Rewiring of Cancer Cell Metabolism by Mitochondrial VDAC1 Depletion Results in Time-Dependent Tumor Reprogramming: Glioblastoma as a Proof of Concept

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Xenograft Experiments
2.2. Immunoblotting and Immunohistochemistry (IHC) and Immunofluorescence (IF) Staining
2.3. RNA Preparation, qRT-PCR, Liquid Chromatography–High–Resolution Mass Spectrometry (LC-HR-MS/MS) and Proteomics Analysis
2.4. Statistical Analyses for Identification of Differentially Expressed Proteins
2.5. Statistics
3. Results
3.1. VDAC1 Depletion Inhibits Cancer Tumor Development and Induced Metabolic Reprograming Following Both Short- and Long-Term Treatment
3.2. Tumor Treatment with si-hVDAC1 Decreases CSCs and Increases Expression of Differentiation-Associated Proteins
3.3. VDAC1 Depletion in GBM Xenografts Does Not Induce Cell Death but Results in an Increase in the Expression of Pro-Apoptotic Proteins, and a Decrease in TSPO Expression
3.4. TSPO Is Localized to Both the Mitochondria and the Nucleus and VDAC1 Depletion in GBM Xenografts Decreases the Expression of TSPO
3.5. Mass Spectrometry Analysis of the GBM Tumor Protein Profile after Short- and Long-Term si-hVDAC1 Treatment
3.6. Differential Expression of Metabolism-, Transport-, and Trafficking-Related Proteins after Short- and Long-Term si-hVDAC1 Treatment of GBM Tumors
3.7. Differential Expression of Signaling-, Development-, Differentiation-, and Human and Mouse Microenvironment-Related Proteins in GBM Tumors after Short- and Long-Term si-hVDAC1 Treatment
3.8. Modified Expression of Proteins Associated with Protein Synthesis and Degradation, DNA Structure and Replication, and Epigenetic Regulation upon si-hVDAC1 Treatment
4. Discussion
4.1. Metabolic Rewiring upon VDAC1 Depletion
4.2. TSPO Over-Expression in GBM, Nuclear Localization and Regulation by VDAC1
4.3. Tumor Cell Differentiation Precedes Metabolic Reprograming
4.4. si-hVDAC1 Tumor Treatment Leads to Microenvironment Rearrangement
4.5. Rewiring Metabolism Alters Signaling Pathways, as Mediated via Regulation of Protein Synthesis and Degradation, Epigenetics, and Gene Transcription
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alexander, B.M.; Cloughesy, T.F. Adult Glioblastoma. J. Clin. Oncol. 2017, 35, 2402–2409. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [PubMed]
- Romani, M.; Pistillo, M.P.; Carosio, R.; Morabito, A.; Banelli, B. Immune Checkpoints and Innovative Therapies in Glioblastoma. Front. Oncol. 2018, 8, 464. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO Classification of Tumours of the Central Nervous System. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Bonavia, R.; Inda, M.M.; Cavenee, W.K.; Furnari, F.B. Heterogeneity maintenance in glioblastoma: A social network. Cancer Res. 2011, 71, 4055–4060. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Dick, J.E. Stem cell concepts renew cancer research. Blood 2008, 112, 4793–4807. [Google Scholar] [CrossRef] [Green Version]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef]
- Cojoc, M.; Mäbert, K.; Muders, M.H.; Dubrovska, A. A role for cancer stem cells in therapy resistance: Cellular and molecular mechanisms. Semin. Cancer Biol. 2015, 31, 16–27. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891. [Google Scholar] [CrossRef]
- Dhup, S.; Kumar Dadhich, R.; Ettore Porporato, P.; Sonveaux, P. Multiple Biological Activities of Lactic Acid in Cancer: Influences on Tumor Growth, Angiogenesis and Metastasis. Curr. Pharm. Des. 2012, 18, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, S.; Zadeh, G. Metabolic reprogramming in glioblastoma: The influence of cancer metabolism on epigenetics and unanswered questions. Neuro Oncol. 2016, 18, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Puzio-Kuter, A.M. The Control of the Metabolic Switch in Cancers by Oncogenes and Tumor Suppressor Genes. Science 2010, 330, 1340–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Wolf, A.; Agnihotri, S.; Micallef, J.; Mukherjee, J.; Sabha, N.; Cairns, R.; Hawkins, C.; Guha, A. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J. Exp. Med. 2011, 208, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Arif, T.; Krelin, Y.; Nakdimon, I.; Benharroch, D.; Paul, A.; Dadon-Klein, D.; Shoshan-Barmatz, V. VDAC1 is a molecular target in glioblastoma, with its depletion leading to reprogrammed metabolism and reversed oncogenic properties. Neuro Oncol. 2017, 19, 951–964. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Ben-Hail, D.; Admoni, L.; Krelin, Y.; Tripathi, S.S. The mitochondrial voltage-dependent anion channel 1 in tumor cells. Biochim. Biophys. Acta 2015, 1848, 2547–2575. [Google Scholar] [CrossRef] [Green Version]
- Arif, T.; Vasilkovsky, L.; Refaely, Y.; Konson, A.; Shoshan-Barmatz, V. Silencing VDAC1 Expression by siRNA Inhibits Cancer Cell Proliferation and Tumor Growth In Vivo. Mol. Ther. Nucleic Acids 2014, 3, e159. [Google Scholar] [CrossRef]
- Arif, T.; Paul, A.; Krelin, Y.; Shteinfer-Kuzmine, A.; Shoshan-Barmatz, V.J.C. Mitochondrial VDAC1 Silencing Leads to Metabolic Rewiring and the Reprogramming of Tumour Cells into Advanced Differentiated States. Cancers 2018, 10, 499. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med. 2010, 31, 227–285. [Google Scholar] [CrossRef]
- Arif, T.; Amsalem, Z.; Shoshan-Barmatz, V. Metabolic Reprograming Via Silencing of Mitochondrial VDAC1 Expression Encourages Differentiation of Cancer Cells. Mol. Ther. Nucleic Acids 2019, 17, 24–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Ghesquiere, B.; Wong, B.W.; Kuchnio, A.; Carmeliet, P.J.N. Metabolism of stromal and immune cells in health and disease. Nature 2014, 511, 167. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Majeed, R.; Hamid, A.; Qurishi, Y.; Qazi, A.K.; Hussain, A.; Ahmed, M.; Najar, R.A.; Bhat, J.A.; Singh, S.K.; Saxena, A.K. Therapeutic Targeting of Cancer Cell Metabolism: Role of Metabolic Enzymes, Oncogenes and Tumor Suppressor Genes. J. Mol. Med. 2012, 4, 205–212. [Google Scholar]
- Xavier, J.M.; Morgado, A.L.; Sola, S.; Rodrigues, C.M. Mitochondrial translocation of p53 modulates neuronal fate by preventing differentiation-induced mitochondrial stress. Antioxid. Redox Signal. 2014, 21, 1009–1024. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Pedini, F.; Mollinari, C.; Condorelli, G.; Bonci, D.; Bez, A.; Colombo, A.; Parati, E.; Peschle, C.; De Maria, R. Absence of caspase 8 and high expression of PED protect primitive neural cells from cell death. J. Exp. Med. 2004, 200, 1257–1266. [Google Scholar] [CrossRef]
- Krelin, Y.; Zhang, L.; Kang, T.B.; Appel, E.; Kovalenko, A.; Wallach, D. Caspase-8 deficiency facilitates cellular transformation in vitro. Cell Death Differ. 2008, 15, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Fernando, P.; Kelly, J.F.; Balazsi, K.; Slack, R.S.; Megeney, L.A. Caspase 3 activity is required for skeletal muscle differentiation. Proc. Natl. Acad. Sci. USA 2002, 99, 11025–11030. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Kepp, O.; Trojel-Hansen, C.; Kroemer, G. Non-apoptotic functions of apoptosis-regulatory proteins. EMBO Rep. 2012, 13, 322–330. [Google Scholar] [CrossRef]
- Murray, T.V.; McMahon, J.M.; Howley, B.A.; Stanley, A.; Ritter, T.; Mohr, A.; Zwacka, R.; Fearnhead, H.O. A non-apoptotic role for caspase-9 in muscle differentiation. J. Cell Sci. 2008, 121, 3786–3793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, E.M.; Wilkinson, A.S.; Jackson, J.S.; Mehra, R.; Varambally, S.; Chinnaiyan, A.M.; Wilkinson, J.C. The enzymatic activity of apoptosis-inducing factor supports energy metabolism benefiting the growth and invasiveness of advanced prostate cancer cells. J. Biol. Chem. 2012, 287, 43862–43875. [Google Scholar] [CrossRef] [PubMed]
- Sevrioukova, I.F. Apoptosis-inducing factor: Structure, function, and redox regulation. Antioxid. Redox Signal. 2011, 14, 2545–2579. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, V.; Baraldi, M.; Guilarte, T.R.; Knudsen, T.B.; Lacapere, J.J.; Lindemann, P.; Norenberg, M.D.; Nutt, D.; Weizman, A.; Zhang, M.R.; et al. Translocator protein (18 kDa): New nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol. Sci. 2006, 27, 402–409. [Google Scholar] [CrossRef]
- Carayon, P.; Portier, M.; Dussossoy, D.; Bord, A.; Petitpretre, G.; Canat, X.; Le Fur, G.; Casellas, P. Involvement of peripheral benzodiazepine receptors in the protection of hematopoietic cells against oxygen radical damage. Blood 1996, 87, 3170–3178. [Google Scholar] [CrossRef]
- Zheng, J.; Boisgard, R.; Siquier-Pernet, K.; Decaudin, D.; Dolle, F.; Tavitian, B. Differential Expression of the 18 kDa Translocator Protein (TSPO) by Neoplastic and Inflammatory Cells in Mouse Tumors of Breast Cancer. Mol. Pharm. 2011, 8, 823–832. [Google Scholar] [CrossRef]
- Corsi, L.; Geminiani, E.; Baraldi, M. Peripheral benzodiazepine receptor (PBR) new insight in cell proliferation and cell differentiation review. Curr. Clin. Pharmacol. 2008, 3, 38–45. [Google Scholar] [CrossRef]
- Batarseh, A.; Papadopoulos, V. Regulation of translocator protein 18 kDa (TSPO) expression in health and disease states. Mol. Cell Endocrinol. 2010, 327, 1–12. [Google Scholar] [CrossRef]
- Yasin, N.; Veenman, L.; Singh, S.; Azrad, M.; Bode, J.; Vainshtein, A.; Caballero, B.; Marek, I.; Gavish, M. Classical and Novel TSPO Ligands for the Mitochondrial TSPO Can Modulate Nuclear Gene Expression: Implications for Mitochondrial Retrograde Signaling. Int. J. Mol. Sci. 2017, 18, 786. [Google Scholar] [CrossRef]
- Gene Ontology Consortium. Gene Ontology Consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.C.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-Inducible Regulator of Glycolysis and Apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldeck-Weiermair, M.; Jean-Quartier, C.; Rost, R.; Khan, M.J.; Vishnu, N.; Bondarenko, A.I.; Imamura, H.; Malli, R.; Graier, W.F. Leucine zipper EF hand-containing transmembrane protein 1 (Letm1) and uncoupling proteins 2 and 3 (UCP2/3) contribute to two distinct mitochondrial Ca2+ uptake pathways. J. Biol. Chem. 2011, 286, 28444–28455. [Google Scholar] [CrossRef] [PubMed]
- Hong, E.-H.; Kim, J.-Y.; Kim, J.-H.; Lim, D.-S.; Kim, M.; Kim, J.-Y.J.M.N. BIG2-ARF1-RhoA-mDia1 Signaling Regulates Dendritic Golgi Polarization in Hippocampal Neurons. Mol. Neurobiol. 2018, 55, 7701–7716. [Google Scholar] [CrossRef]
- Shin, H.-W.; Nakayama, K. Guanine nucleotide-exchange factors for arf GTPases: Their diverse functions in membrane traffic. J. Biochem. 2004, 136, 761–767. [Google Scholar] [CrossRef]
- Guo, W.; Sacher, M.; Barrowman, J.; Ferro-Novick, S.; Novick, P. Protein complexes in transport vesicle targeting. Trends Cell Biol. 2000, 10, 251–255. [Google Scholar] [CrossRef]
- Agudo, D.; Gómez-Esquer, F.; Martínez-Arribas, F.; Núñez-Villar, M.J.; Pollán, M.; Schneider, J. Nup88 mRNA overexpression is associated with high aggressiveness of breast cancer. Int. J. Cancer 2004, 109, 717–720. [Google Scholar] [CrossRef]
- Emterling, A.; Skoglund, J.; Arbman, G.; Schneider, J.; Evertsson, S.; Carstensen, J.; Zhang, H.; Sun, X.-F. Clinicopathological significance of Nup88 expression in patients with colorectal cancer. Oncology 2003, 64, 361–369. [Google Scholar] [CrossRef]
- Barr, F.; Lambright, D.G. Rab gefs and gaps. Curr. Opin. Cell Biol. 2010, 22, 461–470. [Google Scholar] [CrossRef]
- Koliwer, J.; Park, M.; Bauch, C.; von Zastrow, M.; Kreienkamp, H.-J. The Golgi-associated PDZ domain protein PIST/GOPC stabilizes the β1-adrenergic receptor in intracellular compartments after internalization. J. Biol. Chem. 2015, 290, 6120–6129. [Google Scholar] [CrossRef]
- Shang, R.; Wang, J.; Sun, W.; Dai, B.; Ruan, B.; Zhang, Z.; Yang, X.; Gao, Y.; Qu, S.; Lv, X.; et al. RRAD inhibits aerobic glycolysis, invasion, and migration and is associated with poor prognosis in hepatocellular carcinoma. Tumour Biol. 2016, 37, 5097–5105. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Darnell, J., Jr. Signalling: Stats: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.P.; Evans, R.L.; Egland, K.A. Multiple functions of sushi domain containing 2 (SUSD2) in breast tumorigenesis. Mol. Cancer Res. 2013, 11, 74–85. [Google Scholar] [CrossRef]
- Etienne, S.; Adamson, P.; Greenwood, J.; Strosberg, A.D.; Cazaubon, S.; Couraud, P.-O. ICAM-1 signaling pathways associated with Rho activation in microvascular brain endothelial cells. J. Immunol. 1998, 161, 5755–5761. [Google Scholar] [PubMed]
- Huang, C.; Li, N.; Li, Z.; Chang, A.; Chen, Y.; Zhao, T.; Li, Y.; Wang, X.; Zhang, W.; Wang, Z. Tumour-derived Interleukin 35 promotes pancreatic ductal adenocarcinoma cell extravasation and metastasis by inducing ICAM1 expression. Nat. Commun. 2017, 8, 14035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrot, V.; Vázquez-Prado, J.; Gutkind, J.S. Plexin B regulates Rho through the guanine nucleotide exchange factors leukemia-associated Rho GEF (LARG) and PDZ-RhoGEF. J. Biol. Chem. 2002, 277, 43115–43120. [Google Scholar] [CrossRef]
- Xiang, G.; Cheng, Y. MiR-126-3p inhibits ovarian cancer proliferation and invasion via targeting PLXNB2. Reprod. Biol. 2018, 18, 218–224. [Google Scholar] [CrossRef]
- Yu, W.; Goncalves, K.A.; Li, S.; Kishikawa, H.; Sun, G.; Yang, H.; Vanli, N.; Wu, Y.; Jiang, Y.; Hu, M.G. Plexin-B2 Mediates Physiologic and Pathologic Functions of Angiogenin. Cell 2017, 171, 849–864. [Google Scholar] [CrossRef]
- Mayer, H.; Pongratz, M.; Prohaska, R. Molecular cloning, characterization, and tissue-specific expression of human LANCL2, a novel member of the LanC-like protein family. DNA Sequence 2001, 12, 161–166. [Google Scholar] [CrossRef]
- Chen, C.; Li, M.; Chai, H.; Yang, H.; Fisher, W.E.; Yao, Q. Roles of neuropilins in neuronal development, angiogenesis, and cancers. World J. Surg. 2005, 29, 271–275. [Google Scholar] [CrossRef]
- Shimizu, T.; Liao, J.K. Rho kinases and cardiac remodeling. Circ. J. 2016, 80, 1491–1498. [Google Scholar] [CrossRef] [PubMed]
- Melzer, C.; Yang, Y.; Hass, R. Interaction of MSC with tumor cells. Cell Commun. Signal. 2016, 14, 20. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Manzanares, M.; Ma, X.; Adelstein, R.S.; Horwitz, A.R. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat. Rev. Mol. Cell Biol. 2009, 10, 778. [Google Scholar] [CrossRef] [PubMed]
- Soto-Pantoja, D.R.; Kaur, S.; Roberts, D.D. CD47 signaling pathways controlling cellular differentiation and responses to stress. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 212–230. [Google Scholar] [CrossRef] [PubMed]
- Per-Arne, O. Role of CD47 and Signal Regulatory Protein Alpha (SIRPalpha) in Regulating the Clearance of Viable or Aged Blood Cells. Transfus. Med. Hemother. 2012, 39, 315–320. [Google Scholar] [PubMed]
- Soto-Pantoja, D.R.; Terabe, M.; Ghosh, A.; Ridnour, L.A.; DeGraff, W.G.; Wink, D.A.; Berzofsky, J.A.; Roberts, D.D. CD47 in the Tumor Microenvironment Limits Cooperation between Antitumor T-cell Immunity and Radiotherapy. Cancer Res. 2014, 74, 6771. [Google Scholar] [CrossRef]
- Tachikui, H.; Kurosawa, N.; Kadomatsu, K.; Muramatsu, T.J.G. Genomic organization and promoter activity of embigin, a member of the immunoglobulin superfamily. Gene 1999, 240, 325–332. [Google Scholar] [CrossRef]
- Lain, E.; Carnejac, S.; Escher, P.; Wilson, M.C.; Lomo, T.; Gajendran, N.; Brenner, H.R. A novel role for embigin to promote sprouting of motor nerve terminals at the neuromuscular junction. J. Biol. Chem. 2009, 284, 8930–8939. [Google Scholar] [CrossRef]
- Lee, S.; Nam, Y.; Koo, J.Y.; Lim, D.; Park, J.; Ock, J.; Kim, J.; Suk, K.; Park, S.B. A small molecule binding HMGB1 and HMGB2 inhibits microglia-mediated neuroinflammation. Nat. Chem. Biol. 2014, 10, 1055–1060. [Google Scholar] [CrossRef]
- Cai, X.; Ding, H.; Liu, Y.; Pan, G.; Li, Q.; Yang, Z.; Liu, W. Expression of HMGB2 indicates worse survival of patients and is required for the maintenance of Warburg effect in pancreatic cancer. Acta Biochim. Biophys. Sin. 2017, 49, 119–127. [Google Scholar] [CrossRef]
- Overall, C.M.; Kleifeld, O. Tumour microenvironment—Opinion: Validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat. Rev. Cancer 2006, 6, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Goodison, S.; Urquidi, V.; Tarin, D. CD44 cell adhesion molecules. Mol. Pathol. 1999, 52, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awad, W.; Logan, D.T.; Mani, K. GPC1 (glypican 1). Atlas Genet. Cytogenet. Oncol. Haematol. 2014, 18, 461–464. [Google Scholar] [CrossRef]
- Trockenbacher, A.; Suckow, V.; Foerster, J.; Winter, J.; Krauß, S.; Ropers, H.-H.; Schneider, R.; Schweiger, S. MID1, mutated in Opitz syndrome, encodes an ubiquitin ligase that targets phosphatase 2A for degradation. Nat. Genet. 2001, 29, 287. [Google Scholar] [CrossRef]
- Watkins, G.R.; Wang, N.; Mazalouskas, M.D.; Gomez, R.J.; Guthrie, C.R.; Kraemer, B.C.; Schweiger, S.; Spiller, B.W.; Wadzinski, B.E. Monoubiquitination promotes calpain cleavage of the protein phosphatase 2A (PP2A) regulatory subunit α4, altering PP2A stability and microtubule-associated protein phosphorylation. J. Biol. Chem. 2012, 287, 24207–24215. [Google Scholar] [CrossRef]
- Tan, K.-L.; Pezzella, F. Inhibition of NEDD8 and FAT10 ligase activities through the degrading enzyme NEDD8 ultimate buster 1: A potential anticancer approach. Oncol. Lett. 2016, 12, 4287–4296. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Nag, S.; Zhang, X.; Wang, M.H.; Wang, H.; Zhou, J.; Zhang, R. Ribosomal proteins and human diseases: Pathogenesis, molecular mechanisms, and therapeutic implications. Med. Res. Rev. 2015, 35, 225–285. [Google Scholar] [CrossRef]
- Kumar, K.U.; Srivastava, S.P.; Kaufman, R.J. Double-Stranded RNA-Activated Protein Kinase (PKR) Is Negatively Regulated by 60S Ribosomal Subunit Protein L18. Mol. Cell. Biol. 1999, 19, 1116–1125. [Google Scholar] [CrossRef] [Green Version]
- Loging, W.T.; Reisman, D. Elevated expression of ribosomal protein genes L37, RPP-1, and S2 in the presence of mutant p53. Cancer Epidemiol. Biomark. Prev. 1999, 8, 1011–1016. [Google Scholar]
- Baik, I.H.; Jo, G.H.; Seo, D.; Ko, M.J.; Cho, C.H.; Lee, M.G.; Lee, Y.H. Knockdown of RPL9 expression inhibits colorectal carcinoma growth via the inactivation of Id-1/NF-kappaB signaling axis. Int. J. Oncol. 2016, 49, 1953–1962. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, L.; Leidecker, O.; Prokhorova, E.; Dauben, H.; Matic, I.; Ahel, I.J.E. Serine is the major residue for ADP-ribosylation upon DNA damage. eLife 2018, 7, e34334. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, H.; Garcia-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Craigen, W.J.; Scaglia, F. Mitochondrial DNA maintenance defects. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1539–1555. [Google Scholar] [CrossRef] [PubMed]
- Holohan, B.; Kim, W.; Lai, T.-P.; Hoshiyama, H.; Zhang, N.; Alazami, A.M.; Wright, W.E.; Meyn, M.S.; Alkuraya, F.S.; Shay, J.W. Impaired telomere maintenance in Alazami syndrome patients with LARP7 deficiency. BMC Genomics 2016, 17, 749. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gan, S.; Ren, L.; Yuan, L.; Liu, J.; Wang, W.; Wang, X.; Zhang, Y.; Jiang, J.; Zhang, F. Multifaceted regulation and functions of replication factor C family in human cancers. Am. J. Cancer Res. 2018, 8, 1343–1355. [Google Scholar] [PubMed]
- Dehé, P.-M.; Gaillard, P.-H.L. Control of structure-specific endonucleases to maintain genome stability. Nat. Rev. Mol. Cell Biol. 2017, 18, 315–330. [Google Scholar] [CrossRef]
- Kim, N.; Jinks-Robertson, S. The Top1 paradox: Friend and foe of the eukaryotic genome. DNA Repair 2017, 56, 33–41. [Google Scholar] [CrossRef]
- Pokrzywinski, K.L.; Biel, T.G.; Kryndushkin, D.; Rao, V.A. Therapeutic targeting of the mitochondria initiates excessive superoxide production and mitochondrial depolarization causing decreased mtDNA integrity. PLoS ONE 2016, 11, e0168283. [Google Scholar] [CrossRef]
- Veena, V.; Rajan, K.; Saritha, V.; Preethi Sara George, C.K.; Jayasree, K.; Thara, S. DNA Replication Licensing Proteins for Early Detection of Lung Cancer. Asian Pac. J. Cancer Prev. 2017, 18, 3041–3047. [Google Scholar]
- Arif, T.; Krelin, Y.; Shoshan-Barmatz, V. Reducing VDAC1 expression induces a non-apoptotic role for pro-apoptotic proteins in cancer cell differentiation. Biochim. Biophys. Acta 2016, 1857, 1228–1242. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Pittala, S.; Mizrachi, D. VDAC1 and the TSPO: Expression, Interactions, and Associated Functions in Health and Disease States. Int. J. Mol. Sci. 2019, 20, 3348. [Google Scholar] [CrossRef] [PubMed]
- Wenger, J.B.; Chun, S.Y.; Dang, D.T.; Luesch, H.; Dang, L.H. Combination therapy targeting cancer metabolism. Med. Hypotheses 2011, 76, 169–172. [Google Scholar] [CrossRef] [Green Version]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional addiction in cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the intersections between metabolism and cancer biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef]
- Loo, J.M.; Scherl, A.; Nguyen, A.; Man, F.Y.; Weinberg, E.; Zeng, Z.; Saltz, L.; Paty, P.B.; Tavazoie, S.F. Extracellular metabolic energetics can promote cancer progression. Cell 2015, 160, 393–406. [Google Scholar] [CrossRef]
- Krasnov, D.; Thess, A.; Boeck, T.; Zhao, Y.; Zikanov, O. Patterned turbulence in liquid metal flow: Computational reconstruction of the Hartmann experiment. Phys. Rev. Lett. 2013, 110, 084501. [Google Scholar] [CrossRef]
- Cosenza-Nashat, M.; Zhao, M.L.; Suh, H.S.; Morgan, J.; Natividad, R.; Morgello, S.; Lee, S.C. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol. Appl. Neurobiol. 2009, 35, 306–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, D.; Hight, M.R.; McKinley, E.T.; Fu, A.; Buck, J.R.; Smith, R.A.; Tantawy, M.N.; Peterson, T.E.; Colvin, D.C.; Ansari, M.S. Quantitative preclinical imaging of TSPO expression in glioma using N,N-diethyl-2-(2-(4-(2-18F-fluoroethoxy) phenyl)-5, 7-dimethylpyrazolo [1, 5-a] pyrimidin-3-yl) acetamide. J. Nucl. Med. 2012, 53, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Winkeler, A.; Boisgard, R.; Awde, A.R.; Dubois, A.; Thézé, B.; Zheng, J.; Ciobanu, L.; Dollé, F.; Viel, T.; Jacobs, A.H.; et al. The translocator protein ligand [¹⁸F]DPA-714 images glioma and activated microglia in vivo. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 811–823. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.R.; Kersemans, V.; Tredwell, M.; Checa, B.; Serres, S.; Soto, M.S.; Gouverneur, V.; Leppert, D.; Anthony, D.C.; Sibson, N.R. Glial activation in the early stages of brain metastasis: TSPO as a diagnostic biomarker. J. Nucl. Med. 2014, 55, 275–280. [Google Scholar] [CrossRef]
- Bhoola, H.N.; Mbita, Z.; Hull, R.; Dlamini, Z. Translocator Protein (TSPO) as a Potential Biomarker in Human Cancers. Int. J. Mol. Sci. 2018, 19, 2176. [Google Scholar] [CrossRef]
- Dessi, S.; Batetta, B.; Anchisi, C.; Pani, P.; Costelli, P.; Tessitore, L.; Baccino, F.M. Cholesterol metabolism during the growth of a rat ascites hepatoma (Yoshida AH-130). Br. J. Cancer 1992, 66, 787–793. [Google Scholar] [CrossRef] [Green Version]
- Dessi, S.; Batetta, B.; Pulisci, D.; Spano, O.; Anchisi, C.; Tessitore, L.; Costelli, P.; Baccino, F.M.; Aroasio, E.; Pani, P. Cholesterol content in tumor tissues is inversely associated with high-density lipoprotein cholesterol in serum in patients with gastrointestinal cancer. Cancer 1994, 73, 253–258. [Google Scholar] [CrossRef]
- Kolanjiappan, K.; Ramachandran, C.R.; Manoharan, S. Biochemical changes in tumor tissues of oral cancer patients. Clin. Biochem. 2003, 36, 61–65. [Google Scholar] [CrossRef]
- Rudling, M.; Collins, V.P. Low density lipoprotein receptor and 3-hydroxy-3-methylglutaryl coenzyme A reductase mRNA levels are coordinately reduced in human renal cell carcinoma. Biochim. Biophys. Acta 1996, 1299, 75–79. [Google Scholar] [CrossRef]
- Yoshioka, Y.; Sasaki, J.; Yamamoto, M.; Saitoh, K.; Nakaya, S.; Kubokawa, M. Quantitation by (1)H-NMR of dolichol, cholesterol and choline-containing lipids in extracts of normal and phathological thyroid tissue. NMR Biomed. 2000, 13, 377–383. [Google Scholar] [CrossRef]
- Miettinen, H.; Kononen, J.; Haapasalo, H.; Helen, P.; Sallinen, P.; Harjuntausta, T.; Helin, H.; Alho, H. Expression of peripheral-type benzodiazepine receptor and diazepam binding inhibitor in human astrocytomas: Relationship to cell proliferation. Cancer Res. 1995, 55, 2691–2695. [Google Scholar] [PubMed]
- Beinlich, A.; Strohmeier, R.; Kaufmann, M.; Kuhl, H. Relation of cell proliferation to expression of peripheral benzodiazepine receptors in human breast cancer cell lines. Biochem. Pharmacol. 2000, 60, 397–402. [Google Scholar] [CrossRef]
- Lin, R.; Angelin, A.; Da Settimo, F.; Martini, C.; Taliani, S.; Zhu, S.; Wallace, D.C. Genetic analysis of dTSPO, an outer mitochondrial membrane protein, reveals its functions in apoptosis, longevity, and Ab42-induced neurodegeneration. Aging Cell 2014, 13, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Galiegue, S.; Casellas, P.; Kramar, A.; Tinel, N.; Simony-Lafontaine, J. Immunohistochemical assessment of the peripheral benzodiazepine receptor in breast cancer and its relationship with survival. Clin. Cancer Res. 2004, 10, 2058–2064. [Google Scholar] [CrossRef] [PubMed]
- Campanella, M.; Szabadkai, G.; Rizzuto, R. Modulation of intracellular Ca2+ signalling in HeLa cells by the apoptotic cell death enhancer PK11195. Biochem. Pharmacol. 2008, 76, 1628–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeno, S.; Veenman, L.; Katz, Y.; Bode, J.; Gavish, M.; Zaaroor, M. The 18 kDa mitochondrial translocator protein (TSPO) prevents accumulation of protoporphyrin IX. Involvement of reactive oxygen species (ROS). Curr. Mol. Med. 2012, 12, 494–501. [Google Scholar]
- Decaudin, D. Peripheral benzodiazepine receptor and its clinical targeting. Anticancer Drugs 2004, 15, 737–745. [Google Scholar] [CrossRef]
- Hardwick, M.; Fertikh, D.; Culty, M.; Li, H.; Vidic, B.; Papadopoulos, V. Peripheral-type benzodiazepine receptor (PBR) in human breast cancer: Correlation of breast cancer cell aggressive phenotype with PBR expression, nuclear localization, and PBR-mediated cell proliferation and nuclear transport of cholesterol. Cancer Res. 1999, 59, 831–842. [Google Scholar]
- Joo, H.K.; Lee, Y.R.; Lim, S.Y.; Lee, E.J.; Choi, S.; Cho, E.J.; Park, M.S.; Ryoo, S.; Park, J.B.; Jeon, B.H. Peripheral benzodiazepine receptor regulates vascular endothelial activations via suppression of the voltage-dependent anion channel-1. FEBS Lett. 2012, 586, 1349–1355. [Google Scholar] [CrossRef] [Green Version]
- Gatliff, J.; East, D.; Crosby, J.; Abeti, R.; Harvey, R.; Craigen, W.; Parker, P.; Campanella, M. TSPO interacts with VDAC1 and triggers a ROS-mediated inhibition of mitochondrial quality control. Autophagy 2014, 10, 2279–2296. [Google Scholar] [CrossRef] [Green Version]
- Aghazadeh, Y.; Martinez-Arguelles, D.B.; Fan, J.; Culty, M.; Papadopoulos, V. Induction of androgen formation in the male by a TAT-VDAC1 fusion peptide blocking 14-3-3varepsilon protein adaptor and mitochondrial VDAC1 interactions. Mol. Ther. 2014, 22, 1779–1791. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Kim, L.E.; Shim, H.J.; Kim, E.K.; Hwang, W.C.; Min, D.S.; Yu, S.W. A translocator protein 18 kDa ligand, Ro5-4864, inhibits ATP-induced NLRP3 inflammasome activation. Biochem. Biophys. Res. Commun. 2016, 474, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.C.; Degenhardt, B.; Kotoula, M.; Papadopoulous, V. Location-dependent role of the human glioma cell peripheral-type benzodiazepine receptor in proliferation and steroid biosynthesis. Cancer Lett. 2000, 156, 125–132. [Google Scholar] [CrossRef]
- Alho, H.; Varga, V.; Krueger, K.E. Expression of mitochondrial benzodiazepine receptor and its putative endogenous ligand diazepam binding inhibitor in cultured primary astrocytes and C-6 cells: Relation to cell growth. Cell Growth Differ. 1994, 5, 1005–1014. [Google Scholar]
- Mukherjee, S.; Das, S.K. Translocator protein (TSPO) in breast cancer. Curr. Mol. Med. 2012, 12, 443–457. [Google Scholar]
- Banati, R.B.; Middleton, R.J.; Chan, R.; Hatty, C.R.; Kam, W.W.-Y.; Quin, C.; Graeber, M.B.; Parmar, A.; Zahra, D.; Callaghan, P.; et al. Positron emission tomography and functional characterization of a complete PBR/TSPO knockout. Nat. Commun. 2014, 5, 5452. [Google Scholar] [CrossRef]
- Chen, J.; McKay, R.M.; Parada, L.F. Malignant glioma: Lessons from genomics, mouse models, and stem cells. Cell 2012, 149, 36–47. [Google Scholar] [CrossRef]
- Alcantara Llaguno, S.R.; Parada, L.F. Cell of origin of glioma: Biological and clinical implications. Br. J. Cancer 2016, 115, 1445–1450. [Google Scholar] [CrossRef]
- Fotovati, A.; Abu-Ali, S.; Wang, P.S.; Deleyrolle, L.P.; Lee, C.; Triscott, J.; Chen, J.Y.; Franciosi, S.; Nakamura, Y.; Sugita, Y.; et al. YB-1 bridges neural stem cells and brain tumor-initiating cells via its roles in differentiation and cell growth. Cancer Res. 2011, 71, 5569–5578. [Google Scholar] [CrossRef]
- Van Strien, M.E.; Van Den Berge, S.A.; Hol, E.M. Migrating neuroblasts in the adult human brain: A stream reduced to a trickle. Cell Res. 2011, 21, 1523–1525. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.P.; Nahed, B.V.; Madden, M.W.; Oliveira, S.M.; Springer, S.; Bhere, D.; Chi, A.S.; Wakimoto, H.; Rothenberg, S.M.; Sequist, L.V. Brain tumor cells in circulation are enriched for mesenchymal gene expression. Cancer Discov. 2014, 4, 1299–1309. [Google Scholar] [CrossRef]
- Bulstrode, H.; Johnstone, E.; Marques-Torrejon, M.A.; Ferguson, K.M.; Bressan, R.B.; Blin, C.; Grant, V.; Gogolok, S.; Gangoso, E.; Gagrica, S. Elevated FOXG1 and SOX2 in glioblastoma enforces neural stem cell identity through transcriptional control of cell cycle and epigenetic regulators. Genes Dev. 2017, 31, 757–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roshan, R.; Shridhar, S.; Sarangdhar, M.A.; Banik, A.; Chawla, M.; Garg, M.; Singh, V.P.; Pillai, B. Brain-specific knockdown of miR-29 results in neuronal cell death and ataxia in mice. RNA 2014, 20, 1287–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, M.; Zhang, Y.; He, B.; Xiang, J.; Wang, Z.F.; Zheng, F.M.; Xu, J.; Chen, M.Y.; Zhu, Y.L.; Wen, H.J.; et al. IKKalpha restoration via EZH2 suppression induces nasopharyngeal carcinoma differentiation. Nat. Commun. 2014, 5, 3661. [Google Scholar] [CrossRef] [PubMed]
- Ory, V.; Kietzman, W.B.; Boeckelman, J.; Kallakury, B.V.; Wellstein, A.; Furth, P.A.; Riegel, A.T. The PPARgamma agonist efatutazone delays invasive progression and induces differentiation of ductal carcinoma in situ. Breast Cancer Res. Treat. 2018, 169, 47–57. [Google Scholar] [CrossRef]
- Philchenkov, A.; Zavelevich, M.; Kroczak, T.J.; Los, M. Caspases and cancer: Mechanisms of inactivation and new treatment modalities. Exp. Oncol. 2004, 26, 82–97. [Google Scholar]
- Devarajan, E.; Sahin, A.A.; Chen, J.S.; Krishnamurthy, R.R.; Aggarwal, N.; Brun, A.M.; Sapino, A.; Zhang, F.; Sharma, D.; Yang, X.H.; et al. Down-regulation of caspase 3 in breast cancer: A possible mechanism for chemoresistance. Oncogene 2002, 21, 8843–8851. [Google Scholar] [CrossRef]
- Shen, S.M.; Guo, M.; Xiong, Z.; Yu, Y.; Zhao, X.Y.; Zhang, F.F.; Chen, G.Q. AIF inhibits tumor metastasis by protecting PTEN from oxidation. EMBO Rep. 2015, 16, 1563–1580. [Google Scholar] [CrossRef]
- Porter, A.G.; Urbano, A.G.L. Does apoptosis-inducing factor (AIF) have both life and death functions in cells? BioEssays 2006, 28, 834–843. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Witz, I.P.; Levy-Nissenbaum, O. The tumor microenvironment in the post-PAGET era. Cancer Lett. 2006, 242, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Barrett, A.J.; Bateman, A. MEROPS: The peptidase database. Nucleic Acids Res. 2010, 38, D227–D233. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Komuro, Y.; Hayakawa, T.; Oguchi, H.; Ishida, Y.; Murakami, S.; Noguchi, T.; Kinoshita, H.; Sekine, Y.; Iemura, S.-I. Mitochondrial phosphoglycerate mutase 5 uses alternate catalytic activity as a protein serine/threonine phosphatase to activate ASK1. Proc. Natl. Acad. Sci. USA 2009, 106, 12301–12305. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Qian, D.; Ding, X.; Song, T.; Cai, M.; Xie, D.; Wang, Y.; Zhao, J.; Liu, Z.; Wu, Z. High PGAM5 expression induces chemoresistance by enhancing Bcl-xL-mediated anti-apoptotic signaling and predicts poor prognosis in hepatocellular carcinoma patients. Cell Death Dis. 2018, 9, 991. [Google Scholar] [CrossRef]
- Sheets, J.; Iwanicki, M.; Liu, J.; Howitt, B.; Hirsch, M.; Gubbels, J.; Drapkin, R.; Egland, K. SUSD2 expression in high-grade serous ovarian cancer correlates with increased patient survival and defective mesothelial clearance. Oncogenesis 2016, 5, e264. [Google Scholar] [CrossRef]
- De Oliveira, T.; Ramakrishnan, M.; Diamanti, M.A.; Ziegler, P.K.; Brombacher, F.; Greten, F.R. Loss of Stat6 affects chromatin condensation in intestinal epithelial cells causing diverse outcome in murine models of inflammation-associated and sporadic colon carcinogenesis. Oncogene 2018, 38, 1787–1801. [Google Scholar] [CrossRef] [Green Version]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef]
- Dehan, P.; Kustermans, G.; Guenin, S.; Horion, J.; Boniver, J.; Delvenne, P. DNA methylation and cancer diagnosis: New methods and applications. Exp. Rev. Mol. Diagn. 2009, 9, 651–657. [Google Scholar] [CrossRef]
- Hirst, M.; Marra, M.A. Epigenetics and human disease. Int. J. Biochem. Cell Biol. 2009, 41, 136–146. [Google Scholar] [CrossRef]
- Stefanska, B.; Karlic, H.; Varga, F.; Fabianowska-Majewska, K.; Haslberger, A.G. Epigenetic mechanisms in anti-cancer actions of bioactive food components—The implications in cancer prevention. Br. J. Pharmacol. 2012, 167, 279–297. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wang, L.; Di, L. Compartmentation of metabolites in regulating epigenome of cancer. Mol. Med. 2016, 22, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Carrer, A.; Wellen, K.E. Metabolism and epigenetics: A link cancer cells exploit. Curr. Opin. Biotechnol. 2015, 34, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Etchegaray, J.-P.; Mostoslavsky, R. Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol. Cell 2016, 62, 695–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, P.S.; Thompson, C.B. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaelin, W.G., Jr.; McKnight, S.L. Influence of Metabolism on Epigenetics and Disease. Cell 2013, 153, 56–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaochar, S.; Tu, B.P. Gatekeepers of chromatin: Small metabolites elicit big changes in gene expression. Trends Biochem. Sci. 2012, 37, 477–483. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Thompson, C.B. Metabolic Regulation of Epigenetics. Cell Metab. 2012, 16, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Shi, Y. Metabolic enzymes and coenzymes in transcription–a direct link between metabolism and transcription? Trends Genet. 2004, 20, 445–452. [Google Scholar] [CrossRef]


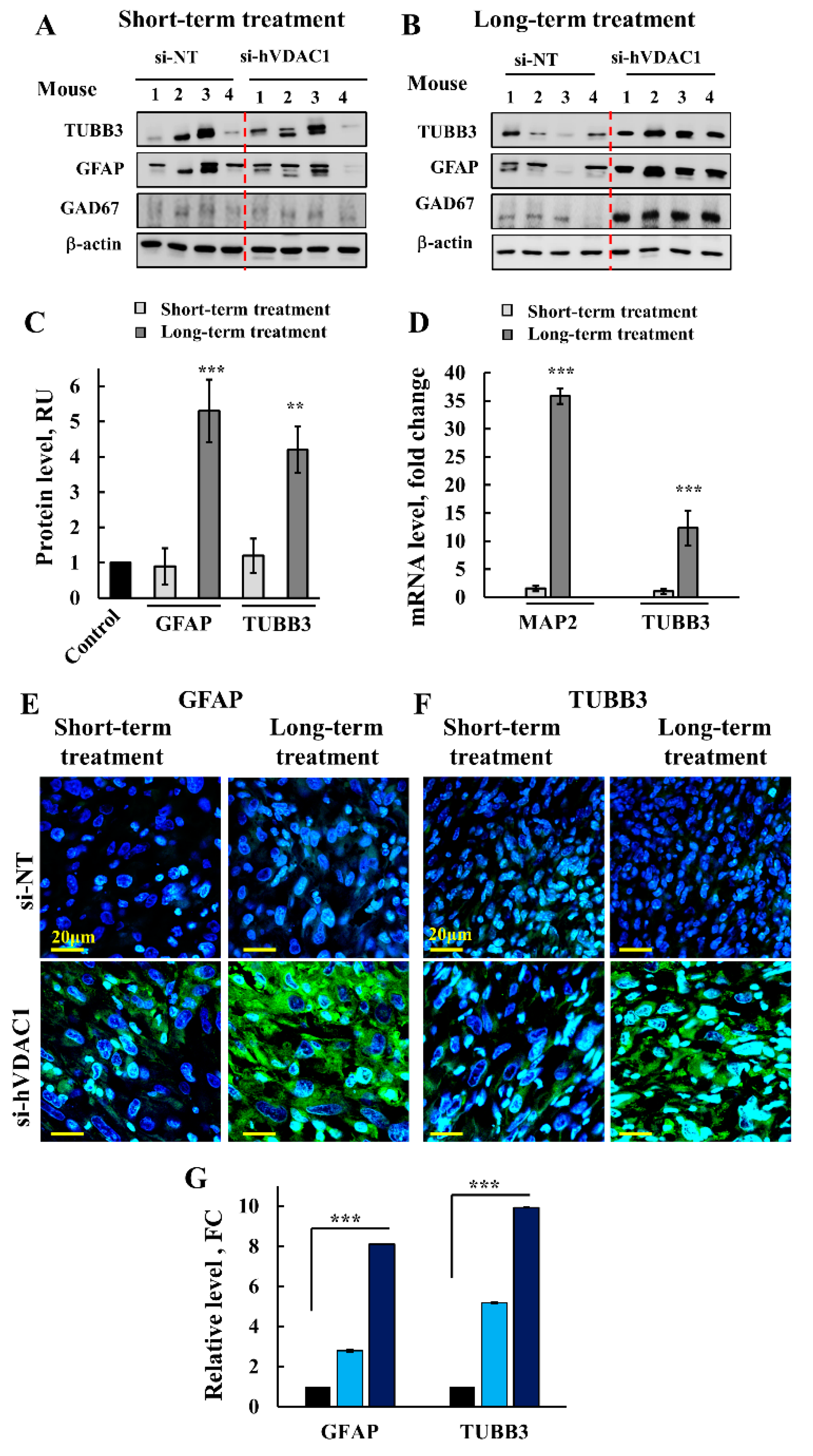


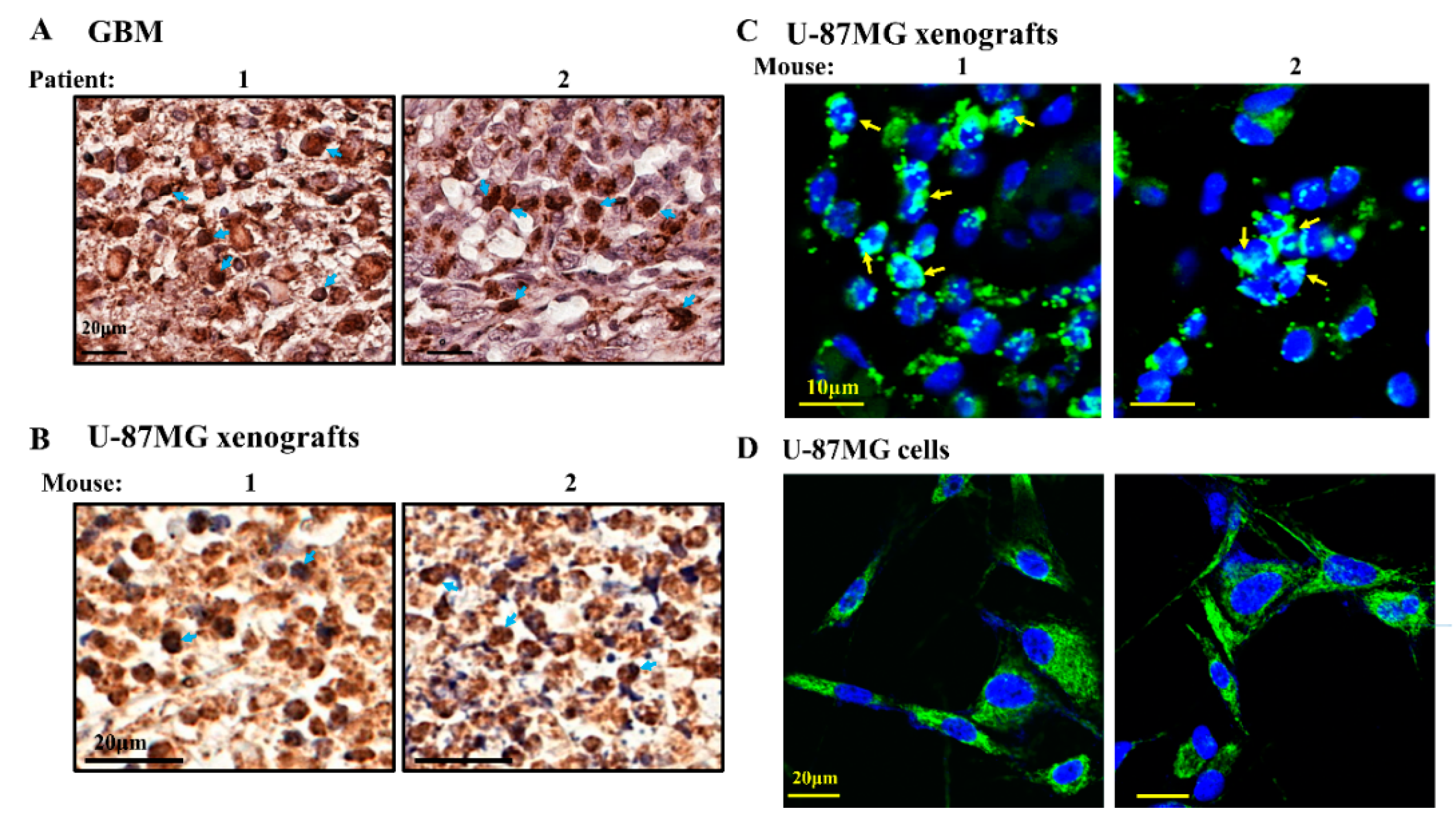

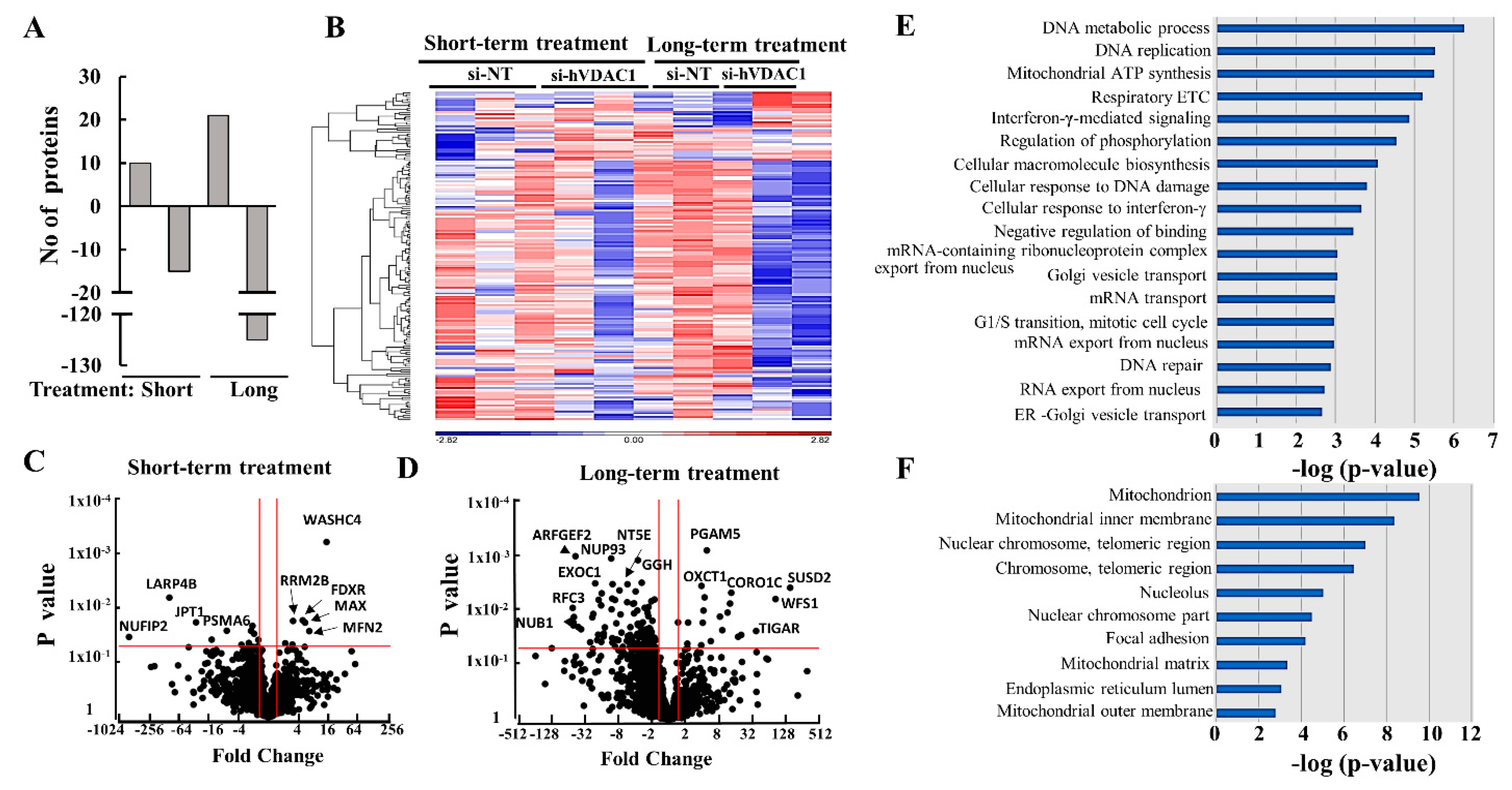
 ) indicate short-term and long-term treatment with si-hVDAC1, respectively.
) indicate short-term and long-term treatment with si-hVDAC1, respectively.
) indicate short-term and long-term treatment with si-hVDAC1, respectively.
) indicate short-term and long-term treatment with si-hVDAC1, respectively. ) indicate short-term and long-term treatment with si-hVDAC1, respectively. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001.
) indicate short-term and long-term treatment with si-hVDAC1, respectively. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001.
) indicate short-term and long-term treatment with si-hVDAC1, respectively. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001.
) indicate short-term and long-term treatment with si-hVDAC1, respectively. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001. ) indicate short-term and long-term treatment with si-hVDAC1, respectively. * p ≤ 0.05, ** p ≤ 0.01.
) indicate short-term and long-term treatment with si-hVDAC1, respectively. * p ≤ 0.05, ** p ≤ 0.01.
) indicate short-term and long-term treatment with si-hVDAC1, respectively. * p ≤ 0.05, ** p ≤ 0.01.
) indicate short-term and long-term treatment with si-hVDAC1, respectively. * p ≤ 0.05, ** p ≤ 0.01.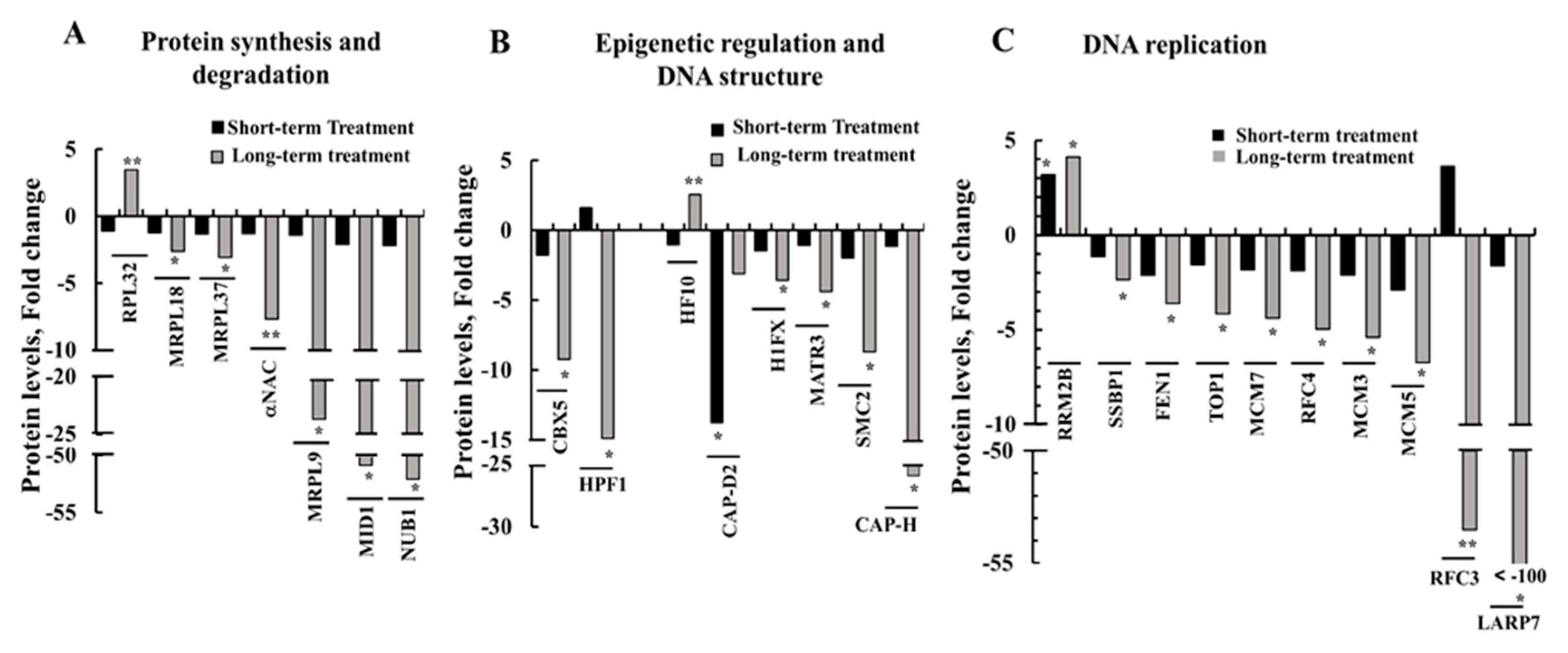
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arif, T.; Stern, O.; Pittala, S.; Chalifa-Caspi, V.; Shoshan-Barmatz, V. Rewiring of Cancer Cell Metabolism by Mitochondrial VDAC1 Depletion Results in Time-Dependent Tumor Reprogramming: Glioblastoma as a Proof of Concept. Cells 2019, 8, 1330. https://doi.org/10.3390/cells8111330
Arif T, Stern O, Pittala S, Chalifa-Caspi V, Shoshan-Barmatz V. Rewiring of Cancer Cell Metabolism by Mitochondrial VDAC1 Depletion Results in Time-Dependent Tumor Reprogramming: Glioblastoma as a Proof of Concept. Cells. 2019; 8(11):1330. https://doi.org/10.3390/cells8111330
Chicago/Turabian StyleArif, Tasleem, Oriel Stern, Srinivas Pittala, Vered Chalifa-Caspi, and Varda Shoshan-Barmatz. 2019. "Rewiring of Cancer Cell Metabolism by Mitochondrial VDAC1 Depletion Results in Time-Dependent Tumor Reprogramming: Glioblastoma as a Proof of Concept" Cells 8, no. 11: 1330. https://doi.org/10.3390/cells8111330
APA StyleArif, T., Stern, O., Pittala, S., Chalifa-Caspi, V., & Shoshan-Barmatz, V. (2019). Rewiring of Cancer Cell Metabolism by Mitochondrial VDAC1 Depletion Results in Time-Dependent Tumor Reprogramming: Glioblastoma as a Proof of Concept. Cells, 8(11), 1330. https://doi.org/10.3390/cells8111330