton and form fibrils with adjacent cells [36,37].
2.3. Bacterial Translocation in Chronic Liver Disease
The intestinal mucosa represents the barrier protecting against luminal microbes while allowing the selective passage of digested nutrients into the circulation. This property, termed intestinal permeability, allows the intestinal tract to fulfill its absorptive function via two pathways [38]. The transcellular pathway enables macromolecules such as glucose, amino acids, or bacterial antigens to move through cells via enterocyte, M cell, and goblet cell transporter-mediated transcytosis or endocytosis. The paracellular pathway allows water and minerals to diffuse through the interspace between adjacent epithelial cells [39]. Passive transport in intestinal epithelium is, however, tightly controlled by the proteins making up the cell TJs [40]. Currently, intestinal permeability can be measured using in vivo or in vitro functional assays [39]. In vivo assays evaluate urinary or blood non-metabolized sugar such as lactulose/mannitol, polyethylene glycols (PEGs), Cr-labeled ethylenediaminetetraacetic acid (51Cr-EDTA), ovalbumin (OVA), or Fluorescein isothiocyanate dextran (FITC-dextran) following ingestion. In vitro assays indirectly measure intestinal permeability by detecting biomarkers such as bacteria, lipopolysaccharides (LPS), endotoxin antibodies, or bacterial fermentation products in lymph/blood or by histologically examining TJ protein localization and expression.
Even in the absence of disease, bacteria can be transported across the intestinal epithelium into the lamina propria where they can interact with mesenteric lymph nodes (MLN) and extra-intestinal organs via the blood [41]. This process was termed bacterial translocation by Berg and Garlington in 1979, and has since been expanded to include other luminal microbes such as viruses and fungi [42]. In fact, sequencing and culture methods have detected low levels of viable and non-viable microbes, microbial DNA, and antigens in the MLN as well as in other “sterile” organs, including the liver [43,44]. Bacteriophages have also been detected in various sites within the human body including the blood, kidney, and liver [45], however their immunogenicity remains uncertain.
Increased bacterial translocation (BT) is associated with gastrointestinal diseases and the extra-intestinal conditions of the liver, kidney, and brain, among others [46,47,48]. As early as 1995, Berg et al. identified three factors that contribute to BT: bacterial growth or dysbiosis, intestinal permeability, and immune deficiency [41]. These factors have been identified in various forms of chronic liver disease, and are outlined below and in Table 1.

Table 1.
Intestinal barrier deficiencies in chronic liver disease.
2.3.1. Alcoholic Liver Disease
In alcoholic fatty liver disease, viable bacteria, endotoxin, and LPS have been observed in the blood of both animal models and ALD patients, for which there are many potential mechanisms [49,50]. Small intestinal bacterial overgrowth (SIBO) in chronic alcoholics is more prevalent than healthy controls [51], perhaps due to extended orocaecal transit time in alcoholics compared to social drinkers [52]. SIBO has been suggested to mediate translocation due to differences in mucosal defense in the small intestine, primarily a reduction in mucus secretion. Increased intestinal permeability has also been reported in alcoholic cirrhosis, ALD, and even in non-cirrhotic alcoholics using in vivo assays [53,54,55].
In human studies, a reduction in the number of small intestinal villi, goblet cells, and TJ protein ZO-1 expression in the colon were observed in chronic alcoholics [56,57,58]. Furthermore, cell culture and animal studies suggest that ethanol and its metabolite, acetaldehyde, can alter intestinal barrier function by (1) inducing epithelial cell apoptosis, (2) disrupting TJs by downregulating ZO-1, occludin, and claudin and redistributing ZO-1 into cytoplasm, and (3) displacing the cytoplasmic skeleton [59,60,61,62].
Lastly, the production of intestinal IgA and the quantity of immune cells have been shown to be significantly altered in cases of chronic alcohol consumption and in ALD patients. While systematic IgA is increased in alcoholic liver diseases due to intestinal permeability, fecal IgA and IgA-producing B cells within the lamina propria are reduced in animal models of ALD [63,64]. Recently, a significant decrease in the number and activation state of mucosa-associated invariant T (MAIT) cells, a key component in antibacterial immune defense, was shown in ALD patients [65].
2.3.2. Non-Alcoholic Fatty Liver Disease
Multiple-pathogen-associated molecular patterns (PAMPs) such as peptidoglycan, LPS, and bacterial DNA have been detected in NALFD/NASH patient blood, and are linked to metabolic syndromes and obesity [66,67,68]. Two key drivers of bacterial translocation in NAFLD are SIBO and intestinal permeability, having been documented in numerous studies [69]. Increased intestinal permeability has been documented in both adult [70] and pediatric [71] NAFLD studies, as measured by 51Cr-EDTA and lactulose–mannitol assays, respectively. Moreover, intestinal permeability was associated with the severity of inflammation and fibrosis in children [71]. Clinical studies by Miele et al. and later by Xin et al. revealed that deregulation of TJ proteins may be responsible for intestinal permeability, demonstrating a reduction of ZO-1 and occludin expression in parallel with increased intestinal permeability in NAFLD patients [70,72]. Both in vitro and animal model studies of obesity further suggest that bile acids and leptin can stimulate intestinal permeability [73,74].
There is also an indication that liver damage as a result of NASH can directly contribute to loss of barrier integrity [67]. A meta-analysis performed by Luther et al. found a higher rate of intestinal permeability in NASH patients compared to NAFLD alone. A further study using a mouse model of NASH indicated that intestinal permeability occurs only after initial liver injury and the induction of pro-inflammatory cytokines [67].
Gut immune alteration is also a factor that can contribute to enhanced bacterial translocation in NAFLD. Luminal IgA and IgA-positive cells within ileal and colonic tissue are decreased in mouse models of NASH fed the methionine/choline-deficient diet [75]. Collective studies of innate and adaptive immunity on animals and patients with obesity have reported an increase in inflammatory cytokine expression and pro-inflammatory cluster differentiation (CD)4+ and CD8+ T cell, but an opposite trend in regulatory T cells [76]. These pro-inflammatory cytokines, such as interferon (IFN)-γ, IFN-α, and IL-6, have been shown to disturb intestinal TJs, allowing the translocation of luminal antigens across the intestinal barrier [77].
2.3.3. Liver Cirrhosis
Due to clinical associations with bacterial infection, microbial translocation is often examined in the context of liver cirrhosis. BT in cirrhosis has been identified using such methods as lymph node homogenate bacterial culture and bacterial DNA sequencing in cirrhotic patient blood [78,79]. Importantly, translocated bacteria, dominated by the Proteobacteria phylum, are abundant in the portal vein, as well as the hepatic and peripheral blood of decompensated cirrhotic patients [80]. When compared to healthy controls, SIBO is also significantly more common in patients with cirrhosis, particularly following decompensation [81,82]. Portal hypertension and abnormal small bowel motility are likely related to prevalent SIBO in decompensated cirrhosis [83].
Intestinal permeability as assessed by dual-sugar ingestion assays has been found to increase in both the small and large intestine of patients with decompensated/advanced cirrhosis [84,85,86]. Reduction in the expression of TJ proteins occludin and claudin-1 in cirrhotic patients may provide a mechanism for this increased permeability [87]. Nonetheless, electron microscopy experiments performed by Such et al. ten years prior demonstrated intact TJs in the duodenal epithelium of cirrhotic patients, but enlarged interspace between enterocytes [88]. More recently, an examination of cirrhotic mice treated with carbon tetrachloride (CCL4) showed a reduction of mucin (MUC)2 expression, mucus thickness, and goblet cell number, as well as an increase in intestinal permeability associated with bacterial overgrowth and translocation. The authors also suggested a modulatory role of the bile acid receptor Farnesoid X receptor (FXR), due to the restoration of TJ protein expression, goblet cell number, and bacterial translocation following FXR agonist treatment [89].
Although clinical associations have yet to be found, alterations in intestinal humoral and cellular immunity within gut-associated lymphoid tissues (GALT) have also been observed in cirrhotic models. An increase in bacterial translocation in cirrhotic rat models can activate monocytes and dendritic cells in GALT, releasing pro-inflammatory cytokines such as tumour necrosis factor (TNF)-α and IL-12 [90,91] that have been shown to increase intestinal permeability by deregulating ZO-1 and claudin 1 within the TJs [92,93]. While dendritic cells have been shown to open TJs to allow microbial sampling of the lumen, an increased incidence of DC sampling has been measured in cirrhotic rats, and is associated with increased translocation of bacterial DNA [91].
Studies in recent years have provided sufficient evidence to suggest that microbial translocation can drive the development and exacerbation of chronic liver diseases. However, the detailed cellular and mechanisms implied by this association will need more efforts to elucidate.
3. Hepatic Recognition of Microbial Ligands: The Role of Pattern Recognition Receptors
Pattern recognition receptors (PRRs) are a group of host sensors that recognize antigens derived from both foreign and endogenous sources. PRRs are essential initiators of the inflammatory and immune responses that defend against foreign microbial invaders as well as endogenous cellular debris known as damage-associated molecular patterns (DAMPs). While these sensors are an essential component of hepatic immunity, they can contribute to chronic inflammation and fibrosis progression in response to prolonged activation. As outlined in the previous section, the intestinal barrier can become impaired in CLD, allowing continuous translocation of microbial antigens into the portal circulation. This section aimed to characterize the hepatic response to these antigens as it relates to inflammation and fibrosis.
The cellular drivers of liver fibrogenesis are the myofibroblast-like hepatic stellate cells (HSCs). They are largely responsible for wound healing in their steady state, but drive fibrogenesis through ECM deposition upon chronic activation [95]. HSC fibrogenesis can be triggered either directly, via induction of PRRs on HSCs, or indirectly via inflammatory signals produced by neighboring cells such as hepatocytes and Kupffer cells (KCs) [95]. This section covers PRRs that contribute to fibrosis development, with in vivo evidence of inflammatory and fibrogenic activity in CLDs, including toll-like receptor (TLR) 2, TLR3, TLR4, TLR5, TLR7 TLR9, nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs), c-type lectin receptors (CLRs), and stimulator of interferon genes (STING). General information regarding PRRs in CLD is summarized in Table 2.
Table 2.
Pattern recognition receptors (PRRs) that contribute to hepatic inflammation and fibrogenesis.
3.1. Toll-Like Receptors
3.1.1. TLR2
Hepatic upregulation of TLR2 has been observed in patients with NAFLD/NASH [98,100] and fibrosis due to chronic viral infection [152]. In contrast, ALD patients show significantly lower expression of hepatic TLR2 compared to healthy controls [101].
In murine models of CLD, TLR2 significantly contributes to hepatic inflammation and fibrosis. In chronic ethanol-binge-fed mice, TLR2 is crucial for hepatic IL-1β, IL-6, and TNF-α-related liver injury and inflammation, as well as neutrophil-mediated hepatic injury [102,103]. In addition, using the choline-deficient L-amino-defined (CDAA)-diet-induced NASH model, TLR2 deficiency improved hepatic inflammation and injury by reducing Kupffer cell inflammasome activation and pro-inflammatory cytokine production, suggesting a KC-dependent inflammatory pathway mediated by TLR2 [96]. In contrast, TLR2 was protective in the NASH methionine/choline-deficient (MCD) diet model, as demonstrated by an increase in ALT and TNF-α in TLR2 KO mice [104].
In mouse models of fibrosis, TLR2 was reported to have limited contribution to fibrogenesis in bile duct ligation (BDL) mice [105]. In contrast, TLR2−/− mice treated with CCL4, possessed significantly impaired HSC activation with reduced collagen deposition, pro-inflammatory cytokine and α-smooth muscle actin (SMA) expression [107]. In addition, they demonstrated attenuated mitogen-activated protein kinase (MAPK) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation compared to wild type (WT) fibrotic mice. A neutrophil-driven mechanism of fibrogenesis in CCL4-induced fibrosis has also been attributed to TLR2-mediated hepatic chemokine C–C motif ligand (CXCL)2 production [106]. Lastly, TLR2 knockout (KO) in CDAA-diet-induced NASH mice significantly dampened HSC activation, collagen deposition, α-SMA expression, and transforming growth factor (TGF)-β expression, thus, ameliorating NASH-associated fibrogenesis [96].
3.1.2. TLR3
The protective and anti-inflammatory role of TLR3 has been reported in mice fed with high-fat diet (HFD) followed by binge drinking to induce liver injury. Stimulating TLR3 using poly I:C resulted in elevated HSC and KC IL-10 expression, as well as reduced hepatic expression of TNF-α, IL-6, CXCL2 and impaired liver injury [113].
TLR3 signaling is well characterized in murine natural killer (NK) cells, where activation of TLR3 results in a potent anti-fibrotic effect in both 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC)-diet- and CCl4-induced murine fibrosis models [153,154]. TLR3 has been shown to work synergistically with IL-18 to activate the p38/PI3K/Akt pathway, thus stimulating NK cells to kill HSCs via TNF-related apoptosis-inducing ligand (TRAIL)-mediated degranulation [114]. In contrast, TLR3 is pro-fibrotic in the CCL4 fibrosis model, where TLR3 KO results in downregulation of IL-6, TNF-α, and pro-fibrotic markers [115]. Interestingly, the authors concluded that CCL4-treated hepatocyte exosomes stimulated HSC TLR3 signaling to drive γδ T cell IL-17 production and fibrosis progression [115].
3.1.3. TLR4
TLR4 is the most thoroughly studied PRR in the setting of CLD. Hepatic and serum TLR4 is significantly upregulated in NASH patients, with elevated levels of circulating LPS in peripheral [100,116] and portal vein blood [155]. High serum levels of TLR4 have also been proposed as a predictive non-invasive marker for liver fibrosis development in NASH patients [156]. Although hepatic expression of TLR4 has not been studied in patients with ALD, peripheral blood mononuclear cells (PBMCs) from patients with ALD showed sensitized responses towards LPS treatment [117].
The role of TLR4 in murine models of liver inflammation has been well studied. TLR4-dependent ROS production and TLR4-dependent interferon regulatory factor (IRF)3 activation in the liver are required to drive hepatic inflammation in mice with alcoholic hepatitis [120]. A similar study showed that hepatic inflammatory cytokines were significantly downregulated in hepatocyte-selective-TLR4-deficient mice fed with a liquid diet containing 5% ethanol [119]. The importance of TLR4 in NASH development was further emphasized in a murine NASH model using high-fat, high-cholesterol (HFHC)-diet fed ApoE KO mice, showing a TLR4-mediated ROS production and triggering pro-inflammatory cytokine expression in KC [122]. Linking TLR4 to NAFLD pathogenesis, fatty acids such as palmitate can also trigger ROS production in a TLR4-dependent manner, inducing IL-1β and TNF-α production from liver macrophages [121].
TLR4-mediated fibrosis has been interrogated in a variety of mouse models. In BDL mice, the TLR4–MyD88–NF-κB pathway in HSCs has been shown to upregulate pro-inflammatory cytokine production, α-SMA, TIMP1, and TGF-β expression, and ECM deposition [105,123]. In addition, TLR4-mediated downregulation of Bambi (a TGFβ pseudoreceptor) was shown to sensitize quiescent HSCs for subsequent activation [105,123]. Using the transforming growth factor beta-activated kinase 1 (TAK1) KO murine model of fibrosis [124], TLR4 and MyD88 double KO mice also demonstrated reduced α-SMA, TIMP1, and TGFβ expression and collagen deposition, supporting the involvement of TLR4–MyD88–NF-κB signaling in hepatic fibrogenesis [124].
3.1.4. TLR5
Peritoneal injection with the TLR5 ligand flagellin has been shown to induce significant TLR5-mediated liver injury in mice, resulting in IL-6, IL-8, and IL-1β production, coupled with neutrophil and macrophage infiltration into the liver [126]. In the context of NASH, however, hepatocyte TLR5 may possess a protective effect. In mice fed with MCD diet to induce NASH, selective hepatocyte deficiency of TLR5 was shown to exacerbate liver inflammation and fibrosis via elevated expression of TNF-α, monocyte chemoattractant protein (MCP)1, and IL-1β, as well as Timp1, Mmp9, Col1, and collagen deposition [125]. These conflicting results may, however, be the result of hepatocyte versus whole-body knockout of TLR5 and require further study.
Unlike hepatocyte-specific TLR5 KO, whole-body KO ameliorates inflammation and fibrogenesis in the CCL4 fibrosis model. TLR5 KO mice demonstrated a significant reduction of inflammatory mediators TNF-α, IL-6, and IL-1β and fibrogenesis, as indicated by downregulation in α-SMA, TGF-β, TIMP1, and collagen deposition compared to WT mice [127]. A significant reduction in NF-κB and MAPK signaling activity was measured in activated HSCs, suggesting a mechanism of hepatic fibrogenesis mediated by the TLR5-activated NF-κB/MAPK signaling pathway in HSCs [127].
3.1.5. TLR7
The role of TLR7 in NASH/NAFLD, liver fibrosis, or liver cirrhosis has been widely overlooked in the clinical setting; however, recent studies suggest that ALD-mediated inflammation and fibrosis is linked to hepatic TLR7 overexpression [130,131]. Moreover, TLR7-mediated IFN production is stimulated by alcohol in primary human hepatocytes and is correlated with patients with more advanced fibrosis as well as higher expression of fibrotic markers α-SMA, collagen I, and Timp1 [130].
The role of TLR7 in ALD, NASH/NAFLD, and fibrosis development has been established primarily in mouse models. A recent study using ethanol (25% w/v) feeding to stimulate alcoholic hepatitis showed that activation of TLR7 significantly upregulated expression of pro-inflammatory cytokines and the endogenous TLR-7 agonist let-7b from hepatocytes, hence exacerbating hepatic inflammation [131]. Roh et al. recently showed that TLR7 deficiency significantly reduced the degree of hepatic steatosis and inflammation in a MCD-diet-induced NASH mouse model, examined by H&E staining, as well as TNF-α and IFN-α production from KC and hepatic dendritic cells, respectively [132]. In contrast, TLR7 has been identified as a protective factor in hepatic fibrosis development in both CCL4 and BDL murine fibrosis models. TLR7 KO mice expressed higher levels of hepatic pro-inflammatory cytokine and fibrosis marker expression as well as exacerbated collagen deposition [129]. Moreover, dendritic cell expressed type I IFNs upon TLR7 stimulation, triggered KC IL-1 receptor antagonist expression and ultimately suppressing IL-1-dependent liver injury and inflammation [129].
3.1.6. TLR9
In humans, hepatic TLR9 expression is upregulated in NASH patients [100,133], while high serum levels of bacterial DNA (TLR9 ligand) have also been linked with liver cirrhosis and liver fibrosis [134,135]. Indeed, an increase in circulating bacterial DNA has been measured in patients with alcoholic and fatty liver disease prior to fibrotic development [136]. In addition, acute binge drinking has been shown to significantly increase serum bacterial DNA and pro-inflammatory cytokines such as IL-6, TNF-α, and IL-1β to indirectly contribute to liver fibrosis [135,139,157].
The role of TLR9 in chronic liver inflammation has been well established in animal models. In mouse models of CDAA-induced NASH, activation of TLR9 can stimulate IL-1β expression in KCs, mediating steatohepatitis and hepatocyte apoptosis driven by lipid accumulation [137,138]. In addition, the pro-inflammatory role of TLR9 in NASH has been further confirmed in atherogenic diet-fed NASH and foz mouse models, demonstrating reduced pro-inflammatory cytokine production and attenuated hepatic neutrophil infiltration in TLR9−/− mice [133]. Similarly, evidence for TLR9-mediated liver injury and inflammation in chronic-binge-ethanol-fed mice was shown to be driven by IL-1β expression as well as TLR9/TLR2-dependent hepatic neutrophil infiltration mediated by CXCL1/2/5 expressed from hepatocytes and HSC [102,139].
Despite the inflammatory role of TLR9 in human NASH, TLR9 KO failed to improve hepatic fibrosis in murine NASH models [133]. TLR9 does, however, seem to influence fibrosis development in other models of fibrosis including murine BDL, where TLR9−/− mice demonstrated significantly alleviated fibrogenesis and HSC activation compared to WT mice [140]. Similar results were found using the spontaneous fibrosis Tak1ΔHep mouse model, also demonstrating a reduction in liver inflammation and fibrosis in TLR9 KO mice [124]. Moreover, stimulation of TLR9 using CpG DNA directly activated the fibrogenic phenotype in primary mouse HSCs and the immortalized human HSC LX-2 cell line [158].
3.2. NOD-Like Receptors
The influence of NLRs on liver inflammation and fibrosis was thoroughly reviewed recently by Xu et al. [145]. NOD1, NOD2, and nucleotide-binding oligomerization domain, leucine-rich repeat- and pyrin-domain-containing (NLRP)3 are the main NLRs driving hepatic inflammation, liver injury, and liver fibrogenesis. NOD1-mediated neutrophil recruitment has been described following acute liver injury and inflammation in a CCL4-treated murine model [159]. In addition, activation of NOD-2 by muramyl dipeptide, a bacterial peptidoglycan motif, can induce NF-kB-dependent hepatic expression of pro-inflammatory cytokines to indirectly orchestrate liver inflammation and fibrosis [145,160]. Furthermore, it has been demonstrated that NLRP3–inflammasome pathway is activated in murine NASH to induce hepatic TNF-a, IL-6, and IL-8 production [144,145].
Hepatocyte stimulation of NOD1 via its ligands can activate the NF-kB and MAPK pathway to induce CXCL1 and CCL5 production, promoting wound healing and fibrogenesis [161]. NLRP3–inflammasome has been demonstrated to be a direct contributor in hepatic fibrogenesis. Activation of NLRP3 undoubtedly triggers direct activation of HSC to enhance matrix deposition, TGF-β expression, and fibrosis progression [142].
3.3. Anti-Fungal PRRs and Liver Fibrosis
The role of the CLR dectin-1 in liver inflammation and fibrosis development has been thoroughly studied. Ethanol-containing-diet-fed mice were found to have elevated serum β-glucan level and hepatic injury, which was significantly reduced upon treatment with anti-fungal agent [147]. More importantly, plasma β-glucans enhanced IL-1β expression in KC to drive hepatic inflammation that was absent in dectin-1 deficient mice [147]. A further study showed that hepatic expression of dectin-1 was upregulated in a thioacetamide (TAA)/CCL4 fibrosis mouse model, and that dectin-1 negatively regulated the expression of TLR4 and its co-receptor CD14 to mitigate fibrosis development and hepatic inflammation [146]. Knocking out dectin-1 exacerbated liver fibrosis and inflammation, as demonstrated by increased expression of TNF-α, IL-6, and MCP-1, neutrophil and macrophage influx, and fibrosis progression [146].
3.4. STING
In view of its recent discovery, there have been a limited number of human studies exploring the role of STING in CLD. Luo et al. reported an overexpression of STING in the non-parenchymal cells within the liver tissue of NAFLD patients, albeit with no examination of their relationship to disease activity [148]. By utilizing the alcohol-fed mouse ALD model, Petrasek et al. demonstrated that activation of the STING–IRF3 pathway stimulates pro-inflammatory cytokine production in the liver [149]. In the same study, alcohol-induced liver injury was also shown to trigger the STING–IRF3 pathway by endoplasmic reticulum (ER) stress, promoting the mitochondrial apoptotic pathway in hepatocytes [149]. Knocking out STING in HFD-induced NAFLD and MCD-induced NASH murine models attenuated hepatic activation of IRF3 and NF-κB pathways, and significantly downregulated expression of pro-inflammatory cytokines to alleviate NAFLD/NASH severity [148]. Similar findings were reported by Yu et al. using HFD- and MCD-diet-induced NASH mice; hepatocytes releasing mitochondrial DNA during NASH development led to the activation of STING–IRF3 pathway on KC to trigger pro-inflammatory cytokine production and hepatic inflammation in NASH [150].
Taken together, the above studies suggest that STING plays an important role in both hepatic inflammation and hepatic fibrosis in NASH [148,150]. Similar findings were elucidated in a CCL4-induced liver fibrosis murine model by Iracheta-Vellve et al. Hepatocytes were shown to undergo significant ER stress resulting in STING–IRF3 activation and induction of the mitochondria-dependent apoptosis pathway. Hepatocyte cell death was shown to activate HSC expression of α-SMA and COL1A2, collagen deposition, and fibrosis progression [151].
4. Therapies to Prevent Microbe-Driven Liver Inflammation and Fibrosis
The primary interventions for alcoholic and fatty liver disease have focused on lifestyle modifications: abstaining from alcohol and improving patient diet and exercise regimens. In more severe cases of obesity, surgical interventions (e.g., bariatric surgery) are required in combination with significant lifestyle adjustments. Unfortunately, even in the case of surgical interventions, patient compliance towards dietary and alcohol restrictions is often lacking, rendering the development of novel therapeutics a top priority.
In cirrhotic patients, dietary interventions and abstinence from alcohol drastically improve survival [162]. These patients also generally require treatment of portal hypertension, which is the contributing factor to intestinal permeability. Unfortunately, however, in decompensated cirrhosis, treatment of portal hypertension using beta-blockers and anti-hypertensives can dangerously reduce mean arterial pressure [163]. In addition, there is an increased risk of susceptibility to infections due to poor phagocytic capacity in the liver and increased likelihood of ascites: the accumulation of fluid in the peritoneal cavity [3]. These aspects further underline the vital need for novel therapies to prevent the progression of fibrosis, particularly in end-stage liver disease where there is currently a lack of effective treatment. Limiting the exacerbation of inflammation and fibrosis by microbial translocation into the liver is therefore an avenue that must be explored. We aimed to summarize current and potential therapies directed at (1) reducing microbial translocation, and (2) limiting the harmful response to microbial antigens in the liver.
4.1. Therapies to Reduce Intestinal Permeability
Studies examining intestinal permeability in ALD [24,53,54], NAFLD/NASH [67,70,164], and fibrotic liver disease/cirrhosis [86,87,165] have focused primarily on the reduction in TJ proteins such as ZO-1 and Claudin-1 [70,87,94], though the mechanisms by which this loss occurs remains largely unknown. Loss of intestinal mucus or mucosal IgA production [166] can also significantly increase intestinal permeability; however, these mechanisms have been largely overlooked in CLD.
Expansion of pathogenic bacterial species both within the colon and into the SIBO have been documented in CLD [70,167], motivating the examination of antibiotics and prebiotics as potential therapies. Antibiotic studies commonly use a broad spectrum and poorly absorbed antibiotics such as neomycin or rifaximin to achieve a gut-targeted intestinal decontamination. In rodent models of obesity [168], long-term ethanol exposure [169], NASH [170], and fibrosis [105], antibiotic treatment has been shown to reduce intestinal permeability and subsequent liver injury. In humans, a number of clinical trials are underway to assess the efficacy of antibiotics for the treatment of AH, NASH, and cirrhosis (reviewed in [171]). Early data suggests that short-term rifaximin treatment can reduce serum endotoxin and liver inflammation in NAFLD/NASH patients [172], whereas rifamixin prophylaxis over 24 weeks can significantly reduce hospitalization and mortality among cirrhotic patients [173]. These data support previous findings that rifaximin can reduce the risk of complications associated with cirrhosis, including hepatic encephalopathy, variceal bleeding, and SBP [174]. Importantly, antibiotics such as levofloxacin and metronidazole, but not rifaximin, can significantly increase gut proteases, thus contributing to intestinal permeability in both humans [175] and rats [176]. An antibiotic-mediated reduction in anti-proteolytic bacterial richness and abundance in the colon is thought to contribute to gut permeability.
Significant microbial perturbations caused by broad spectrum antibiotics have stimulated interest in probiotics (beneficial microbes) and prebiotics (beneficial microbial substrates) for the improvement of intestinal health. In mouse models of ethanol-induced liver injury, probiotic Lactobacillus rhamnosus strains have been shown to reduce serum endotoxin, hepatic oxidative stress, and inflammatory TNFα production [177,178]. While intestinal permeability was not assessed in these studies, Wang et al. demonstrated that Lactobacillus probiotics could prevent the alcohol-induced loss of intestinal ZO-1, Caludin-1, and Occludin-1, keeping the intestinal epithelium intact [179]. In rats, a similar reduction in ZO-1, intestinal permeability, steatosis, and fibrosis was observed in the choline-deficient/L-amino-acid-defined NASH diet, and was significantly attenuated by probiotic treatment with Clostridium butyricum MIYAIRI 588 strain. Interestingly, in mouse models of CCL4-induced fibrosis, different probiotics achieve improved intestinal permeability via unique mechanisms: Bifidobacterium probiotics have been shown to increase intestinal TJ expression [180] whereas Lactobacillus-paracasei-fermented milk reduces intestinal permeability by increasing antimicrobial β-defensin expression [181].
The study of prebiotics allows researchers to understand the relationship between a substrate, microbial metabolism, gut health, and permeability. The majority of prebiotics used today are indigestible carbohydrate polymers (fibers) that become fermented by gut bacteria to produce, among other things, short chain fatty acids (SCFAs) consisting of acetate, propionate, and butyrate [182]. SCFAs are significant homeostatic and anti-inflammatory signaling molecules in the gut (reviewed in Reference [183]), but can also significantly alter intestinal permeability. In vitro, butyrate has been shown to increase ZO-1 expression in Caco-2 cells [184], and stimulate mucous secretion in E12 human colon cells [185]. In vivo, rats supplemented with oats rich in fermentable β-glucans were significantly more resistant to alcohol-induced oxidative stress and intestinal permeability [186]. In humans, increased fiber intake has also been associated with improved permeability (reduced circulating ZO-1) and reduced ALT/AST in NALFD patients [187]. These data are supported by two recent meta-analyses finding that both prebiotics and probiotics reduce liver enzymes ALT, AST, and GGT in NAFLD patients [188], with probiotics also reducing serum ammonia and hepatic encephalopathy in cirrhotic patients [189].
4.2. Therapies to Dampen the Hepatic Immune Response
Direct inhibition of PRRs for the treatment of chronic liver disease has not been a research priority due to their peripheral contribution to disease pathogenesis. It is, however, now becoming evident that PRR activation from both microbial and self-ligands can significantly contribute to liver inflammation and fibrogenesis. TLR-agonistic therapies are currently being actively pursued for cancer therapies/adjuvants (reviewed in Reference [190]). TLR inhibitors, on the other hand, have been studied to a lesser degree in a handful of studies assessing their efficacy in inflammatory and autoimmune disease. Even fewer therapies are being assessed in the context of chronic liver disease, with TLR2/4/9, NLRP3, and STING pathway inhibition garnering some interest from pharmaceutical companies [190].
The humanized TLR2 mAb OPN-305 can achieve significant blockade of TLR2 inflammatory signaling in response to bacterial stimuli in healthy subjects [191], and improved overall response rate in patients with myelodysplastic syndromes that had previously failed hypomethylating agent therapy [192]. While TLR2 blockade has not been assessed in human CLD, the pleiotropic nature of TLR2 suggests that it may benefit inflammatory and fibrotic progression.
Small molecule inhibitors of TLR4, i.e., NI-0101 [193] and Ibudilast [194], are being assessed in extrahepatic disease, and JKB-121, a weak TLR4 antagonist, is currently being assessed in NASH [195]: preliminary results do not support a significant therapeutic benefit of JKB-121, though the study was confounded by a notable improvement in liver inflammation within the placebo group.
TLR9 blockade represents perhaps the most exciting TLR-targeting therapy. The TLR9 inhibitor hydroxychloroquine is currently used as treatment for autoimmune diseases such as rheumatoid arthritis and lupus, allowing potential repurposing for CLD. Furthermore, the novel TLR9 antagonist COV08-0064 has shown promise in animal models of sterile liver inflammation, particularly in the context of fatty liver where it limits inflammasome activation [196,197]. While TLR9 inhibition has yet to be assessed in human CLD, its diversity of microbial ligands, including bacterial, fungal, and phage DNA, will surely make it a candidate for future trials.
Research targeting the NLRP3–inflammasome pathway is undergoing rapid expansion alongside the number of inflammasome-associated diseases. While the NLRP3 inhibitor CP-456,773 was removed from phase II trials for rheumatoid arthritis due to supposed liver toxicity, other inhibitors are in different stages of development for the treatment of gout and Parkinson’s disease, among others [198]. In 2018, Genentec acquired San Diego-based Jecure Therapeutics and their portfolio of preclinical NLRP3 inhibitors as a treatment for NASH and hepatic fibrosis [199].
Lastly, the intracellular DNA sensor STING has become a popular target due to its association with autoimmune disease. STING inhibitors have been recently developed [200], and are being actively generated by a number of pharmaceutical companies with the aim of targeting STING-related genetic disease. This compound would have the potential to move towards therapies for CLDs such as NASH and fibrosis in the future [201].
5. Conclusions and Future Perspectives
In summary, this review has highlighted the strong connection between the liver and gut in the context of liver disease. Indeed, CLD does not occur in isolation, but is accompanied by disturbances of the complex balance of gastrointestinal microbiota, architecture, and immunity. Similarly, gut microbiota can play a significant role in liver health by altering host metabolism and immunity. While such associations are strong, it is much less certain whether the distinct disease states in CLD and fibrosis stimulate changes in microbiota, or if gut microbiota exacerbate inflammatory and fibrotic progression.
Host, microbiomic, and lifestyle factors that influence intestinal permeability are beginning to be understood, but there is still much to be learned. Specifically, there is poor understanding of the exact mechanisms by which intestinal permeability is altered and the physiological reasons for it. TJ expression and localization are often examined in isolation, while overlooking the numerous additional barriers that prevent microbial translocation in the gut. Antimicrobial peptides, IgA, and mucus abundance and localization are rarely examined in this context, and a better understanding of their regulation in health and disease is warranted.
Lastly, along with public health initiatives aimed at reducing the causative lifestyle factors of fibrosis, i.e., alcohol and obesity, we must focus research on the development of novel PRR-antagonizing therapies. Murine studies have highlighted the contribution of individual PRRs to the development and progression of liver inflammation and fibrosis (as summarized in Figure 2); however, it remains unclear how multiple sensors collectively drive disease and may potentiate each signal. Human trials are pending to examine microbial sensors such as TLR9 and STING as well as inflammasome components to determine their contribution, whether alone or in concert, to fibrosis progression.
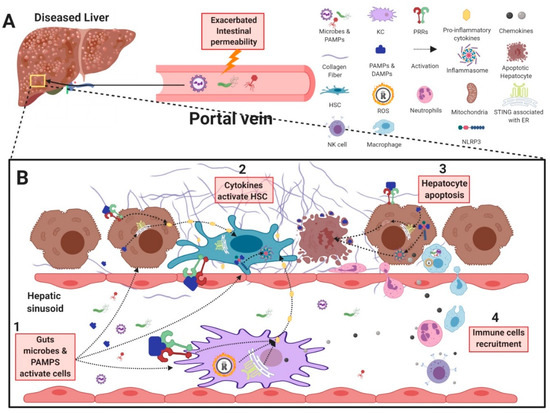
Figure 2.
Gastrointestinal microbes and their contribution to liver inflammation and fibrosis. (A) In chronic liver disease, gut microbes and PAMPs can cross the intestinal barrier due to an increase in intestinal permeability, resulting in their transport into the liver through the hepatic portal vein. (B1) Gut-derived antigens are recognized by, and activate HSCs, KCs, and hepatocytes, resulting in the secretion of pro-inflammatory cytokines and chemokines. (B2) Pro-inflammatory cytokines produced from KCs and hepatocytes further activate HSCs to further exacerbate fibrogenesis. (B3) In addition, activation of STING– and NLRP3–inflammasome pathways in hepatocytes can trigger apoptosis and release of DAMPs to further activate HSCs. (B4) Chemokines produced by activated KCs, HSCs, and hepatocytes recruit immune cells such as neutrophils, NK cells, and monocytes to further exacerbate liver inflammation and injury. PAMP: pathogen associated molecular pattern; KC: Kupffer cell; HSC: hepatic stellate cell; NK cell: natural killer cell; PRR: pattern recognition receptor; TLR: Toll-like receptor; NOD: nucleotide-binding oligomerization domain-containing protein 1; NLRP3: nucleotide-binding oligomerization domain, leucine-rich repeat- and pyrin-domain-containing 3; STING: stimulator of interferon genes; DAMP: damage associate molecular pattern.
Author Contributions
Writing—original draft preparation (S.A.R., T.H.L., D.C.); writing—review and editing (S.A.R., H.S., G.A.); supervision (S.A.R., G.A.).
Funding
This research received no external funding.
Acknowledgments
Figures were created with Biorender.com (©BioRender-biorender.com).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Loomba, R.; Sanyal, A.J. The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Debelius, J.; Brenner, D.A.; Karin, M.; Loomba, R.; Schnabl, B.; Knight, R. The gut-liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Iebba, V.; Guerrieri, F.; Di Gregorio, V.; Levrero, M.; Gagliardi, A.; Santangelo, F.; Sobolev, A.P.; Circi, S.; Giannelli, V.; Mannina, L.; et al. Combining amplicon sequencing and metabolomics in cirrhotic patients highlights distinctive microbiota features involved in bacterial translocation, systemic inflammation and hepatic encephalopathy. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Doré, J.; Simrén, M.; Buttle, L.; Guarner, F. Hot topics in gut microbiota. United Eur. Gastroenterol. J. 2013, 1, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Sekirov, I.; Russell, S.L.; Antunes, L.C.M.; Finlay, B.B. Gut Microbiota in Health and Disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar] [CrossRef]
- Cani, P.D. Human gut microbiome: Hopes, threats and promises. Gut 2018, 67, 1716. [Google Scholar] [CrossRef]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinal microbial flora. Science (New York, NY) 2005, 308, 1635–1638. [Google Scholar] [CrossRef]
- Saffarian, A.; Mulet, C.; Regnault, B.; Amiot, A.; Tran-Van-Nhieu, J.; Ravel, J.; Sobhani, I.; Sansonetti, P.J.; Pédron, T. Crypt- and Mucosa-Associated Core Microbiotas in Humans and Their Alteration in Colon Cancer Patients. MBio 2019, 10, e01315-19. [Google Scholar] [CrossRef]
- Hillman, E.T.; Lu, H.; Yao, T.; Nakatsu, C.H. Microbial Ecology along the Gastrointestinal Tract. Microbes Envrion. 2017, 32, 300–313. [Google Scholar] [CrossRef]
- Reyes, A.; Semenkovich, N.P.; Whiteson, K.; Rohwer, F.; Gordon, J.I. Going viral: Next-generation sequencing applied to phage populations in the human gut. Nat. Rev. Microbiol. 2012, 10, 607–617. [Google Scholar] [CrossRef]
- Cash, H.L.; Whitham, C.V.; Behrendt, C.L.; Hooper, L.V. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science (New York, NY) 2006, 313, 1126–1130. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, M.; Cerda-Cuellar, M.; Martinez, V. Antibiotic-induced dysbiosis alters host-bacterial interactions and leads to colonic sensory and motor changes in mice. Gut Microbes 2015, 6, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Yanagibashi, T.; Hosono, A.; Oyama, A.; Tsuda, M.; Hachimura, S.; Takahashi, Y.; Itoh, K.; Hirayama, K.; Takahashi, K.; Kaminogawa, S. Bacteroides induce higher IgA production than Lactobacillus by increasing activation-induced cytidine deaminase expression in B cells in murine Peyer’s patches. Biosci. Biotechnol. Biochem. 2009, 73, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, A.J.; Gatto, D.; Sainsbury, E.; Harriman, G.R.; Hengartner, H.; Zinkernagel, R.M. A primitive T cell-independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science 2000, 288, 2222–2226. [Google Scholar] [CrossRef]
- Mack, D.R.; Michail, S.; Wei, S.; McDougall, L.; Hollingsworth, M.A. Probiotics inhibit enteropathogenic E. coli adherence in vitro by inducing intestinal mucin gene expression. Am. J. Physiol. 1999, 276, G941–G950. [Google Scholar] [CrossRef]
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Nageshwar Reddy, D. Role of the normal gut microbiota. World J. Gastroentero 2015, 21, 8787–8803. [Google Scholar] [CrossRef]
- Barr, J.J.; Auro, R.; Sam-Soon, N.; Kassegne, S.; Peters, G.; Bonilla, N.; Hatay, M.; Mourtada, S.; Bailey, B.; Youle, M.; et al. Subdiffusive motion of bacteriophage in mucosal surfaces increases the frequency of bacterial encounters. Proc. Natl. Acad. Sci. USA 2015, 112, 13675. [Google Scholar] [CrossRef]
- Henao-Mejia, J.; Elinav, E.; Thaiss, C.A.; Flavell, R.A. Chapter Three—The Intestinal Microbiota in Chronic Liver Disease. In Advances in Immunology; Alt, F.W., Ed.; Academic Press: Cambridge, MA, USA, 2013; Volume 117, pp. 73–97. [Google Scholar]
- Adawi, D.; Molin, G.; Jeppsson, B. Gut–liver axis. HPB 1999, 1, 173–186. [Google Scholar] [CrossRef]
- Mouzaki, M.; Comelli, E.M.; Arendt, B.M.; Bonengel, J.; Fung, S.K.; Fischer, S.E.; McGilvray, I.D.; Allard, J.P. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology 2013, 58, 120–127. [Google Scholar] [CrossRef]
- Da Silva, H.E.; Teterina, A.; Comelli, E.M.; Taibi, A.; Arendt, B.M.; Fischer, S.E.; Lou, W.; Allard, J.P. Nonalcoholic fatty liver disease is associated with dysbiosis independent of body mass index and insulin resistance. Sci Rep. 2018, 8, 1466. [Google Scholar] [CrossRef]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Del Chierico, F.; Nobili, V.; Vernocchi, P.; Russo, A.; De Stefanis, C.; Gnani, D.; Furlanello, C.; Zandonà, A.; Paci, P.; Capuani, G.; et al. Gut microbiota profiling of pediatric nonalcoholic fatty liver disease and obese patients unveiled by an integrated meta-omics-based approach. Hepatology 2017, 65, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, S.; Matamoros, S.; Cani, P.D.; Neyrinck, A.M.; Jamar, F.; Stärkel, P.; Windey, K.; Tremaroli, V.; Bäckhed, F.; Verbeke, K.; et al. Intestinal permeability, gut-bacterial dysbiosis, and behavioral markers of alcohol-dependence severity. Proc. Natl. Acad. Sci. USA 2014, 111, E4485–E4493. [Google Scholar] [CrossRef] [PubMed]
- Yan, A.W.; Fouts, D.E.; Brandl, J.; Stärkel, P.; Torralba, M.; Schott, E.; Tsukamoto, H.; Nelson, K.E.; Brenner, D.A.; Schnabl, B. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology 2011, 53, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, C.; Donders, G.G.; Palmeira-de-Oliveira, R.; Queiroz, J.A.; Tomaz, C.; Martinez-de-Oliveira, J.; Palmeira-de-Oliveira, A. Bacteriocin production of the probiotic Lactobacillus acidophilus KS400. Amb Express 2018, 8, 153. [Google Scholar] [CrossRef] [PubMed]
- Schwenger, K.J.P.; Clermont-Dejean, N.; Allard, J.P. The role of the gut microbiome in chronic liver disease: The clinical evidence revised. Jhep Rep. 2019. [Google Scholar] [CrossRef]
- Bull-Otterson, L.; Feng, W.; Kirpich, I.; Wang, Y.; Qin, X.; Liu, Y.; Gobejishvili, L.; Joshi-Barve, S.; Ayvaz, T.; Petrosino, J.; et al. Metagenomic Analyses of Alcohol Induced Pathogenic Alterations in the Intestinal Microbiome and the Effect of Lactobacillus rhamnosus GG Treatment. PLoS ONE 2013, 8, e53028. [Google Scholar] [CrossRef]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef]
- Bastian, W.P.; Hasan, I.; Lesmana, C.R.A.; Rinaldi, I.; Gani, R.A. Gut Microbiota Profiles in Nonalcoholic Fatty Liver Disease and Its Possible Impact on Disease Progression Evaluated with Transient Elastography: Lesson Learnt from 60 Cases. Case Rep. Gastroenterol. 2019, 13, 125–133. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Matin, P.; White, M.B.; Fagan, A.; Deeb, J.G.; Acharya, C.; Dalmet, S.S.; Sikaroodi, M.; Gillevet, P.M.; Sahingur, S.E. Periodontal therapy favorably modulates the oral-gut-hepatic axis in cirrhosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G824–G837. [Google Scholar] [CrossRef]
- Umar, S. Intestinal stem cells. Curr. Gastroenterol. Rep. 2010, 12, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.V.; Phillipson, M.; Petersson, J.; Velcich, A.; Holm, L.; Hansson, G.C. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 15064–15069. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, A.; Rescigno, M. The biology of intestinal immunoglobulin A responses. Immunity 2008, 28, 740–750. [Google Scholar] [CrossRef] [PubMed]
- Farquhar, M.G.; Palade, G.E. JUNCTIONAL COMPLEXES IN VARIOUS EPITHELIA. J. Cell Biol. 1963, 17, 375–412. [Google Scholar] [CrossRef] [PubMed]
- Ulluwishewa, D.; Anderson, R.C.; McNabb, W.C.; Moughan, P.J.; Wells, J.M.; Roy, N.C. Regulation of Tight Junction Permeability by Intestinal Bacteria and Dietary Components. J. Nutr. 2011, 141, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Fanning, A.S.; Jameson, B.J.; Jesaitis, L.A.; Anderson, J.M. The Tight Junction Protein ZO-1 Establishes a Link between the Transmembrane Protein Occludin and the Actin Cytoskeleton. J. Biol. Chem. 1998, 273, 29745–29753. [Google Scholar] [CrossRef] [PubMed]
- Chelakkot, C.; Ghim, J.; Ryu, S.H. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp. Mol. Med. 2018, 50, 103. [Google Scholar] [CrossRef]
- Galipeau, H.J.; Verdu, E.F. The complex task of measuring intestinal permeability in basic and clinical science. Neurogastroenterol. Motil. 2016, 28, 957–965. [Google Scholar] [CrossRef]
- Groschwitz, K.R.; Hogan, S.P. Intestinal barrier function: Molecular regulation and disease pathogenesis. J. Allergy Clin. Immunol. 2009, 124, 3–22. [Google Scholar] [CrossRef]
- Berg, R.D. Bacterial translocation from the gastrointestinal tract. Trends Microbiol. 1995, 3, 149–154. [Google Scholar] [CrossRef]
- Berg, R.D.; Garlington, A.W. Translocation of certain indigenous bacteria from the gastrointestinal tract to the mesenteric lymph nodes and other organs in a gnotobiotic mouse model. Infect. Immun. 1979, 23, 403–411. [Google Scholar] [PubMed]
- Garcia-Tsao, G.; Lee, F.-Y.; Barden, G.E.; Cartun, R.; Brian West, A. Bacterial translocation to mesenteric lymph nodes is increased in cirrhotic rats with ascites. Gastroenterology 1995, 108, 1835–1841. [Google Scholar] [CrossRef]
- MacFie, J.; O’Boyle, C.; Mitchell, C.J.; Buckley, P.M.; Johnstone, D.; Sudworth, P. Gut origin of sepsis: A prospective study investigating associations between bacterial translocation, gastric microflora, and septic morbidity. Gut 1999, 45, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Barr, J.J. A bacteriophages journey through the human body. Immunol. Rev. 2017, 279, 106–122. [Google Scholar] [CrossRef] [PubMed]
- Wiest, R.; Rath, H.C. Bacterial translocation in the gut. Best Pract. Res. Clin. Gastroenterol. 2003, 17, 397–425. [Google Scholar] [CrossRef]
- Lehto, M.; Groop, P.H. The Gut-Kidney Axis: Putative Interconnections Between Gastrointestinal and Renal Disorders. Front. Endocrinol. (Lausanne) 2018, 9, 553. [Google Scholar] [CrossRef]
- Kohler, C.A.; Maes, M.; Slyepchenko, A.; Berk, M.; Solmi, M.; Lanctot, K.L.; Carvalho, A.F. The Gut-Brain Axis, Including the Microbiome, Leaky Gut and Bacterial Translocation: Mechanisms and Pathophysiological Role in Alzheimer’s Disease. Curr. Pharm. Des. 2016, 22, 6152–6166. [Google Scholar] [CrossRef]
- Yan, A.W.; Schnabl, B. Bacterial translocation and changes in the intestinal microbiome associated with alcoholic liver disease. World J. Hepatol. 2012, 4, 110–118. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhong, W. Targeting the gut barrier for the treatment of alcoholic liver disease. Liver Res. 2017, 1, 197–207. [Google Scholar] [CrossRef]
- Bode, J.C.; Bode, C.; Heidelbach, R.; Durr, H.K.; Martini, G.A. Jejunal microflora in patients with chronic alcohol abuse. Hepatogastroenterology 1984, 31, 30–34. [Google Scholar]
- Hartmann, P.; Seebauer, C.T.; Schnabl, B. Alcoholic liver disease: The gut microbiome and liver cross talk. Alcohol. Clin. Exp. Res. 2015, 39, 763–775. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, S.; Cani, P.D.; Neyrinck, A.M.; Starkel, P.; Jamar, F.; Mikolajczak, M.; Delzenne, N.M.; de Timary, P. Role of intestinal permeability and inflammation in the biological and behavioral control of alcohol-dependent subjects. Brain Behav. Immun. 2012, 26, 911–918. [Google Scholar] [CrossRef] [PubMed]
- Llopis, M.; Cassard, A.M.; Wrzosek, L.; Boschat, L.; Bruneau, A.; Ferrere, G.; Puchois, V.; Martin, J.C.; Lepage, P.; Le Roy, T.; et al. Intestinal microbiota contributes to individual susceptibility to alcoholic liver disease. Gut 2016, 65, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Keshavarzian, A.; Holmes, E.W.; Patel, M.; Iber, F.; Fields, J.Z.; Pethkar, S. Leaky gut in alcoholic cirrhosis: A possible mechanism for alcohol-induced liver damage. Am. J. Gastroenterol. 1999, 94, 200–207. [Google Scholar] [CrossRef]
- Bode, J.C.; Knuppel, H.; Schwerk, W.; Lorenz-Meyer, H.; Durr, H.K. Quantitative histomorphometric study of the jejunal mucosa in chronic alcoholics. Digestion 1982, 23, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Brozinsky, S.; Fani, K.; Grosberg, S.J.; Wapnick, S. Alcohol ingestion-induced changes in the human rectal mucosa: Light and electron microscopic studies. Dis. Colon Rectum 1978, 21, 329–335. [Google Scholar] [CrossRef]
- Tang, Y.; Banan, A.; Forsyth, C.B.; Fields, J.Z.; Lau, C.K.; Zhang, L.J.; Keshavarzian, A. Effect of Alcohol on miR-212 Expression in Intestinal Epithelial Cells and Its Potential Role in Alcoholic Liver Disease. Alcohol. Clin. Exp. Res. 2008, 32, 355–364. [Google Scholar] [CrossRef]
- Zhong, W.; McClain, C.J.; Cave, M.; Kang, Y.J.; Zhou, Z. The role of zinc deficiency in alcohol-induced intestinal barrier dysfunction. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G625–G633. [Google Scholar] [CrossRef]
- Ma, T.Y.; Nguyen, D.; Bui, V.; Nguyen, H.; Hoa, N. Ethanol modulation of intestinal epithelial tight junction barrier. Am. J. Physiol. 1999, 276, G965–G974. [Google Scholar] [CrossRef]
- Asai, K.; Buurman, W.A.; Reutelingsperger, C.P.; Schutte, B.; Kaminishi, M. Low concentrations of ethanol induce apoptosis in human intestinal cells. Scand. J. Gastroenterol. 2003, 38, 1154–1161. [Google Scholar] [CrossRef]
- Elamin, E.E.; Masclee, A.A.; Dekker, J.; Jonkers, D.M. Ethanol metabolism and its effects on the intestinal epithelial barrier. Nutr. Rev. 2013, 71, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Moro-Sibilot, L.; Blanc, P.; Taillardet, M.; Bardel, E.; Couillault, C.; Boschetti, G.; Traverse-Glehen, A.; Defrance, T.; Kaiserlian, D.; Dubois, B. Mouse and Human Liver Contain Immunoglobulin A-Secreting Cells Originating From Peyer’s Patches and Directed Against Intestinal Antigens. Gastroenterology 2016, 151, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Inamine, T.; Yang, A.M.; Wang, L.; Lee, K.C.; Llorente, C.; Schnabl, B. Genetic Loss of Immunoglobulin A Does Not Influence Development of Alcoholic Steatohepatitis in Mice. Alcohol. Clin. Exp. Res. 2016, 40, 2604–2613. [Google Scholar] [CrossRef] [PubMed]
- Riva, A.; Patel, V.; Kurioka, A.; Jeffery, H.C.; Wright, G.; Tarff, S.; Shawcross, D.; Ryan, J.M.; Evans, A.; Azarian, S.; et al. Mucosa-associated invariant T cells link intestinal immunity with antibacterial immune defects in alcoholic liver disease. Gut 2018, 67, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Volynets, V.; Kuper, M.A.; Strahl, S.; Maier, I.B.; Spruss, A.; Wagnerberger, S.; Konigsrainer, A.; Bischoff, S.C.; Bergheim, I. Nutrition, intestinal permeability, and blood ethanol levels are altered in patients with nonalcoholic fatty liver disease (NAFLD). Dig. Dis. Sci. 2012, 57, 1932–1941. [Google Scholar] [CrossRef]
- Luther, J.; Garber, J.J.; Khalili, H.; Dave, M.; Bale, S.S.; Jindal, R.; Motola, D.L.; Luther, S.; Bohr, S.; Jeoung, S.W.; et al. Hepatic Injury in Nonalcoholic Steatohepatitis Contributes to Altered Intestinal Permeability. Cell Mol. Gastroenterol. Hepatol. 2015, 1, 222–232. [Google Scholar] [CrossRef]
- Hawkesworth, S.; Moore, S.E.; Fulford, A.J.C.; Barclay, G.R.; Darboe, A.A.; Mark, H.; Nyan, O.A.; Prentice, A.M. Evidence for metabolic endotoxemia in obese and diabetic Gambian women. Nutr. Diabetes 2013, 3, e83. [Google Scholar] [CrossRef]
- Nazim, M.; Stamp, G.; Hodgson, H.J. Non-alcoholic steatohepatitis associated with small intestinal diverticulosis and bacterial overgrowth. Hepatogastroenterology 1989, 36, 349–351. [Google Scholar]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Masciana, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased Intestinal Permeability and Tight Junction Alterations in Nonalcoholic Fatty Liver Disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef]
- Giorgio, V.; Miele, L.; Principessa, L.; Ferretti, F.; Villa, M.P.; Negro, V.; Grieco, A.; Alisi, A.; Nobili, V. Intestinal permeability is increased in children with non-alcoholic fatty liver disease, and correlates with liver disease severity. Dig. Liver Dis. 2014, 46, 556–560. [Google Scholar] [CrossRef]
- Xin, D.; Zong-Shun, L.; Bang-Mao, W.; Lu, Z. Expression of intestinal tight junction proteins in patients with non-alcoholic fatty liver disease. Hepatogastroenterology 2014, 61, 136–140. [Google Scholar] [PubMed]
- Suzuki, T.; Hara, H. Dietary fat and bile juice, but not obesity, are responsible for the increase in small intestinal permeability induced through the suppression of tight junction protein expression in LETO and OLETF rats. Nutr. Metab. (Lond.) 2010, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Rah, B.; Bastola, D.; Dhawan, P.; Singh, A.B. Obesity-induces Organ and Tissue Specific Tight Junction Restructuring and Barrier Deregulation by Claudin Switching. Sci. Rep. 2017, 7, 5125. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Ichimura, M.; Tsuneyama, K.; Moritoki, Y.; Tsunashima, H.; Omagari, K.; Hara, M.; Yasuda, I.; Miyakawa, H.; Kikuchi, K. Fructo-oligosaccharides and intestinal barrier function in a methionine-choline-deficient mouse model of nonalcoholic steatohepatitis. PLoS ONE 2017, 12, e0175406. [Google Scholar] [CrossRef]
- Winer, D.A.; Luck, H.; Tsai, S.; Winer, S. The Intestinal Immune System in Obesity and Insulin Resistance. Cell Metab. 2016, 23, 413–426. [Google Scholar] [CrossRef]
- Al-Sadi, R.; Boivin, M.; Ma, T. Mechanism of cytokine modulation of epithelial tight junction barrier. Front. Biosci. (Landmark Ed.) 2009, 14, 2765–2778. [Google Scholar] [CrossRef]
- Teltschik, Z.; Wiest, R.; Beisner, J.; Nuding, S.; Hofmann, C.; Schoelmerich, J.; Bevins, C.L.; Stange, E.F.; Wehkamp, J. Intestinal bacterial translocation in rats with cirrhosis is related to compromised Paneth cell antimicrobial host defense. Hepatology 2012, 55, 1154–1163. [Google Scholar] [CrossRef]
- Alvarez-Silva, C.; Schierwagen, R.; Pohlmann, A.; Magdaleno, F.; Uschner, F.E.; Ryan, P.; Vehreschild, M.J.G.T.; Claria, J.; Latz, E.; Lelouvier, B.; et al. Compartmentalization of Immune Response and Microbial Translocation in Decompensated Cirrhosis. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Schierwagen, R.; Alvarez-Silva, C.; Madsen, M.S.A.; Kolbe, C.C.; Meyer, C.; Thomas, D.; Uschner, F.E.; Magdaleno, F.; Jansen, C.; Pohlmann, A.; et al. Circulating microbiome in blood of different circulatory compartments. Gut 2019, 68, 578–580. [Google Scholar] [CrossRef]
- Casafont Morencos, F.; de las Heras Castano, G.; Martin Ramos, L.; Lopez Arias, M.J.; Ledesma, F.; Pons Romero, F. Small bowel bacterial overgrowth in patients with alcoholic cirrhosis. Dig. Dis. Sci. 1996, 41, 552–556. [Google Scholar] [CrossRef]
- Maslennikov, R.; Pavlov, C.; Ivashkin, V. Small intestinal bacterial overgrowth in cirrhosis: Systematic review and meta-analysis. Hepatol. Int. 2018, 12, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Gunnarsdottir, S.A.; Sadik, R.; Shev, S.; Simren, M.; Sjovall, H.; Stotzer, P.O.; Abrahamsson, H.; Olsson, R.; Bjornsson, E.S. Small intestinal motility disturbances and bacterial overgrowth in patients with liver cirrhosis and portal hypertension. Am. J. Gastroenterol. 2003, 98, 1362–1370. [Google Scholar] [CrossRef] [PubMed]
- Campillo, B.; Pernet, P.; Bories, P.N.; Richardet, J.P.; Devanlay, M.; Aussel, C. Intestinal permeability in liver cirrhosis: Relationship with severe septic complications. Eur. J. Gastroenterol. Hepatol. 1999, 11, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Pascual, S.; Such, J.; Esteban, A.; Zapater, P.; Casellas, J.A.; Aparicio, J.R.; Girona, E.; Gutierrez, A.; Carnices, F.; Palazon, J.M.; et al. Intestinal permeability is increased in patients with advanced cirrhosis. Hepatogastroenterology 2003, 50, 1482–1486. [Google Scholar] [PubMed]
- Pijls, K.E.; Koek, G.H.; Elamin, E.E.; de Vries, H.; Masclee, A.A.; Jonkers, D.M. Large intestine permeability is increased in patients with compensated liver cirrhosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G147–G153. [Google Scholar] [CrossRef]
- Assimakopoulos, S.F.; Tsamandas, A.C.; Tsiaoussis, G.I.; Karatza, E.; Triantos, C.; Vagianos, C.E.; Spiliopoulou, I.; Kaltezioti, V.; Charonis, A.; Nikolopoulou, V.N.; et al. Altered intestinal tight junctions’ expression in patients with liver cirrhosis: A pathogenetic mechanism of intestinal hyperpermeability. Eur. J. Clin. Investig. 2012, 42, 439–446. [Google Scholar] [CrossRef]
- Such, J.; Guardiola, J.V.; de Juan, J.; Casellas, J.A.; Pascual, S.; Aparicio, J.R.; Sola-Vera, J.; Perez-Mateo, M. Ultrastructural characteristics of distal duodenum mucosa in patients with cirrhosis. Eur. J. Gastroenterol. Hepatol. 2002, 14, 371–376. [Google Scholar] [CrossRef]
- Sorribas, M.; Jakob, M.O.; Yilmaz, B.; Li, H.; Stutz, D.; Noser, Y.; De Gottardi, A.; Moghadamrad, S.; Hassan, M.; Albillos, A.; et al. FXR-modulates the gut-vascular barrier by regulating the entry sites for bacterial translocation in experimental cirrhosis. J. Hepatol. 2019. [Google Scholar] [CrossRef]
- Muñoz, L.; Albillos, A.; Nieto, M.; Reyes, E.; Lledó, L.; Monserrat, J.; Sanz, E.; de la Hera, A.; Alvarez-Mon, M. Mesenteric Th1 polarization and monocyte TNF-α production: First steps to systemic inflammation in rats with cirrhosis. Hepatology 2005, 42, 411–419. [Google Scholar] [CrossRef]
- Munoz, L.; Jose Borrero, M.; Ubeda, M.; Lario, M.; Diaz, D.; Frances, R.; Monserrat, J.; Pastor, O.; Aguado-Fraile, E.; Such, J.; et al. Interaction between intestinal dendritic cells and bacteria translocated from the gut in rats with cirrhosis. Hepatology 2012, 56, 1861–1869. [Google Scholar] [CrossRef]
- Ma, T.Y.; Iwamoto, G.K.; Hoa, N.T.; Akotia, V.; Pedram, A.; Boivin, M.A.; Said, H.M. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G367–G376. [Google Scholar] [CrossRef] [PubMed]
- Al-Sadi, R.; Ye, D.; Boivin, M.; Guo, S.; Hashimi, M.; Ereifej, L.; Ma, T.Y. Interleukin-6 modulation of intestinal epithelial tight junction permeability is mediated by JNK pathway activation of claudin-2 gene. PLoS ONE 2014, 9, e85345. [Google Scholar] [CrossRef] [PubMed]
- Elamin, E.; Masclee, A.; Troost, F.; Pieters, H.J.; Keszthelyi, D.; Aleksa, K.; Dekker, J.; Jonkers, D. Ethanol impairs intestinal barrier function in humans through mitogen activated protein kinase signaling: A combined in vivo and in vitro approach. PLoS ONE 2014, 9, e107421. [Google Scholar] [CrossRef] [PubMed]
- Elpek, G.O. Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: An update. World J. Gastroenterol. 2014, 20, 7260–7276. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Yang, L.; Van Rooijen, N.; Brenner, D.A.; Ohnishi, H.; Seki, E. Toll-like receptor 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation in mice. Hepatology 2013, 57, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Kiziltas, S. Toll-like receptors in pathophysiology of liver diseases. World J. Hepatol. 2016, 8, 1354–1369. [Google Scholar] [CrossRef]
- Thuy, S.; Ladurner, R.; Volynets, V.; Wagner, S.; Strahl, S.; KöNigsrainer, A.; Maier, K.-P.; Bischoff, S.C.; Bergheim, I. Nonalcoholic Fatty Liver Disease in Humans Is Associated with Increased Plasma Endotoxin and Plasminogen Activator Inhibitor 1 Concentrations and with Fructose Intake. J. Nutr. 2008, 138, 1452–1455. [Google Scholar] [CrossRef]
- Singh, R.; Bullard, J.; Kalra, M.; Assefa, S.; Kaul, A.K.; Vonfeldt, K.; Strom, S.C.; Conrad, R.S.; Sharp, H.L.; Kaul, R. Status of bacterial colonization, Toll-like receptor expression and nuclear factor-kappa B activation in normal and diseased human livers. Clin. Immunol. 2011, 138, 41–49. [Google Scholar] [CrossRef]
- Aragonès, G.; Colom-Pellicer, M.; Aguilar, C.; Guiu-Jurado, E.; Martínez, S.; Sabench, F.; Antonio Porras, J.; Riesco, D.; Del Castillo, D.; Richart, C.; et al. Circulating microbiota-derived metabolites: A “liquid biopsy? Int. J. Obes. 2019. [Google Scholar] [CrossRef]
- Stärkel, P.; De Saeger, C.; Strain, A.J.; Leclercq, I.; Horsmans, Y. NFκB, cytokines, TLR 3 and 7 expression in human end-stage HCV and alcoholic liver disease. Eur. J. Clin. Investig. 2010, 40, 575–584. [Google Scholar] [CrossRef]
- Roh, Y.S.; Zhang, B.; Loomba, R.; Seki, E. TLR2 and TLR9 contribute to alcohol-mediated liver injury through induction of CXCL1 and neutrophil infiltration. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G30–G41. [Google Scholar] [CrossRef] [PubMed]
- Goodridge, H.S.; Underhill, D.M. Fungal Recognition by TLR2 and Dectin-1. In Toll-like Receptors (TLRs) and Innate Immunity; Stefan Bauer, G.H., Ed.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 87–109. [Google Scholar] [CrossRef]
- Szabo, G.; Velayudham, A.; Romics, L.; Mandrekar, P. Modulation of non-alcoholic steatohepatitis by pattern recognition receptors in mice: The role of toll-like receptors 2 and 4. Alcohol. Clin. Exp. Res. 2005, 29, 140S–145S. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; De Minicis, S.; Österreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-β signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Moles, A.; Murphy, L.; Wilson, C.L.; Chakraborty, J.B.; Fox, C.; Park, E.J.; Mann, J.; Oakley, F.; Howarth, R.; Brain, J.; et al. A TLR2/S100A9/CXCL-2 signaling network is necessary for neutrophil recruitment in acute and chronic liver injury in the mouse. J. Hepatol. 2014, 60, 782–791. [Google Scholar] [CrossRef]
- Ji, L.; Xue, R.; Tang, W.; Wu, W.; Hu, T.; Liu, X.; Peng, X.; Gu, J.; Chen, S.; Zhang, S. Toll like receptor 2 knock-out attenuates carbon tetrachloride (CCl4)-induced liver fibrosis by downregulating MAPK and NF-κB signaling pathways. Febs Lett. 2014, 588, 2095–2100. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Ohira, M.; Nakano, R.; Tanaka, Y.; Ohdan, H. Hepatectomy leads to loss of TRAIL-expressing liver NK cells via downregulation of the CXCL9-CXCR3 axis in mice. PLoS ONE 2017, 12, e0186997. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Chen, Z.; Kato, N.; Gale, M.; Lemon, S.M. Distinct Poly(I-C) and Virus-activated Signaling Pathways Leading to Interferon- Production in Hepatocytes. J. Biol. Chem. 2005, 280, 16739–16747. [Google Scholar] [CrossRef]
- Wang, B.; Trippler, M.; Pei, R.; Lu, M.; Broering, R.; Gerken, G.; Schlaak, J.F. Toll-like receptor activated human and murine hepatic stellate cells are potent regulators of hepatitis C virus replication. J. Hepatol. 2009, 51, 1037–1045. [Google Scholar] [CrossRef]
- Tu, Z.; Bozorgzadeh, A.; Pierce, R.H.; Kurtis, J.; Crispe, I.N.; Orloff, M.S. TLR-dependent cross talk between human Kupffer cells and NK cells. JEM 2008, 205, 233–244. [Google Scholar] [CrossRef]
- Faure-Dupuy, S.; Vegna, S.; Aillot, L.; Dimier, L.; Esser, K.; Broxtermann, M.; Bonnin, M.; Bendriss-Vermare, N.; Rivoire, M.; Passot, G.; et al. Characterization of Pattern Recognition Receptor Expression and Functionality in Liver Primary Cells and Derived Cell Lines. J. Innate Immun. 2018, 10, 339–348. [Google Scholar] [CrossRef]
- Byun, J.-S.; Suh, Y.-G.; Yi, H.-S.; Lee, Y.-S.; Jeong, W.-I. Activation of toll-like receptor 3 attenuates alcoholic liver injury by stimulating Kupffer cells and stellate cells to produce interleukin-10 in mice. J. Hepatol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yang, Y.; Song, H.; Li, H.; Cui, A.; Liu, Y.; Su, L.; Crispe, I.N.; Tu, Z. Activated NK cells kill hepatic stellate cells via p38/PI3K signaling in a TRAIL-involved degranulation manner. J. Leukoc. Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Seo, W.; Eun, H.S.; Kim, S.Y.; Yi, H.-S.; Lee, Y.-S.; Park, S.-H.; Jang, M.-J.; Jo, E.; Kim, S.C.; Han, Y.-M.; et al. Exosome-mediated activation of toll-like receptor 3 in stellate cells stimulates interleukin-17 production by γδ T cells in liver fibrosis. Hepatology 2016, 64, 616–631. [Google Scholar] [CrossRef] [PubMed]
- Sharifnia, T.; Antoun, J.; Verriere, T.G.C.; Suarez, G.; Wattacheril, J.; Wilson, K.T.; Peek, R.M.; Abumrad, N.N.; Flynn, C.R. Hepatic TLR4 signaling in obese NAFLD. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G270–G278. [Google Scholar] [CrossRef]
- Saikia, P.; Roychowdhury, S.; Bellos, D.; Pollard, K.A.; McMullen, M.R.; McCullough, R.L.; McCullough, A.J.; Gholam, P.; De La Motte, C.; Nagy, L.E. Hyaluronic acid 35 normalizes TLR4 signaling in Kupffer cells from ethanol-fed rats via regulation of microRNA291b and its target Tollip. Sci. Rep. 2017, 7, 15671. [Google Scholar] [CrossRef]
- Yi, M.; Zhou, J.; Sun, H.; Shi, L.; Wu, Y.; Zhang, J.; Chen, Q. Correlation between TLR4 expression and gene polymorphism in peripheral blood mononuclear cells and condition of nonalcoholic fatty acid disease in Han people of Shaanxi. Int. J. Clin. Exp. Pathol. 2017, 10, 3496–3502. [Google Scholar]
- Jia, L.; Chang, X.; Qian, S.; Liu, C.; Lord, C.C.; Ahmed, N.; Lee, C.E.; Lee, S.; Gautron, L.; Mitchell, M.C.; et al. Hepatocyte toll-like receptor 4 deficiency protects against alcohol-induced fatty liver disease. Mol. Metab. 2018, 14, 121–129. [Google Scholar] [CrossRef]
- Hritz, I.; Mandrekar, P.; Velayudham, A.; Catalano, D.; Dolganiuc, A.; Kodys, K.; Kurt-Jones, E.; Szabo, G. The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology 2008, 48, 1224–1231. [Google Scholar] [CrossRef]
- Kim, S.Y.; Jeong, J.-M.; Kim, S.J.; Seo, W.; Kim, M.-H.; Choi, W.-M.; Yoo, W.; Lee, J.-H.; Shim, Y.-R.; Yi, H.-S.; et al. Pro-inflammatory hepatic macrophages generate ROS through NADPH oxidase 2 via endocytosis of monomeric TLR4–MD2 complex. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Ye, D.; Li, F.Y.L.; Lam, K.S.L.; Li, H.; Jia, W.; Wang, Y.; Man, K.; Lo, C.M.; Li, X.; Xu, A. Toll-like receptor-4 mediates obesity-induced non-alcoholic steatohepatitis through activation of X-box binding protein-1 in mice. Gut 2012, 61, 1058–1067. [Google Scholar] [CrossRef]
- Dattaroy, D.; Seth, R.K.; Sarkar, S.; Kimono, D.; Albadrani, M.; Chandrashekaran, V.; Hasson, F.A.; Singh, U.P.; Seth, R.K.; Scappinie, E.; et al. Sparstolonin B (SSnB) attenuates liver fibrosisvia a parallel conjugate pathway involving P53-P21 axis, TGF-beta signaling and focal adhesion that is TLR4 dependent. Eur. J. Pharmacol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Song, I.J.; Yang, Y.M.; Inokuchi-Shimizu, S.; Roh, Y.S.; Yang, L.; Seki, E. The contribution of toll-like receptor signaling to the development of liver fibrosis and cancer in hepatocyte-specific TAK1-deleted mice. Int. J. Cancer 2018, 142, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Etienne-Mesmin, L.; Vijay-Kumar, M.; Gewirtz, A.T.; Chassaing, B. Hepatocyte Toll-Like Receptor 5 Promotes Bacterial Clearance and Protects Mice Against High-Fat Diet–Induced Liver Disease. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 584–604. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Liu, F.; Yang, J.; Zhong, M.; Zhang, E.; Li, Y.; Zhou, D.; Cao, Y.; Li, W.; Yu, J.; et al. Over-activation of TLR5 signaling by high-dose flagellin induces liver injury in mice. Cell. Mol. Immunol. 2015, 12, 729–742. [Google Scholar] [CrossRef] [PubMed]
- Shu, M.; Huang, D.-D.; Hung, Z.-a.; Hu, X.-R.; Zhang, S. Inhibition of MAPK and NF-κB signaling pathways alleviate carbon tetrachloride (CCl4)-induced liver fibrosis in Toll-like receptor 5 (TLR5) deficiency mice. Biochem. Biophys. Res. Commun. 2016, 471, 233–239. [Google Scholar] [CrossRef]
- Kim, S.; Park, S.; Kim, B.; Kwon, J. Toll-like receptor 7 affects the pathogenesis of non-alcoholic fatty liver disease. Sci. Rep. 2016, 6, 27849. [Google Scholar] [CrossRef]
- Roh, Y.S.; Park, S.; Kim, J.W.; Lim, C.W.; Seki, E.; Kim, B. Toll-like receptor 7-mediated type I interferon signaling prevents cholestasis- and hepatotoxin-induced liver fibrosis. Hepatology 2014, 60, 237–249. [Google Scholar] [CrossRef]
- Stärkel, P.; Schnabl, B.; Leclercq, S.; Komuta, M.; Bataller, R.; Argemi, J.; Palma, E.; Chokshi, S.; Hellerbrand, C.; Maccioni, L.; et al. Deficient IL-6/Stat3 Signaling, High TLR7, and Type I Interferons in Early Human Alcoholic Liver Disease: A Triad for Liver Damage and Fibrosis. Hepatol. Commun. 2019, 3, 867–882. [Google Scholar] [CrossRef]
- Massey, V.L.; Qin, L.; Cabezas, J.; Caballeria, J.; Sancho-Bru, P.; Bataller, R.; Crews, F.T. TLR7-let-7 Signaling Contributes to Ethanol-Induced Hepatic Inflammatory Response in Mice and in Alcoholic Hepatitis. Alcohol. Clin. Exp. Res. 2018. [Google Scholar] [CrossRef]
- Roh, Y.S.; Kim, J.W.; Park, S.; Shon, C.; Kim, S.; Eo, S.K.; Kwon, J.K.; Lim, C.W.; Kim, B. Toll-Like Receptor-7 Signaling Promotes Nonalcoholic Steatohepatitis by Inhibiting Regulatory T Cells in Mice. Am. J. Pathol. 2018, 188, 2574–2588. [Google Scholar] [CrossRef]
- Mridha, A.R.; Haczeyni, F.; Yeh, M.M.; Haigh, W.G.; Ioannou, G.N.; Barn, V.; Ajamieh, H.; Adams, L.; Hamdorf, J.M.; Teoh, N.C.; et al. TLR9 is up-regulated in human and murine NASH: Pivotal role in inflammatory recruitment and cell survival. Clin. Sci. (London, England: 1979) 2017, 131, 2145. [Google Scholar] [CrossRef] [PubMed]
- Wiest, R.; Lawson, M.; Geuking, M. Pathological bacterial translocation in liver cirrhosis. J. Hepatol. 2014, 60, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Koutsounas, I.; Kaltsa, G.; Siakavellas, S.I.; Bamias, G. Markers of bacterial translocation in end-stage liver disease. World J. Hepatol. 2015, 7, 2264–2273. [Google Scholar] [CrossRef] [PubMed]
- Vergis, N.; Atkinson, S.R.; Knapp, S.; Maurice, J.; Allison, M.; Austin, A.; Forrest, E.H.; Masson, S.; McCune, A.; Patch, D.; et al. In Patients With Severe Alcoholic Hepatitis, Prednisolone Increases Susceptibility to Infection and Infection-Related Mortality, and Is Associated With High Circulating Levels of Bacterial DNA. Gastroenterology 2017, 152, 1068–1077.e4. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Kodama, Y.; Inokuchi, S.; Schnabl, B.; Aoyama, T.; Ohnishi, H.; Olefsky, J.M.; Brenner, D.A.; Seki, E. Toll-Like Receptor 9 Promotes Steatohepatitis by Induction of Interleukin-1β in Mice. Gastroenterology 2010, 139, 323–334.e7. [Google Scholar] [CrossRef]
- Gieling, R.; Wallace, K.; Han, Y.-P. Interleukin-1 participates in the progression from liver injury to fibrosis. Am. J. Physiol. 2009, 296, G1324. [Google Scholar] [CrossRef]
- Bala, S.; Marcos, M.; Gattu, A.; Catalano, D.; Szabo, G. Acute Binge Drinking Increases Serum Endotoxin and Bacterial DNA Levels in Healthy Individuals. PLoS ONE 2014, 9, e96864. [Google Scholar] [CrossRef]
- Gabele, E.; Muhlbauer, M.; Dorn, C.; Weiss, T.S.; Froh, M.; Schnabl, B.; Wiest, R.; Scholmerich, J.; Obermeier, F.; Hellerbrand, C. Role of TLR9 in hepatic stellate cells and experimental liver fibrosis. Biochem. Biophys. Res. Commun. 2008, 376, 271–276. [Google Scholar] [CrossRef]
- Boaru, S.G.; Borkham-Kamphorst, E.; Tihaa, L.; Haas, U.; Weiskirchen, R. Expression analysis of inflammasomes in experimental models of inflammatory and fibrotic liver disease. J. Inflamm. 2012, 9, 49. [Google Scholar] [CrossRef]
- Watanabe, A.; Sohail, M.A.; Gomes, D.A.; Hashmi, A.; Nagata, J.; Sutterwala, F.S.; Mahmood, S.; Jhandier, M.N.; Shi, Y.; Flavell, R.A.; et al. Inflammasome-mediated regulation of hepatic stellate cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G1248–G1257. [Google Scholar] [CrossRef]
- Nicoletti, F.; Zaccone, P.; Xiang, M.; Magro, G.; Di Mauro, M.; Di Marco, R.; Garotta, G.; Meroni, P. ESSENTIAL PATHOGENETIC ROLE FOR INTERFERON (IFN-)γ IN CONCANAVALIN A-INDUCED T CELL-DEPENDENT HEPATITIS: EXACERBATION BY EXOGENOUS IFN-γ AND PREVENTION BY IFN-γ RECEPTOR-IMMUNOGLOBULIN FUSION PROTEIN. Cytokine 2000, 12, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-G.; Fang, W.-L.; Wei, J.; Wang, T.; Wang, N.; Ma, J.-L.; Shi, M. The involvement of NLRX1 and NLRP3 in the development of nonalcoholic steatohepatitis in mice. J. Chin. Med. Assoc. 2013, 76, 686–692. [Google Scholar] [CrossRef]
- Xu, T.; Du, Y.; Fang, X.-B.; Chen, H.; Zhou, D.-D.; Wang, Y.; Zhang, L. New insights into Nod-like receptors (NLRs) in liver diseases. Int. J. Physiol. Pathophysiol. Pharmacol. 2018, 10, 1–16. [Google Scholar] [PubMed]
- Seifert, L.; Deutsch, M.; Alothman, S.; Alqunaibit, D.; Werba, G.; Pansari, M.; Pergamo, M.; Ochi, A.; Torres-Hernandez, A.; Levie, E.; et al. Dectin-1 Regulates Hepatic Fibrosis and Hepatocarcinogenesis by Suppressing TLR4 Signaling Pathways. Cell Rep. 2015, 13, 1909–1921. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.M.; Inamine, T.; Hochrath, K.; Chen, P.; Wang, L.; Llorente, C.; Bluemel, S.; Hartmann, P.; Xu, J.; Koyama, Y.; et al. Intestinal fungi contribute to development of alcoholic liver disease. J. Clin. Investig. 2017, 127, 2829–2841. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Li, H.; Ma, L.; Zhou, J.; Guo, X.; Woo, S.-L.; Pei, Y.; Knight, L.R.; Deveau, M.; Chen, Y.; et al. Expression of STING Is Increased in Liver Tissues From Patients With NAFLD and Promotes Macrophage-Mediated Hepatic Inflammation and Fibrosis in Mice. Gastroenterology 2018, 155, 1971–1984.e4. [Google Scholar] [CrossRef]
- Petrasek, J.; Iracheta-Vellve, A.; Csak, T.; Satishchandran, A.; Kodys, K.; Kurt-Jones, E.; Fitzgerald, K.; Szabo, G. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc. Natl. Acad. Sci. USA 2013, 110, 16544–16549. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, Y.; An, W.; Song, J.; Zhang, Y.; Zhao, X. STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis.(RESEARCH ARTICLE). J. Clin. Investig. 2019, 129, 546. [Google Scholar] [CrossRef]
- Iracheta-Vellve, A.; Petrasek, J.; Gyongyosi, B.; Satishchandran, A.; Lowe, P.; Kodys, K.; Catalano, D.; Calenda, C.D.; Kurt-Jones, E.A.; Fitzgerald, K.A.; et al. Endoplasmic Reticulum Stress-induced Hepatocellular Death Pathways Mediate Liver Injury and Fibrosis via Stimulator of Interferon Genes. J. Biol. Chem. 2016, 291, 26794–26805. [Google Scholar] [CrossRef]
- Soares, J.-B.; Pimentel-Nunes, P.; Afonso, L.; Rolanda, C.; Lopes, P.; Roncon-Albuquerque, R.; Gonçalves, N.; Boal-Carvalho, I.; Pardal, F.; Lopes, S.; et al. Increased hepatic expression of TLR2 and TLR4 in the hepatic inflammation-fibrosis-carcinoma sequence. Innate Immun. 2012, 18, 700–708. [Google Scholar] [CrossRef]
- Radaeva, S.; Sun, R.; Jaruga, B.; Nguyen, V.T.; Tian, Z.; Gao, B. Natural Killer Cells Ameliorate Liver Fibrosis by Killing Activated Stellate Cells in NKG2D-Dependent and Tumor Necrosis Factor–Related Apoptosis-Inducing Ligand–Dependent Manners. Gastroenterology 2006, 130, 435–452. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.-I.; Park, O.; Radaeva, S.; Gao, B. STAT1 inhibits liver fibrosis in mice by inhibiting stellate cell proliferation and stimulating NK cell cytotoxicity. Hepatology 2006, 44, 1441–1451. [Google Scholar] [CrossRef]
- Queck, A.; Carnevale, R.; Uschner, F.E.; Schierwagen, R.; Klein, S.; Jansen, C.; Meyer, C.; Praktiknjo, M.; Thomas, D.; Strassburg, C.; et al. Role of portal venous platelet activation in patients with decompensated cirrhosis and TIPS. Gut 2019. [Google Scholar] [CrossRef] [PubMed]
- Cengiz, M.; Ozenirler, S.; Elbeg, S. Role of serum toll-like receptors 2 and 4 in non-alcoholic steatohepatitis and liver fibrosis. J. Gastroenterol. Hepatol. 2015, 30, 1190–1196. [Google Scholar] [CrossRef] [PubMed]
- González-Navajas, J.M.; Bellot, P.; Francés, R.; Zapater, P.; Muñoz, C.; García-Pagán, J.C.; Pascual, S.; Pérez-Mateo, M.; Bosch, J.; Such, J. Presence of bacterial-DNA in cirrhosis identifies a subgroup of patients with marked inflammatory response not related to endotoxin. J. Hepatol. 2008, 48, 61–67. [Google Scholar] [CrossRef]
- Watanabe, A.; Hashmi, A.; Gomes, D.A.; Town, T.; Badou, A.; Flavell, R.A.; Mehal, W.Z. Apoptotic hepatocyte DNA inhibits hepatic stellate cell chemotaxis via toll-like receptor 9. Hepatology 2007, 46, 1509–1518. [Google Scholar] [CrossRef]
- Dharancy, S.; Body-Malapel, M.; Louvet, A.; Berrebi, D.; Gantier, E.; Gosset, P.; Viala, J.; Hollebecque, A.; Moreno, C.; Philpott, D.J.; et al. Neutrophil Migration During Liver Injury Is Under Nucleotide-Binding Oligomerization Domain 1 Control. Gastroenterology 2010, 138, 1546–1556.e5. [Google Scholar] [CrossRef]
- Kobayashi, K.S.; Chamaillard, M.; Ogura, Y.; Henegariu, O.; Inohara, N.; Nuñez, G.; Flavell, R.A. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science (New York, NY) 2005, 307, 731–734. [Google Scholar] [CrossRef]
- Seki, E.; De Minicis, S.; Gwak, G.-Y.; Kluwe, J.; Inokuchi, S.; Bursill, C.A.; Llovet, J.M.; Brenner, D.A.; Schwabe, R.F. CCR1 and CCR5 promote hepatic fibrosis in mice.(Research article)(Chemokine (C-C motif) receptor)(Report). J. Clin. Investig. 2009, 119, 1858. [Google Scholar] [CrossRef]
- Veldt, B.J.; Laine, F.; Guillygomarc’h, A.; Lauvin, L.; Boudjema, K.; Messner, M.; Brissot, P.; Deugnier, Y.; Moirand, R. Indication of liver transplantation in severe alcoholic liver cirrhosis: Quantitative evaluation and optimal timing. J. Hepatol. 2002, 36, 93–98. [Google Scholar] [CrossRef]
- Ge, P.S.; Runyon, B.A. Treatment of Patients with Cirrhosis. N. Engl. J. Med. 2016, 375, 2104–2105. [Google Scholar] [CrossRef] [PubMed]
- Nier, A.; Engstler, A.J.; Maier, I.B.; Bergheim, I. Markers of intestinal permeability are already altered in early stages of non-alcoholic fatty liver disease: Studies in children. PLoS ONE 2017, 12, e0183282. [Google Scholar] [CrossRef] [PubMed]
- Tuomisto, S.; Pessi, T.; Collin, P.; Vuento, R.; Aittoniemi, J.; Karhunen, P.J. Changes in gut bacterial populations and their translocation into liver and ascites in alcoholic liver cirrhotics. BMC Gastroenterol. 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Floreani, A.; Baragiotta, A.; Pizzuti, D.; Martines, D.; Cecchetto, A.; Chiarelli, S. Mucosal IgA defect in primary biliary cirrhosis. Am. J. Gastroenterol. 2002, 97, 508–510. [Google Scholar] [CrossRef]
- Schwimmer, J.B.; Johnson, J.S.; Angeles, J.E.; Behling, C.; Belt, P.H.; Borecki, I.; Bross, C.; Durelle, J.; Goyal, N.P.; Hamilton, G.; et al. Microbiome Signatures Associated with Steatohepatitis and Moderate to Severe Fibrosis in Children With Nonalcoholic Fatty Liver Disease. Gastroenterology 2019. [Google Scholar] [CrossRef]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef]
- Adachi, Y.; Moore, L.E.; Bradford, B.U.; Gao, W.; Thurman, R.G. Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology 1995, 108, 218–224. [Google Scholar] [CrossRef]
- Douhara, A.; Moriya, K.; Yoshiji, H.; Noguchi, R.; Namisaki, T.; Kitade, M.; Kaji, K.; Aihara, Y.; Nishimura, N.; Takeda, K.; et al. Reduction of endotoxin attenuates liver fibrosis through suppression of hepatic stellate cell activation and remission of intestinal permeability in a rat non-alcoholic steatohepatitis model. Mol. Med. Rep. 2015, 11, 1693–1700. [Google Scholar] [CrossRef]
- Wiest, R.; Albillos, A.; Trauner, M.; Bajaj, J.S.; Jalan, R. Targeting the gut-liver axis in liver disease. J. Hepatol. 2017, 67, 1084–1103. [Google Scholar] [CrossRef]
- Gangarapu, V.; Ince, A.T.; Baysal, B.; Kayar, Y.; Kilic, U.; Gok, O.; Uysal, O.; Senturk, H. Efficacy of rifaximin on circulating endotoxins and cytokines in patients with nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 2015, 27, 840–845. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Heimanson, Z.; Israel, R.; Sanyal, A. Efficacy of rifaximin soluble solid dispersion in patients with early decompensated cirrhosis and a Conn score of 0: A post hoc analysis of a randomized, double-blind, placebo-controlled trial. J. Hepatol. 2019, 70, E631. [Google Scholar] [CrossRef]
- Kang, S.H.; Lee, Y.B.; Lee, J.H.; Nam, J.Y.; Chang, Y.; Cho, H.; Yoo, J.J.; Cho, Y.Y.; Cho, E.J.; Yu, S.J.; et al. Rifaximin treatment is associated with reduced risk of cirrhotic complications and prolonged overall survival in patients experiencing hepatic encephalopathy. Aliment. Pharmacol. Ther. 2017, 46, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Schaubeck, M.; Lagkouvardos, I.; Blesl, A.; Heinzlmeir, S.; Hahne, H.; Clavel, T.; Panda, S.; Ludwig, C.; Kuster, B.; et al. Increased Pancreatic Protease Activity in Response to Antibiotics Impairs Gut Barrier and Triggers Colitis. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 370. [Google Scholar] [CrossRef] [PubMed]
- Tulstrup, M.V.L.; Christensen, E.G.; Carvalho, V.; Linninge, C.; Ahrne, S.; Hojberg, O.; Licht, T.R.; Bahl, M.I. Antibiotic Treatment Affects Intestinal Permeability and Gut Microbial Composition in Wistar Rats Dependent on Antibiotic Class. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Liu, Y.L.; Kirpich, I.; Ma, Z.H.; Wang, C.L.; Zhang, M.; Suttles, J.; McClain, C.; Feng, W.K. Lactobacillus rhamnosus GG reduces hepatic TNF alpha production and inflammation in chronic alcohol-induced liver injury. J. Nutr. Biochem. 2013, 24, 1609–1615. [Google Scholar] [CrossRef]
- Tian, F.W.; Chi, F.F.; Wang, G.; Liu, X.M.; Zhang, Q.X.; Chen, Y.Q.; Zhang, H.; Chen, W. Lactobacillus rhamnosus CCFM1107 treatment ameliorates alcohol -induced liver injury in a mouse model of chronic alcohol feeding. J. Microbiol. 2015, 53, 856–863. [Google Scholar] [CrossRef]
- Wang, Y.H.; Liu, Y.L.; Kirpich, I.; McClain, C.; Feng, W.K. Lactobacillus rhamnosus GG treatment potentiates intestinal hypoxia-inducible factor, promotes intestinal integrity, prevents inflammation, and ameliorates alcohol-induced liver injury. Faseb J. 2012, 26. [Google Scholar]
- Moratalla, A.; Gomez-Hurtado, I.; Santacruz, A.; Moya, A.; Peiro, G.; Zapater, P.; Gonzalez-Navajas, J.M.; Gimenez, P.; Such, J.; Sanz, Y.; et al. Protective effect of Bifidobacterium pseudocatenulatum CECT7765 against induced bacterial antigen translocation in experimental cirrhosis. Liver Int. 2014, 34, 850–858. [Google Scholar] [CrossRef]
- Sanchez, E.; Nieto, J.C.; Vidal, S.; Santiago, A.; Martinez, X.; Sancho, F.J.; Sancho-Bru, P.; Mirelis, B.; Corominola, H.; Juarez, C.; et al. Fermented milk containing Lactobacillus paracasei subsp paracasei CNCM I-1518 reduces bacterial translocation in rats treated with carbon tetrachloride. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Holscher, H.D. Dietary fiber and prebiotics and the gastrointestinal microbiota. Gut Microbes 2017, 8, 172–184. [Google Scholar] [CrossRef]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Backhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.Y.; Li, Z.R.; Green, R.S.; Holzman, I.R.; Lin, J. Butyrate Enhances the Intestinal Barrier by Facilitating Tight Junction Assembly via Activation of AMP-Activated Protein Kinase in Caco-2 Cell Monolayers. J. Nutr. 2009, 139, 1619–1625. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, D.S.G.; Jensen, B.B.; Theil, P.K.; Nielsen, T.S.; Knudsen, K.E.B.; Purup, S. Effect of butyrate and fermentation products on epithelial integrity in a mucus-secreting human colon cell line. J. Funct. Foods 2018, 40, 9–17. [Google Scholar] [CrossRef]
- Tang, Y.M.; Forsyth, C.B.; Banan, A.; Fields, J.Z.; Keshavarzian, A. Oats Supplementation Prevents Alcohol-Induced Gut Leakiness in Rats by Preventing Alcohol-Induced Oxidative Tissue Damage. J. Pharm. Exp. 2009, 329, 952–958. [Google Scholar] [CrossRef]
- Krawczyk, M.; Maciejewska, D.; Ryterska, K.; Czerwinka-Rogowska, M.; Jamiol-Milc, D.; Skonieczna-Zydecka, K.; Milkiewicz, P.; Raszeja-Wyszomirska, J.; Stachowska, E. Gut Permeability Might be Improved by Dietary Fiber in Individuals with Nonalcoholic Fatty Liver Disease (NAFLD) Undergoing Weight Reduction. Nutrients 2018, 10. [Google Scholar] [CrossRef]
- Loman, B.R.; Hernandez-Saavedra, D.; An, R.P.; Rector, R.S. Prebiotic and probiotic treatment of nonalcoholic fatty liver disease: A systematic review and meta-analysis. Nutr. Rev. 2018, 76, 822–839. [Google Scholar] [CrossRef]
- Cao, Q.; Yu, C.B.; Yang, S.G.; Cao, H.C.; Chen, P.; Deng, M.; Li, L.J. Effect of probiotic treatment on cirrhotic patients with minimal hepatic encephalopathy: A meta-analysis. Hepatob. Pancreat Dis. 2018, 17, 9–16. [Google Scholar] [CrossRef]
- Anwar, M.A.; Shah, M.; Kim, J.; Choi, S. Recent clinical trends in Toll-like receptor targeting therapeutics. Med. Res. Rev. 2019, 39, 1053–1090. [Google Scholar] [CrossRef]
- Reilly, M.; Miller, R.M.; Thomson, M.H.; Patris, V.; Ryle, P.; McLoughlin, L.; Mutch, P.; Gilboy, P.; Miller, C.; Broekema, M.; et al. Randomized, Double-Blind, Placebo-Controlled, Dose-Escalating Phase I, Healthy Subjects Study of Intravenous OPN-305, a Humanized Anti-TLR2 Antibody. Clin. Pharm. 2013, 94, 593–600. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Jabbour, E.J.; Konopleva, M.Y.; Daver, N.G.; Borthakur, G.; DiNardo, C.D.; Bose, P.; Patel, P.; Komrokji, R.S.; Shastri, A.; et al. A Clinical Study of Tomaralimab (OPN-305), a Toll-like Receptor 2 (TLR-2) Antibody, in Heavily Pre-Treated Transfusion Dependent Patients with Lower Risk Myelodysplastic Syndromes (MDS) That Have Received and Failed on Prior Hypomethylating Agent (HMA) Therapy. Blood 2018, 132. [Google Scholar] [CrossRef]
- Monnet, E.; Lapeyre, G.; van Poelgeest, E.; Jacqmin, P.; de Graaf, K.; Reijers, J.; Moerland, M.; Burggraaf, J.; de Min, C. Evidence of NI-0101 Pharmacological Activity, an Anti-TLR4 Antibody, in a Randomized Phase I Dose Escalation Study in Healthy Volunteers Receiving LPS. Clin. Pharm. 2017, 101, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.J.; Coffey, C.S.; Conwit, R.; Cudkowicz, M.E.; Gleason, T.; Goodman, A.; Klawiter, E.C.; Matsuda, K.; McGovern, M.; Naismith, R.T.; et al. Phase 2 Trial of Ibudilast in Progressive Multiple Sclerosis. N. Engl. J. Med. 2018, 379, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Diehl, A.M.; Harrison, S.; Caldwell, S.; Rinella, M.; Paredes, A.; Moylan, C.; Guy, C.; Bashir, M.; Wang, Y.; Miller, L.; et al. JKB-121 in patients with nonalcoholic steatohepatitis: A phase 2 double blind randomized placebo control study. J. Hepatol. 2018, 68, S103. [Google Scholar] [CrossRef]
- Hoque, R.; Farooq, A.; Malik, A.; Trawick, B.N.; Berberich, D.W.; McClurg, J.P.; Galen, K.P.; Mehal, W. A Novel Small-Molecule Enantiomeric Analogue of Traditional (-)-Morphinans Has Specific TLR9 Antagonist Properties and Reduces Sterile Inflammation-Induced Organ Damage. J. Immunol. 2013, 190, 4297–4304. [Google Scholar] [CrossRef] [PubMed]
- Shaker, M.E.; Trawick, B.N.; Mehal, W.Z. The novel TLR9 antagonist COV08-0064 protects from ischemia/reperfusion injury in non-steatotic and steatotic mice livers. Biochem Pharm. 2016, 112, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Jansen, T.L.; Kluck, V.; Janssen, M.; Comarniceanu, A.; Efde, M.; Scribner, C.L.; Barrow, R.B.; Skouras, D.B.; Dinarello, C.A.; Joosten, L.A. The First Phase 2a Proof-of-Concept Study of a Selective Nlrp3 Inflammasome Inhibitor, Dapansutrile (Tm) (Olt1177 (Tm)), in Acute Gout. Ann. Rheum. Dis. 2019, 78, A70–A71. [Google Scholar] [CrossRef]
- Mullard, A. NLRP3 inhibitors stoke anti-inflammatory ambitions. Nat. Rev. Drug Discov. 2019, 18, 405–410. [Google Scholar] [CrossRef]
- Haag, S.M.; Gulen, M.F.; Reymond, L.; Gibelin, A.; Abrami, L.; Decout, A.; Heymann, M.; van der Goot, F.G.; Turcatti, G.; Behrendt, R.; et al. Targeting STING with covalent small-molecule inhibitors. Nature 2018, 559, 269–273. [Google Scholar] [CrossRef]
- Sheridan, C. Drug developers switch gears to inhibit STING. Nat. Biotechnol. 2019, 37, 199–201. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).