Functions and Implications of Autophagy in Colon Cancer

1
Cellular and Molecular Biology, University of Michigan, Ann Arbor, MI 48109, USA
2
Molecular and Integrative Physiology, Internal Medicine, Division of Gastroenterology, and Rogel Cancer Center, University of Michigan, Ann Arbor, MI 48109, USA
*
Author to whom correspondence should be addressed.
Cells 2019, 8(11), 1349; https://doi.org/10.3390/cells8111349
Submission received: 27 September 2019
/
Revised: 24 October 2019
/
Accepted: 28 October 2019
/
Published: 30 October 2019
(This article belongs to the Special Issue Molecular and Cellular Mechanisms of Cancers: Colorectal Cancer)
Abstract
:Autophagy is an essential function to breakdown cellular proteins and organelles to recycle for new nutrient building blocks. In colorectal cancer, the importance of autophagy is becoming widely recognized as it demonstrates both pro- and anti-tumorigenic functions. In colon cancer, cell autonomous and non-autonomous roles for autophagy are essential in growth and progression. However, the mechanisms downstream of autophagy (to reduce or enhance tumor growth) are not well known. Additionally, the signals that activate and coordinate autophagy for tumor cell growth and survival are not clear. Here, we highlight the context- and cargo-dependent role of autophagy in proliferation, cell death, and cargo breakdown.
1. Introduction
Colorectal cancer (CRC) is the third leading cause of cancer-related deaths in the United States. It is estimated that in 2018, over 140,000 people were diagnosed with CRC [1]. Survival rates have improved in the last three decades due to early detection, however patients diagnosed at later stages of disease have a 5-year survival rate of 14% [2,3]. Treatments available for these patients are limited, therefore it is important to better understand tumor development and identify targeted therapies to improve overall patient care. Sporadic CRCs are typically marked by the initial loss of the adenomatous polyposis coli (APC) gene. APC is a scaffold protein that leads to proteasomal degradation of β-catenin. Under active cell states, WNT ligands bind to its receptor frizzled and prevent β-catenin degradation and activate target genes. Loss of APC constitutively activates β-catenin and causes uncontrolled epithelial proliferation. Mutations in APC are typically followed by sequential mutations in tumor protein p53 (p53), and mutations in KRAS leading to spontaneous tumor development and progression [4]. It is well known that chronic inflammatory diseases such as Crohn’s Disease or ulcerative colitis increase risk of developing colon cancer, referred to as colitis-associated cancer (CAC) [5,6]. CACs developed from inflammatory bowel diseases (IBD) are a rare subset. Moreover, p53 mutations occur earlier in the progression of CAC and APC mutations are less frequent and are found in late stage tumors in comparison to sporadic CRC [7]. However, CAC provides a clear link between inflammation and tumor initiation.
Autophagy is a highly regulated process that degrades and recycles cellular components. Dysregulation of autophagy is implicated in many diseases (as reviewed in [8]). Under several different cell stressors, autophagy is activated through kinase signaling and transcriptional activation by serine/threonine protein kinase 1 (ULK1) and transcription factor EB (TFEB). This activates a cascade of autophagy-related genes (ATG) [9], and formation of a spherical double layer membrane termed the autophagosome. The autophagosome delivers key cytoplasmic cargo such as organelles, foreign bodies, and cellular components to the lysosome for degradation into macromolecules that can be utilized by the cell. In CRC, autophagy is known to play tumor promoting and tumor suppressive roles [10,11], but the underlying mechanisms are not well understood. Studies have found conflicting functions of autophagy in tumors. These discrepancies are typically due to differences in the cells and tumor models that are utilized [12,13,14,15]. Further study of autophagy and its prevalence in CRC will uncover its potential therapeutic use [16]. Here we highlight cellular pathways that regulate autophagy, selective forms of autophagy, and how these mechanisms target different cargo for degradation.
2. Autophagy Subtypes
Autophagy can be classified into three major subtypes; macro-autophagy, micro-autophagy, and chaperone-mediated autophagy. There is a need for better understanding of cellular cues and the cell-dependent context by which autophagic subtypes are co-opted in cancer cells for growth and survival.
2.1. Chaperone-Mediated Autophagy
Chaperone-mediated autophagy (CMA) differs from macro-autophagy in that select proteins are targeted for degradation by direct targeting to the lysosome. Proteins are recognized by heat shock cognate protein 70 (HSC70). HSC70 interacts with lysosome-associated membrane protein type 2A (LAMP-2A) to internalize proteins into the lysosome. CMA substrates contain a specific motif, KFERQ, which is essential for HSC70 binding [17]. Relevant to cancer, inhibition of macro-autophagy enhances CMA-dependent degradation of mutant p53 [18]. Increased expression of LAMP-2A demonstrated activated CMA in CRC [19]. These functions highlight a potential role of CMA in tumor development. However, these processes have not been well studied in CRC.
2.2. Micro-Autophagy
Micro-autophagy is the direct engulfment of cellular components by invagination of the late endosome (Figure 1). Broadly, the implications of micro-autophagy have not been studied in many cancer types. There is evidence in lung cancer that amino acid starvation, an important factor in cancer growth (discussed below), induces micro-autophagy [20]. However, the role of micro-autophagy in CRC has not been investigated in detail.
2.3. Macro-Autophagy
Macro-autophagy can be broken down in to two subcategories that target cellular components for degradation; non-selective and selective. Non-selective macro-autophagy engulfs bulk cytosolic components and selective autophagy targets specific cargo for degradation (e.g., organelles and protein aggregates). For macro-autophagy, the phagophore, a precursor to the autophagosome, forms. Several ATG protein complexes are involved in early and late autophagosome formation [21]. As the membrane is forming, microtubule associated protein 1 light chain 3 beta (LC3-I) is conjugated with phosphatidylethanolamine and is processed into LC3-II [22]. Following fusion of the autophagosome with the lysosome, which contains the required enzymes for cargo degradation, LC3-II is broken down by the lysosome [23]. The turnover of LC3-II is often used as a readout of autophagic activity [24]. Here, we have provided a highly simplified overview of the complex process of macro-autophagy. This extremely coordinated event of cytoplasmic engulfment is generally activated in states of cell stress such as starvation. Anding and Baehrecke reviewed the important role of selective-autophagic processes in maintaining cellular homeostasis in response to stress [25]. Below we highlight the known selective-autophagic pathways and their potential role in CRC (Figure 1).
2.4. Mitophagy
Mitophagy is a process that selectively degrades mitochondria [26,27,28,29]. Mitophagy is highly evolutionarily conserved [30] and is known to be activated in yeast under starvation [31]. Therefore, highly proliferative cells under nutrient stress and starvation, such as cancer cells, may activate mitophagy. This process can be modulated by different pathways as discussed below.
2.4.1. Parkin-Mediated Mitophagy
Under normal cell homeostasis, PTEN induced kinase 1 (PINK1) is maintained on the inner mitochondrial membrane. Upon damage or stress, PINK1 moves to the outer membrane and phosphorylates parkin (PRKN). This phosphorylation allows for ubiquitination of PRKN1 and targeting to the autophagosome. Poly-ubiquitination is recognized by adapters that direct mitochondria to the autophagosome. Known adapters include sequestosome (SQSTM1), neighbor of BRCA1 (NBR1), optineurin, nuclear domain 10 protein 52, and TAX1 binding protein; although, NBR1 is found to be non-essential for PRKN-mediated mitophagy [26,32,33]. Through this mechanism, a recent study found that in intestinal cancers, activation of mitophagy increased CD8+ T cells [34]. The upregulation of mitophagy causes an accumulation of iron followed by permeabilization of the lysosome. This permeabilization causes the release of proteases into the cytosol that induces presentation of MHC class I on the cell surface. This presentation elicits and an anti-tumor immune response by induction of CD8+ T-cells. In colitis, pharmacological induction of mitophagy through PRKN is found to inactivate inflammasomes in macrophages and ameliorate the impact of colitis [35,36,37]. While there are implications for PRKN-mediated mitophagy in CRC, one study found around 33% of colon tumors harbor PARK2 (the gene that encodes for PRKN) DNA copy number loss [38]. Interestingly, some colorectal cancer cell lines contain mutated forms of PRKN and may use alternate mechanisms to activate mitophagy [38]. Research is starting to investigate how PRKN may be used as a prognostic marker in CRC as it may correlate with invasion and overall survival [39].
2.4.2. Parkin-Independent Mitophagy
Alternatively to PRKN directed mitophagy, interaction of LC3 to FUN14 domain containing 1 (FUNDC1) protein located on the outer mitochondrial membrane can also initiate mitophagy [40]. Another PRKN-independent mechanism includes BCL2 interacting protein 3 (BNIP3), a critical receptor for mitophagy [41]. In breast cancer, BNIP3 loss promoted tumor progression and metastasis [42]. BNIP3 is induced by hypoxia signaling, a critical micro-environmental stressor in CRC (discussed below) [43]. The impact of BNIP3 and mitophagy in CRC have not been studied in detail. The known function in breast cancer, and BNIP3’s relationship with hypoxia signaling provides a foundation to investigate the role in CRC. Mitochondria may also be recruited to the autophagosome by FKBP8, a member of the FK506-binding protein family. FKBP8 is located on the outer membrane and has an anti-apoptotic role by interacting with Bcl-2 [44]. PRKN-independent mitophagy can be initiated by binding of LC3A to FKBP8 [45]. Additional mechanisms of PRKN-independent mitochondrial control are reviewed by Stockum et al. [46]. PRKN-independent mechanisms, and the evidence of mutated PRKN discussed above in CRC highlight the potential importance of investigating PRKN- independent mitophagy in CRC.
Pharmacological targeting of mitochondria with Mito-CP or Mito-Met10 in KRAS mutant colorectal cancers induced mitophagy and decreased cell proliferation [47]. In colon cancer, treatment with a BH3 mimetic, which inhibits Bcl-2 anti-apoptotic proteins, induced mitophagy. Treatment with the mimetic, in combination with an mitophagy inhibitor, reduced CRC cell growth [48]. However, conflicting roles of mitophagy have been noted in cancer (Reviewed in [49]). Treatment with the mitophagy inhibitor liensinine increased breast cancer cell death [50]. Conversely, activation of mitophagy with ceramide, a molecule involved in sphingolipid metabolism, reduced tumor burden in acute myeloid leukemia [51]. In-depth understanding of mitophagy in CRC is needed in order to develop better therapies that can target mitophagy to reduce tumor growth.
2.5. Ribophagy
Ribophagy is the breakdown of ribosomes in cells, which constitute 10% of total cellular protein. Ribophagy is extremely low basally in cells [52]. Initiation of ribophagy occurs by the binding of nuclear FMR1 interacting protein 1 (NUFIP1) to ribosomes. This interaction leads to autophagosome recruitment by LC3 [53]. Starvation or molecular target of rapamycin complex 1 (mTORC1) inhibition induced NUFIP1 activity and increased ribophagy [53]. Breakdown of ribosomes under starvation underscores the importance of ribophagy for cellular nutrient maintenance. However, non-selective bulk degradation of ribosomes may also be utilized [52]. Little information is known about ribophagy in cancer. However, ribosomes contain a large amount of amino acids and nucleotides and can potentially serve as a nutrient store in the tumor environment.
2.6. Proteophagy
Clearance of proteasomes through autophagy is known as proteophagy. Cross-talk between the proteasome and autophagy is found under nitrogen starvation in cells wherein autophagy degrades ribosomes (and proteasomes) under nutrient starvation [54]. One of the earliest citations of proteophagy suggests that this process occurs through a chaperone-mediated mechanism where the proteasome is targeted to the lysosome by HSC73 [55]. More recent work uncovers the sequestration of the proteasome in autophagosomes under cell stress, suggesting proteophagy can occur in a macro-autophagy or CMA driven fashion [56]. Currently there is no data to suggest that proteophagy is activated in cancer.
2.7. Pexophagy
Peroxisomes are small organelle structures that break down fatty acids in the cytoplasm. The degradation of these products through autophagy requires SQSTM1 and NBR1 [57]. In healthy liver, loss of autophagy through ATG7 led to a buildup of peroxisomes [58]. Under starvation conditions, ubiquitination of peroxisomes occurred by peroxisomal biogenesis factor 2 (PEX2) in HeLa cells and mouse embryonic fibroblasts [59]. To our knowledge, the utilization of pexophagy in CRC has yet to be investigated. However, hypoxia inducible factor-2α (HIF-2α), an important transcription factor in CRC (discussed below), was found to promote pexophagy in hepatocytes [60]. While these findings were not investigated in colon tissue, the activation of pexophagy under starvation and hypoxia highlights the potential importance of studying pexophagy in CRC.
2.8. Ferritinophagy
Iron storage protein ferritin is broken down by the lysosome for iron release and cellular iron utilization. This degradation is directed by the nuclear receptor coactivator 4 (NCOA4) [61]. Interestingly, ferritinophagy is required for induction of ferroptosis, a form of cell death that requires iron [62,63]. Certain cancers have shown a sensitivity to ferroptosis [64] and pharmacological induction of ferroptosis is found to reduce pancreatic and hepatic cancer cell growth [63,65]. While little work has been done to investigate the importance of ferritinophagy in CRC, the essential role of iron in CRC growth [66] and the sensitivity of different cancer types to ferroptosis [67] highlights the importance in studying ferritin turnover in CRC.
2.9. Xenophagy
Xenophagy is a process initiated by the cell for protection against pathogens. Phagophores engulf pathogens and fuse to the autophagosome for breakdown by autophagy. Xenophagy can play a particularly important role in the colon due to the host-microbiome interaction. Protection from intestinal epithelial infection requires the autophagy gene ATG16L1 [68]. Recent screening of xenophagy effectors identified a V-ATPase and ATG16L1 mechanism to specifically activate xenophagy under bacterial infiltration [69]. Certain bacteria can be targeted by SQSTM1, an important protein in autophagy [70]. In Crohn’s Disease, the stimulation of xenophagy using resveratrol reduced Salmonella Typhimurium, an enteric pathogen associated with Crohn’s Disease [71]. When colon cancer cells are treated with two mircoRNAs, MIR106B and MIR93, reduced ATG16L1 prevented removal of intracellular bacteria from epithelial cells via autophagy [72]. As mentioned, patients with Crohn’s Disease have an increased risk of developing CAC and this is partially due to bacterial infiltration. Understanding the role of xenophagy in host-microbiome homeostasis may be essential in characterizing the microbiota-tumor interaction.
3. Role of Autophagy in CRC
It is important to understand the role of autophagy at different stages and under different mutational loads to properly target tumors. A clinical study observed down-regulation of ATG5 in CRC patients. However, increased expression correlated with increased incidence of invasion [73]. Conversely, expression of LC3B and SQSTM1 correlated with poor prognosis [74]. In mouse models, loss of Atg7 in intestinal epithelial cells inhibited tumor growth through an immune response elicited by the microbiome [75]. Additionally, receptor for activated C kinase 1 (RACK1), a commonly found mutation in cancer, induced autophagy and promoted proliferation while inhibiting apoptosis in colon cancer [76]. Autophagy also modulated the degradation of the transcription factor FOXO3a in CRC. Inhibition of autophagy elevated levels of FOXO3a and led to transcriptional upregulation of pro-apoptotic genes [77]. Apoptosis also increased when autophagy was inhibited in CRC cells following activation of p53 and endoplasmic reticulum stress [78]. Conversely, treatment with Brevlin A increased autophagy and decreased tumor size [79]. This brief overview emphasizes the complexity of autophagy in CRC. It remains unclear if autophagy is anti- or pro- tumorigenic and in-depth mechanistic studies are needed. Table 1 outlines some of the opposing roles of autophagy in CRC.
An understanding of mutations in autophagy-associated genes, how autophagy is altered by increased mutational load, or via specific tumor suppressors or oncogenes is essential in assessing its role in tumor development. In CRC, there is low frequency of mutations in autophagy-associated genes [90]. In a small cohort of patient samples, tumors expressed decreased levels of ATG5, however increased expression correlated with invasion into lymphovascular tissue [73]. A study demonstrated that 95% of colon tumors expressed higher Beclin-1 compared to normal tissue [80]. These studies suggest that autophagy is important in cancer development. Similarly, in mutant KRAS cancers, autophagy induction occurred under starvation. Inhibition of KRAS reduced autophagy in these cells and inhibited cell growth [84]. In CRC-derived cell lines, p53 promoted the degradation of LC3 allowing for stable autophagic flux [81]. With loss of p53, LC3 accumulated and led to apoptosis. In CRCs with high microsatellite instability, 27% of the cancers harbored at least one mutation in either ATG2B, ATG5, ATG9B, or ATG12 [91]. Additionally, a study aimed to understand responsiveness to therapy in BRAF (a protein involved in RAS/MAPK signaling) mutant colon cancers found that treatment with EGFR antibodies and checkpoint inhibitors induced autophagy and combining these treatments with an autophagy inhibitor reduced CRC cell growth [92]. Patients with Crohn’s Disease have an increased chance of developing CAC. Deficiencies in the response to bacterial sensing and invasion were observed following loss of autophagy through impairment in nucleotide-binding oligomerization domain (NOD1/2) signaling [93]. Loss of autophagy or mutations in autophagic genes may increase bacterial infiltration, which can impact the development of CAC. Mutations in autophagy-associated genes, or regulation of autophagy through mutations in genes such as KRAS and p53, demonstrate the important role of this mechanism in CRC. To begin addressing how autophagy can be used clinically, researchers have found a gene signature based on nine autophagy related genes that can accurately predict survival in colon cancer [94].
Histone deacetylase inhibitors as stand-alone or adjuvant therapies are currently used in several cancers [95]. In colorectal cancer cells, inhibition of autophagy through chloroquine, in combination with the histone deacetylase inhibitor vorinostat, led to an accumulation of ubiquitinated proteins and increased cell death [85]. Additionally, chemo-resistance required decreased autophagy in 5-fluorouracil (a common chemotherapeutic for CRC) resistant cells [86]. The authors speculated, that this observation was due to low autophagy resulting in accumulation of tumor promoting oxidative stress, inflammation, and damaged mitochondria.
CRC consist of multiple epithelial cell types as well as infiltrating immune cells. In IBD it is clear that dysregulation of autophagy in Paneth cells impacts tissue injury and inflammation [96]. However, cell type specificity of autophagy in tumor growth is unclear.
Immune cell autophagy: The anti-tumor immune response can directly kill cancer cells. As the tumor progresses, the microenvironment shifts to a highly immunosuppressive state and many of the immune cells potentiate tumor growth. Immunosuppression is essential in enhancing tumor progression, and immune cells can employ autophagy to perform standard functions including antigen presentation and cytokine production (Reviewed in [97]). In tumor-associated macrophages (TAM)s, upregulation of autophagy reduced tumor growth and increased apoptosis in CRC cells. Moreover, radiosensitization of CRC required increased autophagy in TAMs [98]. Conversely, when autophagy is lost in regulatory T-cells by disrupting Atg7, there was impaired ability of the anti-tumor immune response to CRC cells [99]. This was due in part by increased apoptosis in the T-cells. While not specifically studied in colon cancer, different immune cells including neutrophils, macrophages, B-cells, and natural killer cells rely on autophagy for their development and function (Reviewed in [100]). Immune cell specific autophagy underscore the importance of investigating this pathway in different cell types to better develop strategies for modulating tumor growth.
Epithelial autophagy: Tumor epithelial autophagy in KRAS-driven cancers alters inflammatory mediators to suppress the immune response [82]. Furthermore, inhibition of autophagy in cancer cells blocked interferon gamma-mediated cell death [101]. The role of the immune system and its interaction with the gut microbiota is important in tumor development. Cell autonomous autophagy in healthy epithelial cells altered barrier function by breaking down junctional proteins such as claudin 2 [102]. Impaired barrier function can lead to increased bacterial infiltration to cause inflammation and damage in the gut. Recent work demonstrated tumor stage specific changes in bacterial infiltration, inflammatory signaling and cancer progression and growth in CRC [103]. This suggests a possible role of epithelial xenophagy in CRC. In CRC tumors, regulatory T-cell infiltration inversely correlated with SQSTM1 expression [104]. The utilization of autophagy in epithelial cells may alter recruitment or function of the immune response. In summary there are major differences in the direct impact of autophagy in epithelial cells, immune cells, or the heterocellular cross-talk between these cells that can impact CRC growth and progression (Figure 2). Understanding the changes in autophagy and how it impacts tumor response will allow researchers to further understand these mechanisms in different cell types.
4. Cellular Cues for Autophagic Activation in Cancer
4.1. Starvation
The highly proliferative nature of tumors leads to a reduction in availability of nutrients in the microenvironment. In cancer, hyper-activation of mTORC1, a known pathway of nutrient sensing, contributes to cell proliferation and tumor progression. mTORC1 is activated in about 50% of CRC tumors. Figure 3A outlines the known mechanistic cross-talk in CRC between autophagy and mTORC1. In conditions where amino acids are abundant, mTORC1 is localized to the lysosomal membrane (Reviewed in [105]). Hypoxic induction of DNA damage inducible transcript 4 (REDD1) signaling has been shown to regulate mTORC1 through truncation of the hemartin (Tsc1/Tsc2) complex [106]. Since mTORC1 is activated by available nutrients, a feedback loop exists between these two mechanisms wherein autophagy generates new macromolecules to activate mTORC1. The cross-talk between these two mechanisms are essential in maintaining cell growth and proliferation [106]. Importantly, mTORC1 is integrated to the autophagic pathway via activation of TFEB and ULK1-ATG13-FIP200 (Family kinase-interacting protein of 200 kDa) complex [107,108]. Independent of mTORC1, AMPK activated the ULK1 complex under starvation [109]. Under nutrient rich conditions mTORC1 phosphorylated ULK1 and inhibited the ULK1-AMPK interaction to block autophagy [109]. It is important to consider that the TFEB, ULK1, and AMPK pathways are known to be regulated by amino acids, which contributes to another mode of autophagy regulation [110]. In a model of lung cancer, amino acid starvation led to an induction of non-selective macro-autophagy. However, amino acid starvation has been shown independent of mTORC1 to induce micro-autophagy that directly engulfs receptors of selective autophagy including NCOA4, LC3B, and SQSTM1 into endosomes [20]. The authors suggest these functions may prevent selective macro-autophagy and promote non-selective autophagy under starvation. Interestingly, under leucine starvation, a cleaved form of SQSTM1 is generated by the protease caspase-8. Under starvation when autophagy is active, a portion of the available SQSTM1 is cleaved. In nutrient replete conditions, this cleaved protein activates mTORC1 to increase leucine sensing [111]. The cleaved SQSTM1 is not able to participate in autophagy preventing opposing functions between mTORC1 and autophagy. Moreover, in ovarian cancer cells, arginine deprivation activated autophagy to promote cell survival [112]. Inhibition of autophagy both chemically or genetically significantly reduced cell growth. While these studies were not in CRC, these findings highlight the potential of combinatorial therapeutics with autophagy inhibitors and treatments such as arginase for tumors that rely on arginine for growth [112]. Glucose uptake plays a critical role in the growth of many cancer types, including CRC. In glucose-free conditions, knock-down of autophagy-associated genes increased cell death [113]. Similarly, when colon cancer spheroids were stressed under restricted glucose or serum an increase in autophagy was observed [114]. Under similar starvation conditions, Kras mutant tumors require autophagy for oxidative metabolism [115]. Starvation also affected expression of claudin 1 in colon cancer. Expression of claudin 1 was higher in tumor tissue and showed co-staining with lysosomal markers LAMP1 and 2 with increased autophagy. Under starvation, claudin 1 expression increased mediating a reduction in SQSTM1. This suppression suggests claudin 1 cross-talks with autophagy under starvation [116]. When and how nutrient availability impacts autophagy is essential in understanding its function in CRC tumors (Figure 3B).
4.2. Hypoxia
Hypoxia plays a key role in CRC development and progression. Hypoxia signaling is mediated by two transcription factors, hypoxia-inducible factor (HIF)-1α and HIF-2α, which have overlapping and distinct functions. In CRC, HIF-2α (not HIF-1α) is essential for CRC growth and progression [117]. Hypoxia is a well conserved cell stress that activates autophagy [43]. In tumor hypoxic foci, autophagy levels are highly elevated but rapidly subside upon establishment of a blood supply [118]. In colon cancer there is a known connection between hypoxia and mitophagy. Hypoxia disrupted mitochondrial respiration leading to increased mitophagy (Figure 2) [119]. Moreover, HIF-1α upregulated BNIP3 to induce mitophagy [43]. In patient derived CRC cells, inhibition of autophagy with 3-Methyladenin in combination with hypoxia, increased apoptotic death in cancer cells [120]. Moreover, the micro RNA miR-20a was found to inhibit hypoxia induced autophagy [121]. Additionally, in glioblastoma, HIF-1α induced autophagy and drove tumor growth [122]. HIF-1α does not alter CRC tumorigenesis in mouse models [117], however it will be interesting to assess if HIF-2α has overlapping roles in the context of autophagy.
4.3. Microbiota
As highlighted briefly above, autophagy can play an important role through xenophagy in managing the host–microbiome interaction. Moreover, dysregulation of autophagy is well characterized in IBD. New work studying chronic colitis suggests that autophagy protected cells by reducing apoptosis through upregulation of tumor necrosis factor-α [123]. As discussed above, the importance of autophagy specifically in Paneth cells is known [96]. In healthy tissue, induction of autophagy in Paneth cells induced interferon gamma to protect against microbiota. However, when this mechanism is lost, intestinal inflammation is exacerbated [124]. Consistent with data from IBD, the heterocellular cross-talk with microbiota is a major factor in tumor-elicited inflammation in CRC. When microbiome composition is altered under chronic inflammation or barrier defects, changes in the inflammatory response altered tumorigenesis [125,126]. The cross-talk between the microbiota and immune system highlights the complexity of the tumor microenvironment in the colon [127,128]. The importance of these mechanisms have been studied in depth [129,130]. Loss of autophagy in healthy colon epithelial cells through Atg5 disruption altered the composition of the gut microbiota and the gut immune response suggesting implications in chronic colitis [131]. Similarly, loss of Atg7 in intestinal epithelial cells and tumor tissue led to infiltration of anti-tumor immune cells decreasing tumor burden [75]. Treatment with antibiotics attenuated this response, further supporting a novel integration of microbiota and autophagy in tumor growth. It is important to highlight that this work utilized an Apc model where tumor suppressor p53 was intact. In many cancers, however, p53 is deleted or mutated thus these findings may only be applicable to patients with wild-type p53 [75]. In summary, the above findings highlight the cross-talk between the microbiota and autophagy. Further mechanistic studies may uncover novel therapeutic approaches targeting autophagy and microbiota.
5. Autophagic Substrates
The broad use of autophagy to meet metabolic demands is reviewed by Rabinowitz and White [132]. Autophagy in normal cell physiology is critical to maintain amino acid levels [133]. While it is thought that the products of autophagic degradation are recycled for use in cancer, in CRC the substrates targeted for autophagy and how the degradative products are utilized is not clear. In cancer, autophagy can degrade macromolecules for nutrients, and degrade tumor suppressors or oncogenes to alter growth. Below we outline both of these functions.
Previous literature has found an increase in autophagy in CRC spheroids under glucose or serum restriction [114]. However, the degradative products of this process are unknown. A study investigated this question by studying loss of ATG5 in RAS-driven cancer cells. Loss of ATG5 showed global changes in the proteome. Inhibition of autophagy, in combination with starvation, increased endoplasmic reticulum chaperones, proteins involved in DNA replication, and Rig-I like receptor signaling pathway. However, proteins that are known to be essential in stress survival were not altered with autophagy inhibition under starvation conditions [82]. This work uncovers how autophagy impacts cellular response to stresses such as starvation that are observed in the tumor microenvironment. Additionally, in RAS-driven cancers, autophagy drove glycolysis [83]. Degradation of cellular components into amino acids is essential for cancer utilization. Thomas and colleagues demonstrated that amino acid levels in starved breast cancer cells increased with activated autophagy, whereas normal cells maintained amino acid levels under starvation. It is hypothesized that this is due to the high nutrient demand to maintain the proliferation rates of the cancer cells [134]. While this study was not in CRC this underscores the importance of understanding how autophagy is used for nutrient acquisition.
While autophagy may be employed to acquire nutrients, it has been shown to break down proteins that activate or block tumor growth. Autophagy can cause the degradation of dishevelled in colon cancer and contribute to the activation of Wnt signaling, thus promoting tumor growth [135]. Similarly in CRC, the cancerous inhibitor of protein phosphatase 2a (CIP2A) is overexpressed [136]. CIP2a is involved in Myc protein stability. Temsirolimus, an FDA approved mTORC1 inhibitor that activates autophagy led to degradation of CIP2a and cell death in CRC [87]. In colon cancer, CyclinD1 is highly expressed and contributes to hyper-proliferation. Estrogen receptor beta was shown to activate autophagy and cause the breakdown of CyclinD1 causing cell cycle arrest and tumor death [88]. To prevent growth, treatment with 4-hydroxytamoxifen caused degradation of KRAS through autophagy in colon cancer [89]. As mentioned previously, basal autophagy breaks down FOXO3a to prevent apoptosis in CRC, to promote tumor growth [77]. While some work has been done, the process of breaking down proteins to inhibit or promote tumor growth are not well studied. A thorough understanding of autophagy in the context of CRC is important in targeting these mechanisms.
6. Conclusions and Future Perspectives
In general, non-selective autophagy is used for nutrient stress while selective autophagy is used for cell maintenance. However, in the context of tumor growth in CRC or CAC, these roles may change. Understanding the autophagic substrates that are recycled and how those substrates are utilized in tumor growth and development will identify ideal targets for treatments. While we have discussed mechanisms by which tumor cells may obtain nutrients through autophagy, these mechanisms are not clearly defined in CRC. The cross-talk between hypoxia and mitophagy underscores the importance of these mechanisms in CRC. Identifying the role of selective autophagy for tumor growth will allow the development of targeted therapeutics for CRC. The potential importance of mitophagy in cell stress and nutrient availability highlights a potential target in cancers. Moreover, if tumor cells employ selective autophagy for growth and survival, these mechanisms may be targets for vulnerability in CRC. Some of these approaches are reviewed by Martins and Baptista [137]. Additionally, we have highlighted the cell type specific contributions of autophagy and more precise work on cell type specific dependency on autophagy will shed light on the mechanistic role of autophagy in tumor development. More directly, the pathways activated or inhibited during nutrient stress and how autophagic substrates are being utilized in cancer cells will be critical to understanding the pleiotropic role of autophagy in cancer growth and progression.
Author Contributions
All authors contributed to writing and editing this manuscript.
Funding
This work was supported by the NIH, R01CA148828 and R01DK095201 to Y.M.S, and T32GM007315 and T32CA140044 to S.N.D.
Conflicts of Interest
The authors declare no conflict of interest.
References
- National Cancer Institute. Surveillance, Epidemiology, and End Results (SEER) Program Research Data (1973–2014); S.R.P. National Cancer Institute: Rockville, MD, USA, 2017.
- Siegel, R.L.; Miller, K.D.; Fedewa, S.A.; Ahnen, D.J.; Meester, R.G.; Barzi, A.; Jemal, A. Colorectal cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 177–193. [Google Scholar] [CrossRef]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Bishop, K.; Altekruse, S.F. SEER Cancer Statistics Review, 1975–2013; National Cancer Institute: Rockville, MD, USA, 2016.
- Fearon, E.R. Molecular Genetics of Colorectal Cancer. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 479–507. [Google Scholar] [CrossRef] [PubMed]
- Eaden, J.; Abrams, K.; Mayberry, J. The risk of colorectal cancer in ulcerative colitis: A meta-analysis. Gut 2001, 48, 526–535. [Google Scholar] [CrossRef]
- Canavan, C.; Abrams, K.R.; Mayberry, J. Meta-analysis: Colorectal and small bowel cancer risk in patients with Crohn’s disease. Aliment. Pharm. Ther. 2006, 23, 1097–1104. [Google Scholar] [CrossRef]
- Kameyama, H.; Nagahashi, M.; Shimada, Y.; Tajima, Y.; Ichikawa, H.; Nakano, M.; Sakata, J.; Kobayashi, T.; Narayanan, S.; Takabe, K.; et al. Genomic characterization of colitis-associated colorectal cancer. World J. Surg. Oncol. 2018, 16, 121. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Pyo, J.-O.; Nah, J.; Jung, Y.-K. Molecules and their functions in autophagy. Exp. Mol. Med. 2012, 44, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef]
- Burada, F.; Nicoli, E.R.; Ciurea, M.E.; Uscatu, D.C.; Ioana, M.; Gheonea, D.I. Autophagy in colorectal cancer: An important switch from physiology to pathology. World J. Gastrointest. Oncol. 2015, 7, 271–284. [Google Scholar] [CrossRef]
- Wu, Y.; Yao, J.; Xie, J.; Liu, Z.; Zhou, Y.; Pan, H.; Han, W. The role of autophagy in colitis-associated colorectal cancer. Signal Transduct. Target. Ther. 2018, 3, 31. [Google Scholar] [CrossRef]
- Garbar, C.; Mascaux, C.; Giustiniani, J.; Merrouche, Y.; Bensussan, A. Chemotherapy treatment induces an increase of autophagy in the luminal breast cancer cell MCF7, but not in the triple-negative MDA-MB231. Sci. Rep. 2017, 7, 7201. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yang, M.; Kang, R.; Wang, Z.; Zhao, Y.; Yu, Y.; Xie, M.; Yin, X.; Livesey, K.M.; Lotze, M.T.; et al. HMGB1-induced autophagy promotes chemotherapy resistance in leukemia cells. Leukemia 2011, 25, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef] [PubMed]
- Mokarram, P.; Albokashy, M.; Zarghooni, M.; Moosavi, M.A.; Sepehri, Z.; Chen, Q.M.; Hudecki, A.; Sargazi, A.; Alizadeh, J.; Moghadam, A.R.; et al. New frontiers in the treatment of colorectal cancer: Autophagy and the unfolded protein response as promising targets. Autophagy 2017, 13, 781–819. [Google Scholar] [CrossRef] [PubMed]
- Dice, J.F. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem. Sci. 1990, 15, 305–309. [Google Scholar] [CrossRef]
- Vakifahmetoglu-Norberg, H.; Kim, M.; Xia, H.-G.; Iwanicki, M.P.; Ofengeim, D.; Coloff, J.L.; Pan, L.; Ince, T.A.; Kroemer, G.; Brugge, J.S.; et al. Corrigendum: Chaperone-mediated autophagy degrades mutant p53. Genome Res. 2016, 30, 870. [Google Scholar] [CrossRef]
- Kon, M.; Kiffin, R.; Koga, H.; Chapochnick, J.; Macian, F.; Varticovski, L.; Cuervo, A.M. Chaperone-mediated autophagy is required for tumor growth. Sci. Transl. Med. 2011, 3, 109–117. [Google Scholar] [CrossRef]
- Mejlvang, J.; Olsvik, H.; Svenning, S.; Bruun, J.-A.; Abudu, Y.P.; Larsen, K.B.; Brech, A.; Hansen, T.E.; Brenne, H.; Hansen, T.; et al. Starvation induces rapid degradation of selective autophagy receptors by endosomal microautophagy. J. Cell Boil. 2018, 217, 3640–3655. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The Role of Atg Proteins in Autophagosome Formation. Annu. Rev. Cell Dev. Boil. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef] [Green Version]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar] [PubMed]
- Tanida, I.; Minematsu-Ikeguchi, N.; Ueno, T.; Kominami, E. Lysosomal Turnover, but not a Cellular Level, of Endogenous LC3 is a Marker for Autophagy. Autophagy 2005, 1, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geißler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nature 2010, 12, 119–131. [Google Scholar]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; De Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Hollville, É.; Carroll, R.G.; Cullen, S.P.; Martin, S.J. Bcl-2 Family Proteins Participate in Mitochondrial Quality Control by Regulating Parkin/PINK1-Dependent Mitophagy. Mol. Cell 2014, 55, 451–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Xue, L.; Li, L.; Tang, C.; Wan, Z.; Wang, R.; Tan, J.; Tan, Y.; Han, H.; Tian, R.; et al. BNIP3 Protein Suppresses PINK1 Kinase Proteolytic Cleavage to Promote Mitophagy. J. Boil. Chem. 2016, 291, 21616–21629. [Google Scholar] [CrossRef] [Green Version]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Eiyama, A.; Kondo-Okamoto, N.; Okamoto, K. Mitochondrial degradation during starvation is selective and temporally distinct from bulk autophagy in yeast. FEBS Lett. 2013, 587, 1787–1792. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Fung, G.; Deng, H.; Zhang, J.; Fiesel, F.C.; Springer, W.; Li, X.; Luo, H. NBR1 is dispensable for PARK2-mediated mitophagy regardless of the presence or absence of SQSTM1. Cell Death Dis. 2015, 6, e1943. [Google Scholar] [CrossRef]
- Yoo, S.-M.; Jung, Y.-K. A Molecular Approach to Mitophagy and Mitochondrial Dynamics. Mol. Cells 2018, 41, 18–26. [Google Scholar] [PubMed]
- Ziegler, P.K.; Bollrath, J.; Pallangyo, C.K.; Matsutani, T.; Canli, Ö.; De Oliveira, T.; Diamanti, M.A.; Müller, N.; Gamrekelashvili, J.; Putoczki, T.; et al. Mitophagy in Intestinal Epithelial Cells Triggers Adaptive Immunity during Tumorigenesis. Cell 2018, 174, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Mai, C.T.; Wu, M.M.; Wang, C.L.; Su, Z.R.; Cheng, Y.Y.; Zhang, X.J. Palmatine attenuated dextran sulfate sodium (DSS)-induced colitis via promoting mitophagy-mediated NLRP3 inflammasome inactivation. Mol. Immunol. 2019, 105, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wang, J.; Yang, Y.; Liu, X.; Zhu, Y.; Zou, J.; Peng, S.; Le, T.H.; Chen, Y.; Zhao, S.; et al. Ginsenoside Rd ameliorates colitis by inducing p62-driven mitophagy-mediated NLRP3 inflammasome inactivation in mice. Biochem. Pharm. 2018, 155, 366–379. [Google Scholar] [CrossRef]
- Guo, W.; Sun, Y.; Liu, W.; Wu, X.; Guo, L.; Cai, P.; Wu, X.; Wu, X.; Shen, Y.; Shu, Y.; et al. Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy 2014, 10, 972–985. [Google Scholar] [CrossRef]
- Poulogiannis, G.; McIntyre, R.E.; Dimitriadi, M.; Apps, J.R.; Wilson, C.H.; Ichimura, K.; Luo, F.; Cantley, L.C.; Wyllie, A.H.; Adams, D.J.; et al. PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice. Proc. Natl. Acad. Sci. USA 2010, 107, 15145–15150. [Google Scholar] [CrossRef]
- Da Silva-Camargo, C.C.V.; Baldin, R.K.S.; Polli, N.L.C.; Agostinho, A.P.; Olandosk, M.; de Noronha, L.; Sotomaior, V.S. Parkin protein expression and its impact on survival of patients with advanced colorectal cancer. Cancer Biol. Med. 2018, 15, 61–69. [Google Scholar]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nature 2012, 14, 177–185. [Google Scholar] [CrossRef]
- Zhu, Y.; Massen, S.; Terenzio, M.; Lang, V.; Chen-Lindner, S.; Eils, R.; Novak, I.; Dikic, I.; Hamacher-Brady, A.; Brady, N.R. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J. Biol. Chem. 2013, 288, 1099–1113. [Google Scholar] [CrossRef]
- Chourasia, A.H.; Tracy, K.; Frankenberger, C.; Boland, M.L.; Sharifi, M.N.; Drake, L.; Sachleben, J.R.; Asara, J.M.; Locasale, J.W.; Karczmar, G.S.; et al. Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep. 2015, 16, 1145–1163. [Google Scholar] [CrossRef] [Green Version]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-Induced Autophagy Is Mediated through Hypoxia-Inducible Factor Induction of BNIP3 and BNIP3L via Their BH3 Domains. Mol. Cell. Boil. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirane, M.; Nakayama, K.I. Inherent calcineurin inhibitor FKBP38 targets Bcl-2 to mitochondria and inhibits apoptosis. Nat. Cell Biol. 2003, 5, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Bhujabal, Z.; Birgisdottir, Å.B.; Sjøttem, E.; Brenne, H.B.; Øvervatn, A.; Habisov, S.; Kirkin, V.; Lamark, T.; Johansen, T. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 2017, 18, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Von Stockum, S.; Marchesan, E.; Ziviani, E. Mitochondrial quality control beyond PINK1/Parkin. Oncotarget 2018, 9, 12550–12551. [Google Scholar] [CrossRef] [PubMed]
- Boyle, K.A.; Van Wickle, J.; Hill, R.B.; Marchese, A.; Kalyanaraman, B.; Dwinell, M.B. Mitochondria-targeted drugs stimulate mitophagy and abrogate colon cancer cell proliferation. J. Boil. Chem. 2018, 293, 14891–14904. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhang, C.; Yan, X.; Lan, B.; Wang, J.; Wei, C.; Cao, X.; Wang, R.; Yao, J.; Zhou, T.; et al. A Novel Bioavailable BH3 Mimetic Efficiently Inhibits Colon Cancer via Cascade Effects of Mitochondria. Clin. Cancer Res. 2016, 22, 1445–1458. [Google Scholar] [CrossRef]
- Yan, C.; Li, T.-S. Dual Role of Mitophagy in Cancer Drug Resistance. Anticancer Res. 2018, 38, 617–621. [Google Scholar] [Green Version]
- Zhou, J.; Li, G.; Zheng, Y.; Shen, H.-M.; Hu, X.; Ming, Q.-L.; Huang, C.; Li, P.; Gao, N. A novel autophagy/mitophagy inhibitor liensinine sensitizes breast cancer cells to chemotherapy through DNM1L-mediated mitochondrial fission. Autophagy 2015, 11, 1259–1279. [Google Scholar] [CrossRef] [Green Version]
- Dany, M.; Gencer, S.; Nganga, R.; Thomas, R.J.; Oleinik, N.; Baron, K.D.; Szulc, Z.M.; Ruvolo, P.; Kornblau, S.; Andreeff, M.; et al. Targeting FLT3-ITD signaling mediates ceramide-dependent mitophagy and attenuates drug resistance in AML. Blood 2016, 128, 1944–1958. [Google Scholar] [CrossRef] [Green Version]
- An, H.; Harper, J.W. Systematic analysis of ribophagy in human cells reveals bystander flux during selective autophagy. Nat. Cell Biol. 2018, 20, 135–143. [Google Scholar] [CrossRef]
- Wyant, G.A.; Abu-Remaileh, M.; Frenkel, E.M.; Laqtom, N.N.; Dharamdasani, V.; Lewis, C.A.; Chan, S.H.; Heinze, I.; Ori, A.; Sabatini, D.M. NUFIP1 is a ribosome receptor for starvation-induced ribophagy. Science 2018, 360, 751–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waite, K.A.; Mota-Peynado, A.; Vontz, G.; Roelofs, J. Starvation Induces Proteasome Autophagy with Different Pathways for Core and Regulatory Particles. J. Biol. Chem. 2016, 291, 3239–3253. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Palmer, A.; Rivett, A.J.; Knecht, E. Degradation of Proteasomes by Lysosomes in Rat Liver. JBIC J. Boil. Inorg. Chem. 1995, 227, 792–800. [Google Scholar]
- Dengjel, J.; Høyer-Hansen, M.; Nielsen, M.O.; Eisenberg, T.; Harder, L.M.; Schandorff, S.; Farkas, T.; Kirkegaard, T.; Becker, A.C.; Schroeder, S.; et al. Identification of Autophagosome-associated Proteins and Regulators by Quantitative Proteomic Analysis and Genetic Screens. Mol. Cell. Proteom. 2012, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zientara-Rytter, K.; Subramani, S.; Katarzyna, Z.-R.; Suresh, S. Autophagic degradation of peroxisomes in mammals. Biochem. Soc. Trans. 2016, 44, 431–440. [Google Scholar] [CrossRef] [Green Version]
- Iwata, J.-I.; Ostrovsky, O.; Berman, B.; Gallagher, J.; Mulloy, B.; Fernig, D.G.; Delehedde, M.; Ron, D. Excess Peroxisomes Are Degraded by Autophagic Machinery in Mammals. J. Boil. Chem. 2006, 281, 4035–4041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sargent, G.; Van Zutphen, T.; Shatseva, T.; Zhang, L.; Di Giovanni, V.; Bandsma, R.; Kim, P.K. PEX2 is the E3 ubiquitin ligase required for pexophagy during starvation. J. Cell Boil. 2016, 214, 677–690. [Google Scholar] [CrossRef]
- Walter, K.M.; Schönenberger, M.J.; Trötzmüller, M.; Horn, M.; Elsässer, H.-P.; Moser, A.B.; Lucas, M.S.; Schwarz, T.; Gerber, P.A.; Faust, P.L.; et al. Hif-2α Promotes Degradation of Mammalian Peroxisomes by Selective Autophagy. Cell Metab. 2014, 20, 882–897. [Google Scholar] [CrossRef]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef]
- Sui, S.; Zhang, J.; Xu, S.; Wang, Q.; Wang, P.; Pang, D. Ferritinophagy is required for the induction of ferroptosis by the bromodomain protein BRD4 inhibitor (+)-JQ1 in cancer cells. Cell Death Dis. 2019, 10, 331. [Google Scholar] [CrossRef]
- Louandre, C.; Ezzoukhry, Z.; Godin, C.; Barbare, J.-C.; Mazière, J.-C.; Chauffert, B.; Galmiche, A. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int. J. Cancer 2013, 133, 1732–1742. [Google Scholar] [CrossRef] [PubMed]
- Manz, D.H.; Blanchette, N.L.; Paul, B.T.; Torti, F.M.; Torti, S.V. Iron and cancer: Recent insights. Ann. N. Y. Acad. Sci. 2016, 1368, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Eling, N.; Reuter, L.; Hazin, J.; Hamacher-Brady, A.; Brady, N.R. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2015, 2, 517–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, R.L.; Davis, F.G.; Sutter, E.; Sobin, L.H.; Kikendall, J.W.; Bowen, P. Body Iron Stores and Risk of Colonic Neoplasia. J. Natl. Cancer Inst. 1994, 86, 455–460. [Google Scholar] [CrossRef]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef]
- Conway, K.L.; Kuballa, P.; Song, J.; Patel, K.K.; Castoreno, A.B.; Yilmaz, Ö.H.; Jijon, H.B.; Zhang, M.; Aldrich, L.N.; Villablanca, E.J.; et al. Atg16l1 is required for autophagy in intestinal epithelial cells and protection of mice from Salmonella infection. Gastroenterology 2013, 145, 1347–1357. [Google Scholar] [CrossRef]
- Xu, Y.; Zhou, P.; Cheng, S.; Lu, Q.; Nowak, K.; Hopp, A.K.; Li, L.; Shi, X.; Zhou, Z.; Gao, W.; et al. A Bacterial Effector Reveals the V-ATPase-ATG16L1 Axis that Initiates Xenophagy. Cell 2019, 178, 552–566. [Google Scholar] [CrossRef]
- Zheng, Y.T.; Shahnazari, S.; Brech, A.; Lamark, T.; Johansen, T.; Brumell, J.H. The Adaptor Protein p62/SQSTM1 Targets Invading Bacteria to the Autophagy Pathway. J. Immunol. 2009, 183, 5909–5916. [Google Scholar] [CrossRef] [Green Version]
- Al Azzaz, J.; Rieu, A.; Aires, V.; Delmas, D.; Chluba, J.; Winckler, P.; Bringer, M.A.; Lamarche, J.; Vervandier-Fasseur, D.; Dalle, F.; et al. Resveratrol-Induced Xenophagy Promotes Intracellular Bacteria Clearance in Intestinal Epithelial Cells and Macrophages. Front. Immunol. 2018, 9, 3149. [Google Scholar] [CrossRef]
- Lu, C.; Chen, J.; Xu, H.G.; Zhou, X.; He, Q.; Li, Y.L.; Jiang, G.; Shan, Y.; Xue, B.; Zhao, R.X.; et al. MIR106B and MIR93 prevent removal of bacteria from epithelial cells by disrupting ATG16L1-mediated autophagy. Gastroenterology 2014, 146, 188–199. [Google Scholar] [CrossRef]
- Cho, D.-H.; Jo, Y.K.; Kim, S.C.; Park, I.J.; Kim, J.C. Down-regulated expression of ATG5 in colorectal cancer. Anticancer Res. 2012, 32, 4091–4096. [Google Scholar] [PubMed]
- Niklaus, M.; Adams, O.; Berezowska, S.; Zlobec, I.; Graber, F.; Slotta-Huspenina, J.; Nitsche, U.; Rosenberg, R.; Tschan, M.P.; Langer, R. Expression analysis of LC3B and p62 indicates intact activated autophagy is associated with an unfavorable prognosis in colon cancer. Oncotarget 2017, 8, 54604–54615. [Google Scholar] [CrossRef] [PubMed]
- Lévy, J.; Cacheux, W.; Bara, M.A.; L’Hermitte, A.; Lepage, P.; Fraudeau, M.; Trentesaux, C.; LeMarchand, J.; Durand, A.; Crain, A.-M.; et al. Intestinal inhibition of Atg7 prevents tumour initiation through a microbiome-influenced immune response and suppresses tumour growth. Nature 2015, 17, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Zhu, W.; Huang, W.; Lu, S.-S.; Li, X.-H.; Xiao, Z.-Q.; Yi, H. RACK1 promotes tumorigenicity of colon cancer by inducing cell autophagy. Cell Death Dis. 2018, 9, 1148. [Google Scholar] [CrossRef]
- Fitzwalter, B.E.; Towers, C.G.; Sullivan, K.D.; Andrysik, Z.; Hoh, M.; Ludwig, M.; O’Prey, J.; Ryan, K.M.; Espinosa, J.M.; Morgan, M.J.; et al. Autophagy Inhibition Mediates Apoptosis Sensitization in Cancer Therapy by Relieving FOXO3a Turnover. Dev. Cell 2018, 44, 555–565. [Google Scholar] [CrossRef]
- Sakitani, K.; Hirata, Y.; Hikiba, Y.; Hayakawa, Y.; Ihara, S.; Suzuki, H.; Suzuki, N.; Serizawa, T.; Kinoshita, H.; Sakamoto, K.; et al. Inhibition of autophagy exerts anti-colon cancer effects via apoptosis induced by p53 activation and ER stress. BMC Cancer 2015, 15, 795. [Google Scholar] [CrossRef]
- You, P.; Wu, H.; Deng, M.; Peng, J.; Li, F.; Yang, Y. Brevilin A induces apoptosis and autophagy of colon adenocarcinoma cell CT26 via mitochondrial pathway and PI3K/AKT/mTOR inactivation. Biomed. Pharm. 2018, 98, 619–625. [Google Scholar] [CrossRef]
- Ahn, C.H.; Jeong, E.G.; Lee, J.W.; Kim, M.S.; Kim, S.H.; Kim, S.S.; Yoo, N.J.; Lee, S.H. Expression of beclin-1, an autophagy-related protein, in gastric and colorectal cancers. APMIS 2007, 115, 1344–1349. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Weidberg, H.; Gonen, C.; Wilder, S.; Elazar, Z.; Oren, M. p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc. Natl. Acad. Sci. USA 2010, 107, 18511–18516. [Google Scholar] [CrossRef] [Green Version]
- Mathew, R.; Khor, S.; Hackett, S.R.; Rabinowitz, J.D.; Perlman, D.H.; White, E. Functional role of autophagy-mediated proteome remodeling in cell survival signaling and innate immunity. Mol. Cell 2014, 55, 916–930. [Google Scholar] [CrossRef]
- Lock, R.; Roy, S.; Kenific, C.M.; Su, J.S.; Salas, E.; Ronen, S.M.; Debnath, J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Boil. Cell 2011, 22, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Alves, S.; Castro, L.; Fernandes, M.S.; Francisco, R.; Castro, P.; Priault, M.; Chaves, S.R.; Moyer, M.P.; Oliveira, C.; Seruca, R.; et al. Colorectal cancer-related mutant KRAS alleles function as positive regulators of autophagy. Oncotarget 2015, 6, 30787–30802. [Google Scholar] [CrossRef] [PubMed]
- Carew, J.S.; Medina, E.C.; Esquivel, J.A.; Mahalingam, D.; Swords, R.; Kelly, K.; Zhang, H.; Huang, P.; Mita, A.C.; Mita, M.M.; et al. Autophagy inhibition enhances vorinostat-induced apoptosis via ubiquitinated protein accumulation. J. Cell Mol. Med. 2010, 14, 2448–2459. [Google Scholar] [CrossRef]
- Yao, C.W.; Kang, K.A.; Piao, M.J.; Ryu, Y.S.; Fernando, P.M.D.J.; Oh, M.C. Reduced Autophagy in 5-Fluorouracil Resistant Colon Cancer Cells. Biomol. Ther. 2017, 25, 315–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.-W.; Yang, S.-H.; Huang, G.-D.; Lin, J.-K.; Chen, W.-S.; Jiang, J.-K.; Lan, Y.-T.; Lin, C.-C.; Hwang, W.-L.; Tzeng, C.-H.; et al. Temsirolimus enhances the efficacy of cetuximab in colon cancer through a CIP2A-dependent mechanism. J. Cancer Res. Clin. Oncol. 2014, 140, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Huang, C.; Wu, H.; Huang, J. Estrogen Receptor Beta (ERβ) Mediated-CyclinD1 Degradation via Autophagy Plays an Anti-Proliferation Role in Colon Cells. Int. J. Boil. Sci. 2019, 15, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Kohli, L.; Kaza, N.; Coric, T.; Byer, S.J.; Brossier, N.M.; Klocke, B.J.; Bjornsti, M.-A.; Carroll, S.L.; Roth, K.A. 4-Hydroxytamoxifen induces autophagic death through K-Ras degradation. Cancer Res. 2013, 73, 4395–4405. [Google Scholar] [CrossRef]
- Costa, J.R.; Prak, K.; Aldous, S.; Gewinner, C.A.; Ketteler, R. Autophagy gene expression profiling identifies a defective microtubule-associated protein light chain 3A mutant in cancer. Oncotarget 2016, 7, 41203–41216. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.R.; Kim, M.S.; Oh, J.E.; Kim, Y.R.; Song, S.Y.; Kim, S.S. Frameshift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J. Pathol. 2009, 217, 702–706. [Google Scholar] [CrossRef]
- Koustas, E.; Papavassiliou, A.G.; Karamouzis, M.V. The role of autophagy in the treatment of BRAF mutant colorectal carcinomas differs based on microsatellite instability status. PLoS ONE 2018, 13, e0207227. [Google Scholar] [CrossRef]
- Travassos, L.H.; Carneiro, L.A.; Ramjeet, M.; Hussey, S.; Kim, Y.G.; Magalhães, J.G.; Yuan, L.; Soares, F.; Chea, E.; Bourhis, L.L.; et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 2010, 11, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Mo, S.; Dai, W.; Xiang, W.; Li, Y.; Feng, Y.; Zhang, L.; Li, Q.; Cai, G. Prognostic and predictive value of an autophagy-related signature for early relapse in stages I-III colon cancer. Carcinogenesis 2019, 40, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Tampakis, A.; Tampaki, E.; Nebiker, C.; Kouraklis, G. Histone deacetylase inhibitors and colorectal cancer: What is new? Anticancer Agents Med. Chem. 2014, 14, 1220–1227. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-L.; Shao, B.-Z.; Zhao, S.-B.; Fang, J.; Gu, L.; Miao, C.-Y.; Li, Z.-S.; Bai, Y. Impact of Paneth Cell Autophagy on Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuballa, P.; Nolte, W.M.; Castoreno, A.B.; Xavier, R.J. Autophagy and the immune system. Annu. Rev. Immunol. 2012, 30, 611–646. [Google Scholar] [CrossRef]
- Shao, L.-N.; Xing, C.-G.; Yang, X.-D.; Young, W.; Zhu, B.-S.; Cao, J.-P. Effects of autophagy regulation of tumor-associated macrophages on radiosensitivity of colorectal cancer cells. Mol. Med. Rep. 2016, 13, 2661–2670. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Long, L.; Yang, K.; Guy, C.; Shrestha, S.; Chen, Z.; Wu, C.; Vogel, P.; Neale, G.; Green, D.R.; et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat. Immunol. 2016, 17, 277–285. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.-M.; Tan, Y.; Wang, H.; Peng, L.; Chen, H.-T.; Meng, X.-J.; Li, L.-L.; Liu, Y.; Li, W.-F.; Shan, H. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Mol. Cancer 2019, 18, 17. [Google Scholar] [CrossRef]
- Mathew, R.; White, E. Eat this, not that! How selective autophagy helps cancer cells survive. Mol. Cell. Oncol. 2015, 2, e975638. [Google Scholar] [CrossRef] [Green Version]
- Nighot, P.K.; Hu, C.-A.A.; Ma, T.Y. Autophagy Enhances Intestinal Epithelial Tight Junction Barrier Function by Targeting Claudin-2 Protein Degradation. J. Boil. Chem. 2015, 290, 7234–7246. [Google Scholar] [CrossRef] [Green Version]
- Yachida, S.; Mizutani, S.; Shiroma, H.; Shiba, S.; Nakajima, T.; Sakamoto, T.; Watanabe, H.; Masuda, K.; Nishimoto, Y.; Kubo, M.; et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat. Med. 2019, 25, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Kosumi, K.; Masugi, Y.; Yang, J.; Qian, Z.R.; Kim, S.A.; Li, W.; Shi, Y.; Da Silva, A.; Hamada, T.; Liu, L.; et al. Tumor SQSTM1 (p62) expression and T cells in colorectal cancer. OncoImmunology 2017, 6, e1284720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Jones, E.; Inoki, K. Lysosomal Regulation of mTORC1 by Amino Acids in Mammalian Cells. Biomolecules 2017, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genome Res. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [Green Version]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012, 5, 42. [Google Scholar] [CrossRef]
- Jung, C.H.; Jun, C.B.; Ro, S.-H.; Kim, Y.-M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.-H. ULK-Atg13-FIP200 Complexes Mediate mTOR Signaling to the Autophagy Machinery. Mol. Boil. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Meijer, A.J.; Lorin, S.; Blommaart, E.F.; Codogno, P. Regulation of autophagy by amino acids and MTOR-dependent signal transduction. Amino Acids 2015, 47, 2037–2063. [Google Scholar] [CrossRef]
- Sanchez-Garrido, J.; Sancho-Shimizu, V.; Shenoy, A.R. Regulated proteolysis of p62/SQSTM1 enables differential control of autophagy and nutrient sensing. Sci. Signal. 2018, 11, 6903. [Google Scholar] [CrossRef]
- Shuvayeva, G.; Bobak, Y.; Igumentseva, N.; Titone, R.; Morani, F.; Stasyk, O.; Isidoro, C. Single Amino Acid Arginine Deprivation Triggers Prosurvival Autophagic Response in Ovarian Carcinoma SKOV3. BioMed. Res. Int. 2014, 2014, 505041. [Google Scholar] [CrossRef]
- Schroll, M.M.; Labonia, G.J.; Ludwig, K.R.; Hummon, A.B. Glucose Restriction Combined with Autophagy Inhibition and Chemotherapy in HCT 116 Spheroids Decreases Cell Clonogenicity and Viability Regulated by Tumor Suppressor Genes. J. Proteome Res. 2017, 16, 3009–3018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroll, M.M.; Liu, X.; Herzog, S.K.; Skube, S.B.; Hummon, A.B. Nutrient Restriction of Glucose or Serum Results in Similar Proteomic Expression Changes in 3D Colon Cancer Cell Cultures. Nutr. Res. 2016, 36, 1068–1080. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.Y.; Chen, H.-Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Choi, S.; Kim, J.O.; Kim, K.K. Autophagy-mediated upregulation of cytoplasmic claudin 1 stimulates the degradation of SQSTM1/p62 under starvation. Biochem. Biophys. Res. Commun. 2018, 496, 159–166. [Google Scholar] [CrossRef]
- Xue, X.; Ramakrishnan, S.K.; Shah, Y.M. Activation of HIF-1alpha does not increase intestinal tumorigenesis. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, 187–195. [Google Scholar] [CrossRef]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Sakakibara, K.; Chen, Q.; Okamoto, K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 2014, 24, 787–795. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Wu, Y.; Zhao, G.-L.; Ye, Z.-Y.; Xing, C.-G.; Yang, X.-D. Inhibition of autophagy by 3-MA promotes hypoxia-induced apoptosis in human colorectal cancer cells. Eur. Rev. Med Pharm. Sci. 2019, 23, 1047–1054. [Google Scholar]
- Che, J.; Wang, W.; Huang, Y.; Zhang, L.; Zhao, J.; Zhang, P.; Yuan, X. miR-20a inhibits hypoxia-induced autophagy by targeting ATG5/FIP200 in colorectal cancer. Mol. Carcinog. 2019, 58, 1234–1247. [Google Scholar] [CrossRef]
- Hu, Y.-L.; DeLay, M.; Jahangiri, A.; Molinaro, A.M.; Rose, S.D.; Carbonell, W.S.; Aghi, M.K. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012, 72, 1773–1783. [Google Scholar] [CrossRef]
- Pott, J.; Maloy, K.J. Epithelial autophagy controls chronic colitis by reducing TNF-induced apoptosis. Autophagy 2018, 14, 1460–1461. [Google Scholar] [CrossRef] [PubMed]
- Burger, E.; Araujo, A.; López-Yglesias, A.; Rajala, M.W.; Geng, L.; Levine, B.; Hooper, L.V.; Burstein, E.; Yarovinsky, F. Loss of Paneth Cell Autophagy Causes Acute Susceptibility to Toxoplasma gondii-Mediated Inflammation. Cell Host Microbe 2018, 23, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Elinav, E.; Huber, S.; Strowig, T.; Hao, L.; Hafemann, A.; Jin, C.; Wunderlich, C.; Wunderlich, T.; Eisenbarth, S.C.; et al. Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 9862–9867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grivennikov, S.I.; Wang, K.; Mucida, D.; Stewart, C.A.; Schnabl, B.; Jauch, D.; Taniguchi, K.; Yu, G.-Y.; Österreicher, C.H.; Hung, K.E.; et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012, 491, 254–258. [Google Scholar] [CrossRef] [Green Version]
- Triner, D.; Devenport, S.N.; Ramakrishnan, S.K.; Ma, X.; Frieler, R.A.; Greenson, J.K.; Inohara, N.; Nunez, G.; Colacino, J.A.; Mortensen, R.M.; et al. Neutrophils Restrict Tumor-Associated Microbiota to Reduce Growth and Invasion of Colon Tumors in Mice. Gastroenterology 2018, 156, 1467–1482. [Google Scholar] [CrossRef]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef]
- Cianci, R.; Pagliari, D.; Piccirillo, C.A.; Fritz, J.H.; Gambassi, G. The Microbiota and Immune System Crosstalk in Health and Disease. Mediat. Inflamm. 2018, 2018, 2912539. [Google Scholar] [CrossRef]
- Hooper, L.V.; Littman, D.R.; MacPherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef]
- Yang, L.; Liu, C.; Zhao, W.; He, C.; Ding, J.; Dai, R.; Xu, K.; Xiao, L.; Luo, L.; Liu, S.; et al. Impaired Autophagy in Intestinal Epithelial Cells Alters Gut Microbiota and Host Immune Responses. Appl. Environ. Microbiol. 2018, 84, e00880-18. [Google Scholar] [CrossRef]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef]
- Onodera, J.; Ohsumi, Y. Autophagy Is Required for Maintenance of Amino Acid Levels and Protein Synthesis under Nitrogen Starvation. J. Boil. Chem. 2005, 280, 31582–31586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.; Davis, T.; Loos, B.; Sishi, B.; Huisamen, B.; Strijdom, H.; Engelbrecht, A.-M. Autophagy is essential for the maintenance of amino acids and ATP levels during acute amino acid starvation in MDAMB231 cells. Cell Biochem. Funct. 2018, 36, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Cao, W.; Bao, L.; Zuo, W.; Xie, G.; Cai, T.; Fu, W.; Zhang, J.; Wu, W.; Zhang, X.; et al. Autophagy negatively regulates Wnt signalling by promoting Dishevelled degradation. Nature 2010, 12, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Wiegering, A.; Pfann, C.; Uthe, F.W.; Otto, C.; Rycak, L.; Mäder, U.; Gasser, M.; Waaga-Gasser, A.-M.; Eilers, M.; Germer, C.-T. CIP2A Influences Survival in Colon Cancer and Is Critical for Maintaining Myc Expression. PLoS ONE 2013, 8, e75292. [Google Scholar] [CrossRef] [PubMed]
- Martins, W.K.; Baptista, M.S. Autophagy Modulation for Organelle-Targeting Therapy. In Autophagy in Current Trends in Cellular Physiology and Pathology; IntechOpen: London, UK, 2016. [Google Scholar] [Green Version]
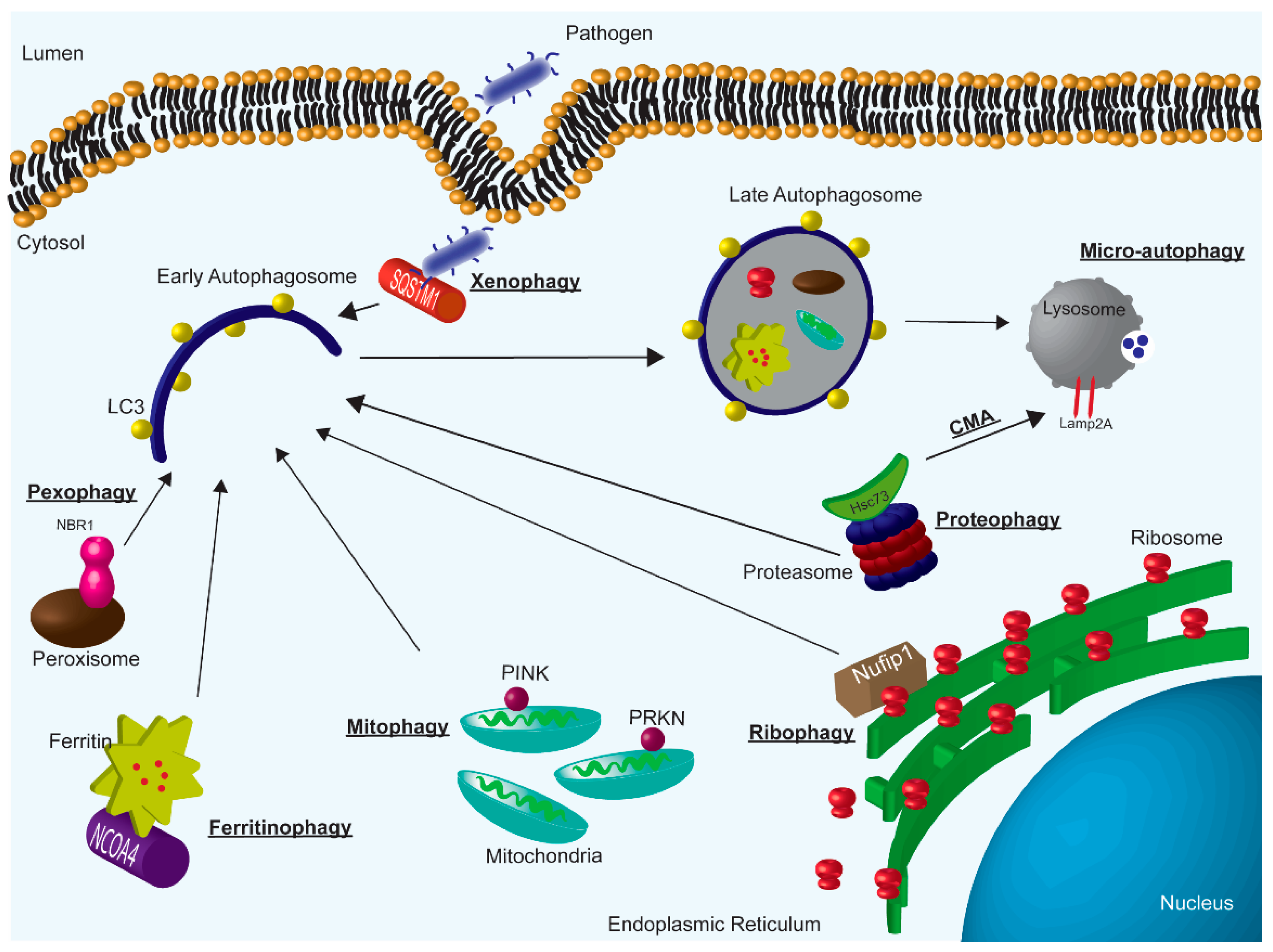
Figure 1.
Overview of autophagy subtypes; macro-autophagy, micro-autophagy, and CMA. Specifically, highlighting examples of selective macro-autophagy.
Figure 1.
Overview of autophagy subtypes; macro-autophagy, micro-autophagy, and CMA. Specifically, highlighting examples of selective macro-autophagy.

Figure 2.
Schematic of the tumor microenvironment highlighting the impact of autophagy. Cell autonomous roles of autophagy in immune, epithelial or, the cross-talk between cell types in colorectal cancer.
Figure 2.
Schematic of the tumor microenvironment highlighting the impact of autophagy. Cell autonomous roles of autophagy in immune, epithelial or, the cross-talk between cell types in colorectal cancer.

Figure 3.
Schematic summarizing (A) Simplified overview of mechanisms of mTORC1 regulation. and (B) how nutrient modulation impacts autophagy. Bolded mechanisms indicate data from non-CRC samples. Please refer to text for detailed mechanisms.
Figure 3.
Schematic summarizing (A) Simplified overview of mechanisms of mTORC1 regulation. and (B) how nutrient modulation impacts autophagy. Bolded mechanisms indicate data from non-CRC samples. Please refer to text for detailed mechanisms.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Functions of autophagy in CRC. A. Observation/Autophagy indicates what mechanisms are observed in CRC tumors and if autophagy is active or inactive. Tumor response is a summary of whether or not the autophagy activity indicated generates a pro- or anti- tumor response. B. Summary of therapies and their modulation of autophagy. Treatment: Which therapy was employed. Autophagy: how the stated therapy modulated autophagy activation. Tumor response: how manipulation of autophagy via therapeutic treatment impacted tumor growth.
Table 1.
Functions of autophagy in CRC. A. Observation/Autophagy indicates what mechanisms are observed in CRC tumors and if autophagy is active or inactive. Tumor response is a summary of whether or not the autophagy activity indicated generates a pro- or anti- tumor response. B. Summary of therapies and their modulation of autophagy. Treatment: Which therapy was employed. Autophagy: how the stated therapy modulated autophagy activation. Tumor response: how manipulation of autophagy via therapeutic treatment impacted tumor growth.
| A | ||||
| Observation | Autophagy | Tumor response | Reference | |
| Activated chaperone-mediated autophagy in tumors | Active | Pro-tumor | [19] | |
| Epithelial mitophagy increases CD8+T-Cells | Active | Anti-tumor | [34] | |
| Loss of PARK2 accelerates tumor development | Inactive | Pro-tumor | [38] | |
| Decreased ATG5 in CRC patients | Inactive | Pro-tumor | [73] | |
| Increased ATG5 yields increased invasion | Active | Pro-tumor | [73] | |
| Active autophagy through LC3B and SQSTM1 | Active | Pro-tumor | [74] | |
| Loss of ATG7 | Inactive | Anti-tumor | [75] | |
| RACK1 induces autophagy | Active | Pro-tumor | [76] | |
| High Beclin-1 in CRC | Active | Pro-tumor | [80] | |
| Increased LC3 with loss of p53 | Active | Anti-tumor | [81] | |
| Autophagy suppresses immune response in KRAS cancer | Active | Pro-tumor | [82] | |
| Autophagy drives glycolysis in RAS cancers | Active | Pro-tumor | [83] | |
| B | ||||
| Treatment | Autophagy | Tumor response | Reference | |
| Mito-CP or Mito-Met10 | Active | Anti-Tumor; Decreased proliferation in KRAS mutant cancers. | [47] | |
| BH3 mimetic and chloroquine | Inactive | Anti-Tumor; Induced apoptosis. | [48] | |
| Bafilomycin A1 or chloroquine | Inactive | Anti-Tumor; Elevated FOXO3a and transcriptional upregulation of pro-apoptotic genes. | [77] | |
| Brevlin A | Active | Anti-Tumor; Promoted expression of LC3-II and induced autophagy. | [79] | |
| KRAS siRNA | Inactive | Anti-Tumor; Inhibiting mutant Kras inhibits autophagy and induces apoptosis. | [84] | |
| Vorinostat with chloroquine | Inactive | Anti-Tumor; Induced apoptosis. | [85] | |
| 5-Fuorouracil and chloroquine | Inactive | Anti-Tumor; 5-FU treatment induced autophagy for resistance. Inhibition of autophagy reduced growth. | [86] | |
| Temsirolimus | Active | Anti-Tumor; Inhibited mTOR to activate autophagy and degrade CIP2A. | [87] | |
| Estrogen Receptor Beta | Active | Anti-Tumor; Autophagy directed CyclinD1 degradation inhibited growth. | [88] | |
| 4-Hydroxytamoxifen | Active | Anti-Tumor; Degradation of KRAS through autophagy induced cel death. | [89] | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Devenport, S.N.; Shah, Y.M. Functions and Implications of Autophagy in Colon Cancer. Cells 2019, 8, 1349. https://doi.org/10.3390/cells8111349
AMA Style
Devenport SN, Shah YM. Functions and Implications of Autophagy in Colon Cancer. Cells. 2019; 8(11):1349. https://doi.org/10.3390/cells8111349
Chicago/Turabian StyleDevenport, Samantha N, and Yatrik M Shah. 2019. "Functions and Implications of Autophagy in Colon Cancer" Cells 8, no. 11: 1349. https://doi.org/10.3390/cells8111349
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.