Metabolic, Physiological, and Transcriptomics Analysis of Batch Cultures of the Green Microalga Chlamydomonas Grown on Different Acetate Concentrations

 , ,
, , 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains and Cultivation Conditions
2.2. Samples and RNA Extraction
2.3. Sequencing
2.4. Read Trimming and Quality Filtering
2.5. Read Mapping
2.6. Surprisal Analysis
2.7. Differential Gene Expression in the Constraint Vector Giα
2.8. Gene Set Enrichment
2.9. Metabolic Analyses
2.10. Oxygen Evolution Measurements
3. Results
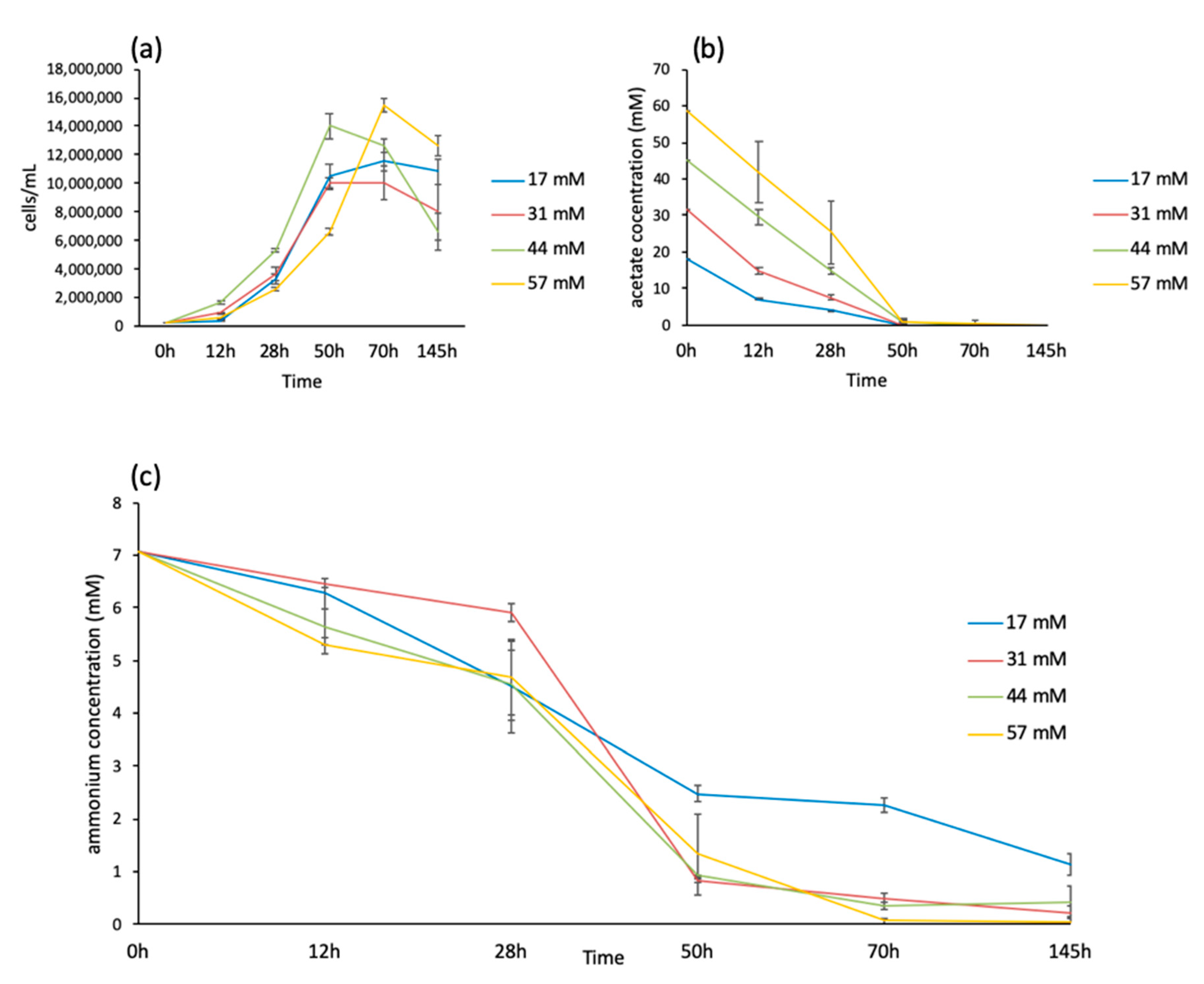
3.1. Growth Curves and Medium Composition of C. Reinhardtii Using Different Acetate Concentrations
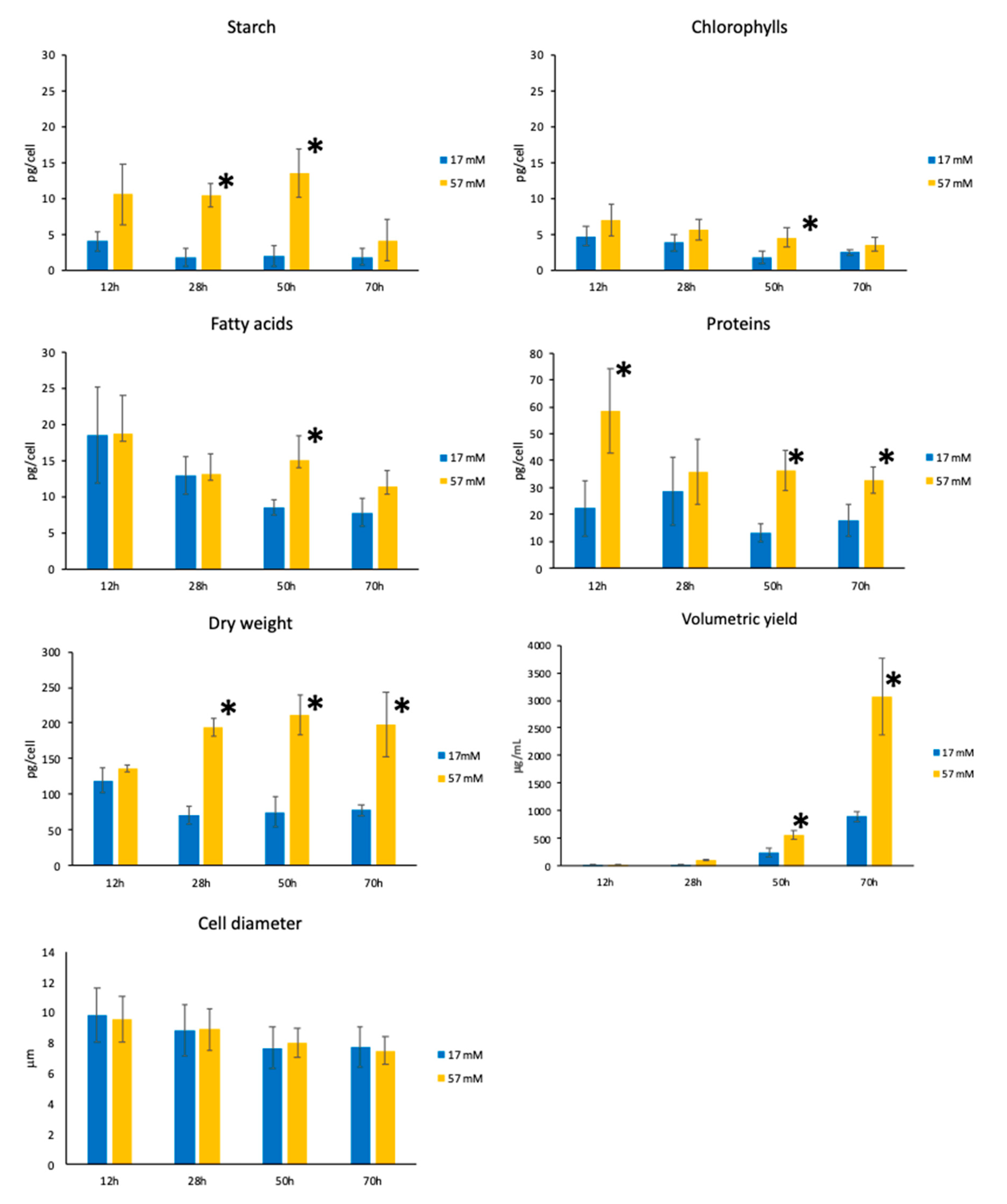
3.2. Biomass Composition Analysis
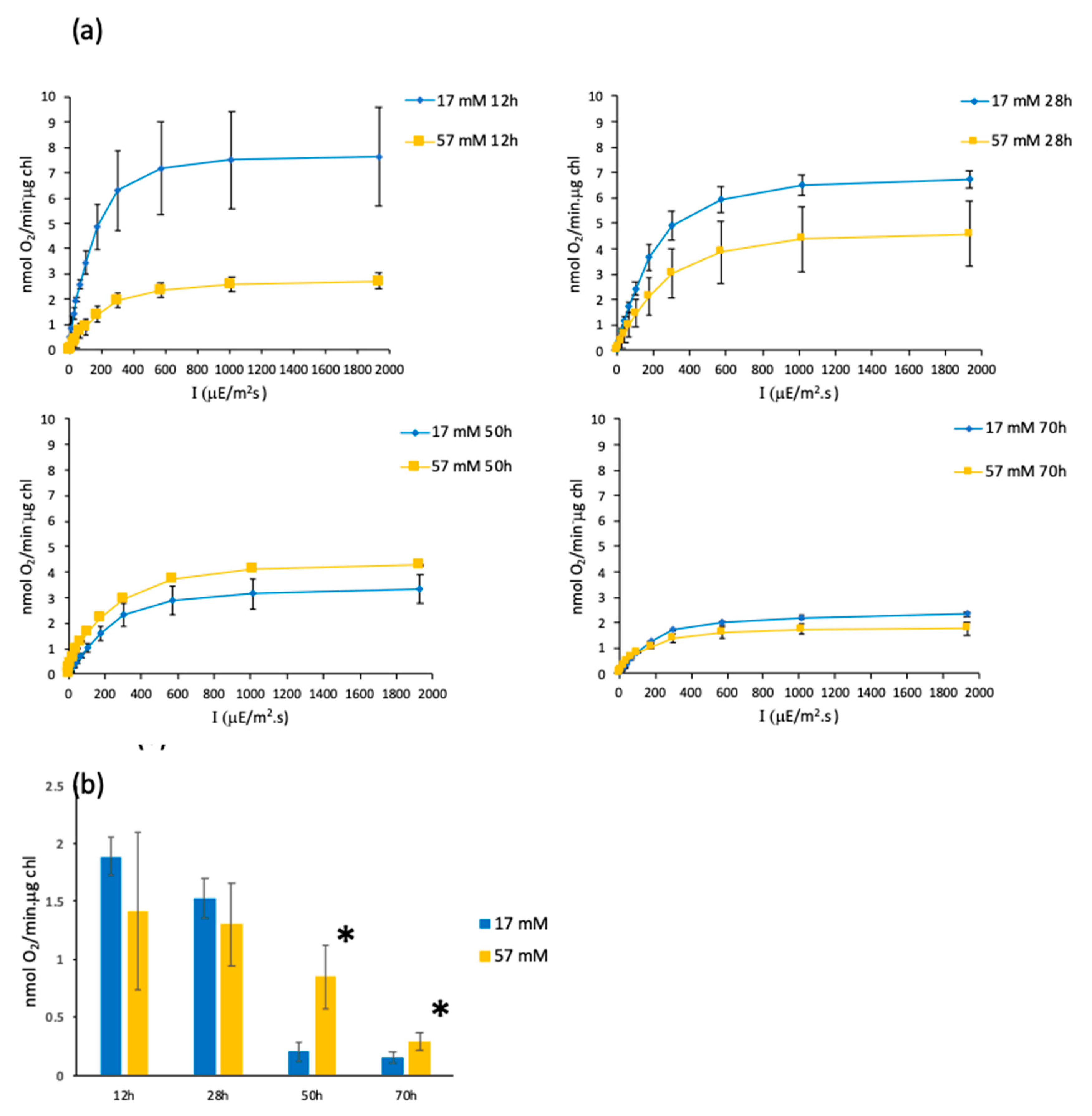
3.3. O2 Evolution
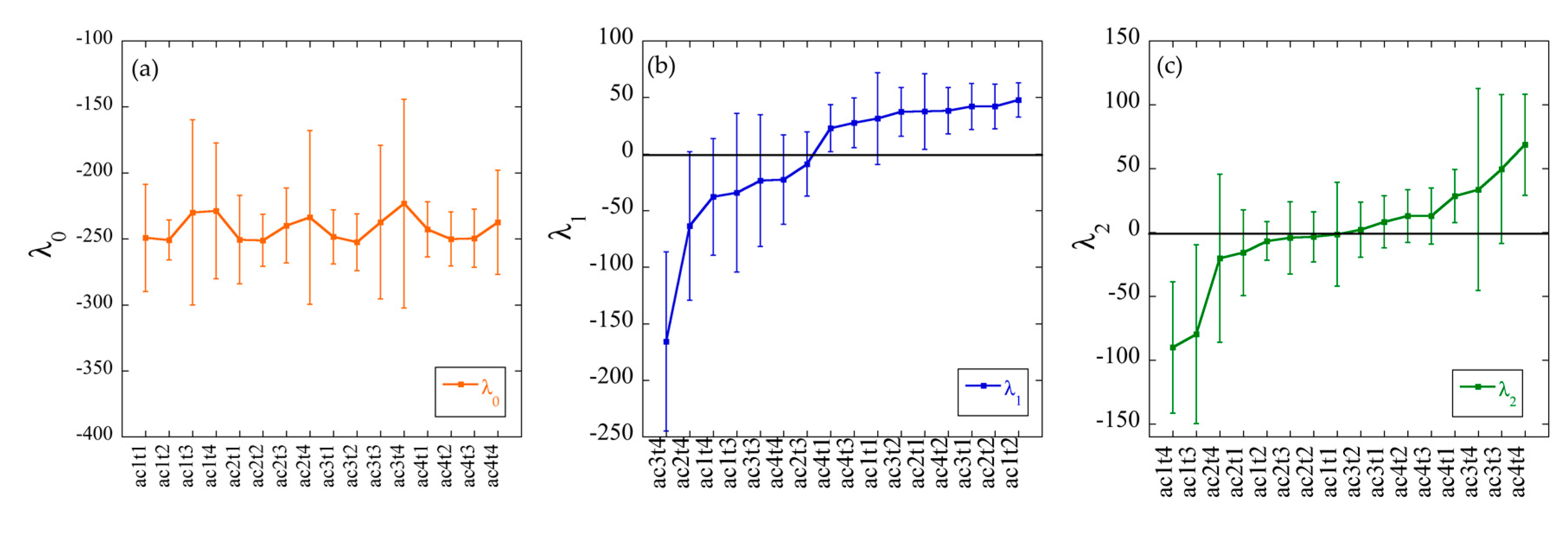
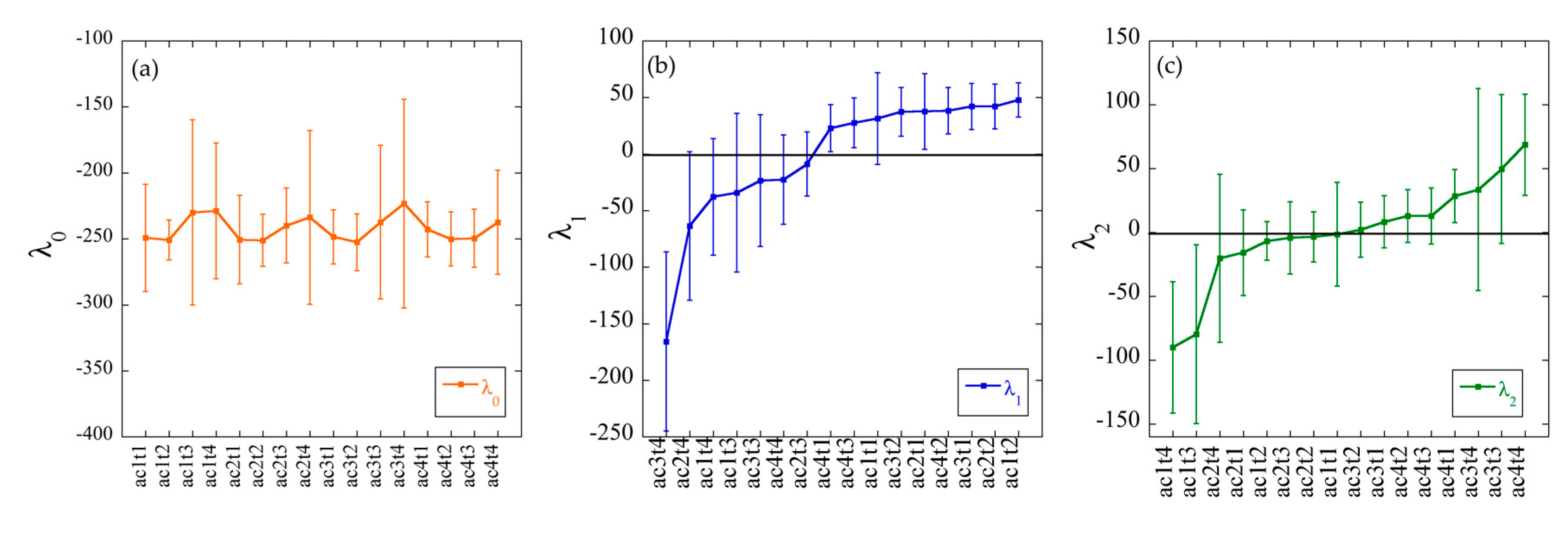
3.4. Surprisal Analysis of Transcriptomics Data
3.4.1. Constraints 1 and 2 Respectively Allow the Grouping of Samples Based on Their Physiological State or the Acetate Concentration
3.4.2. Gene Set Enrichment Analysis Allows for the Description of the Biological Pathways Contributing to the Balanced State and to the First and Second Constraints
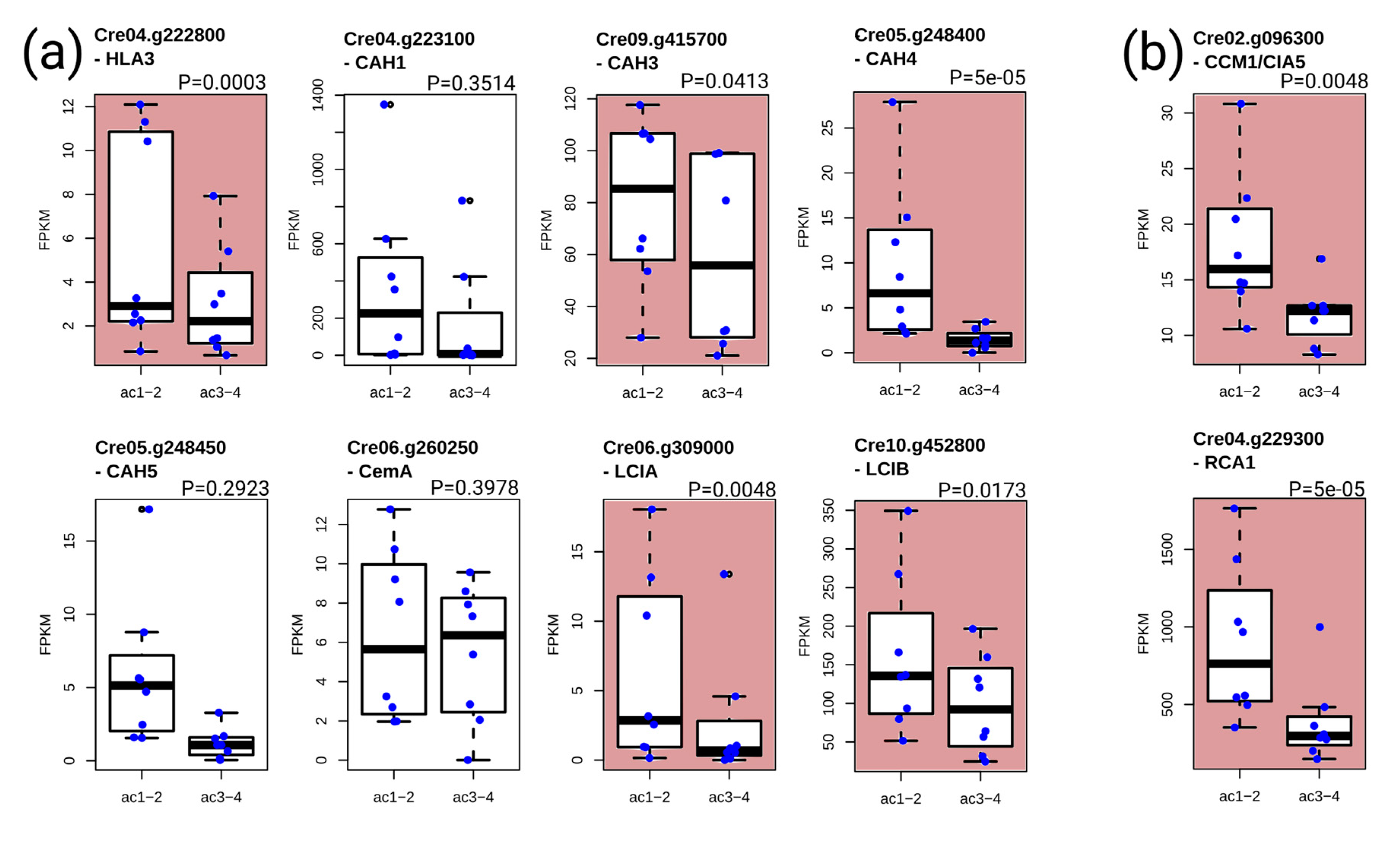
3.5. Analysis of the Transcripts Encoding Components of the Carbon-Concentrating Mechanism (CCM)
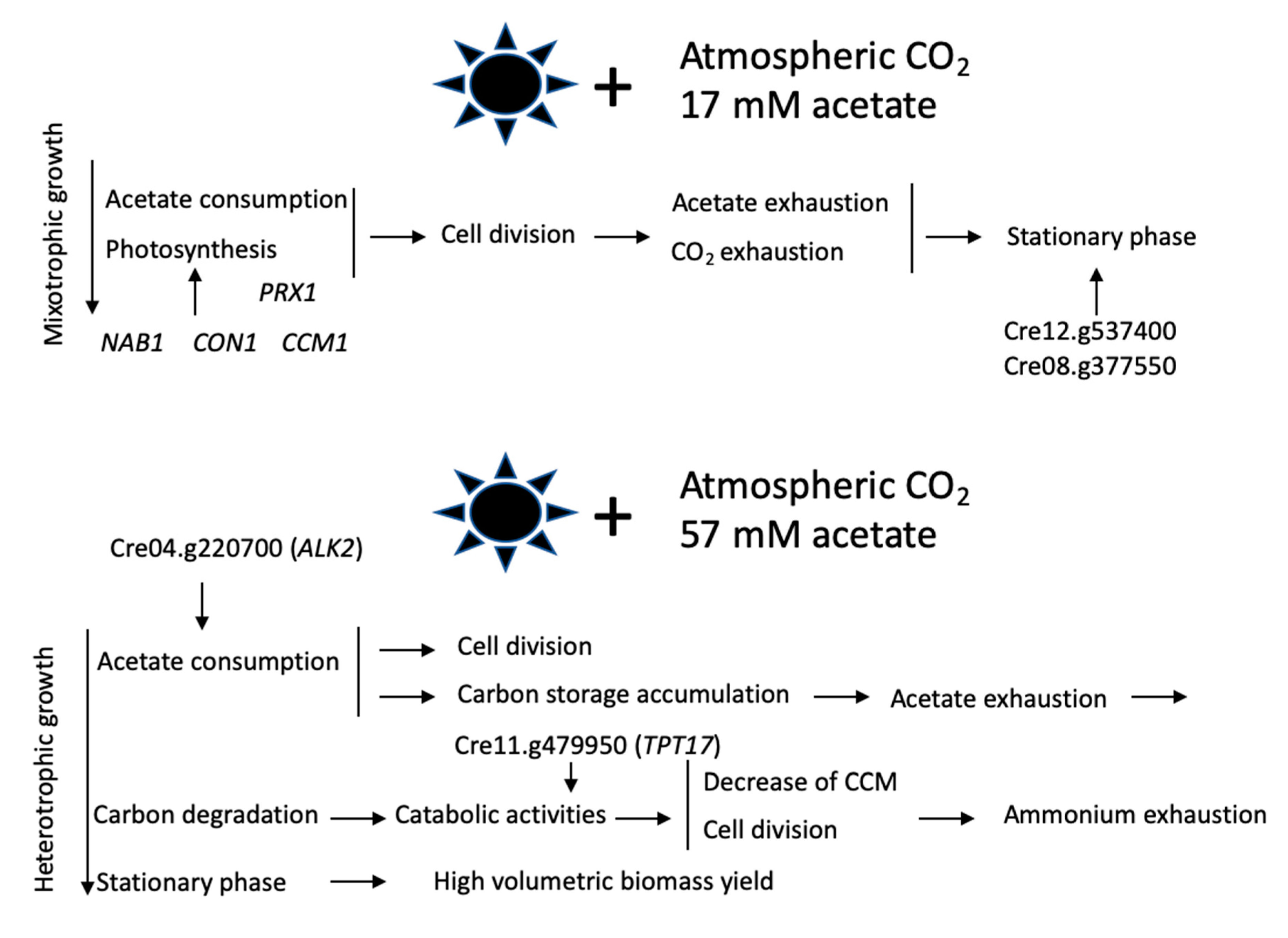
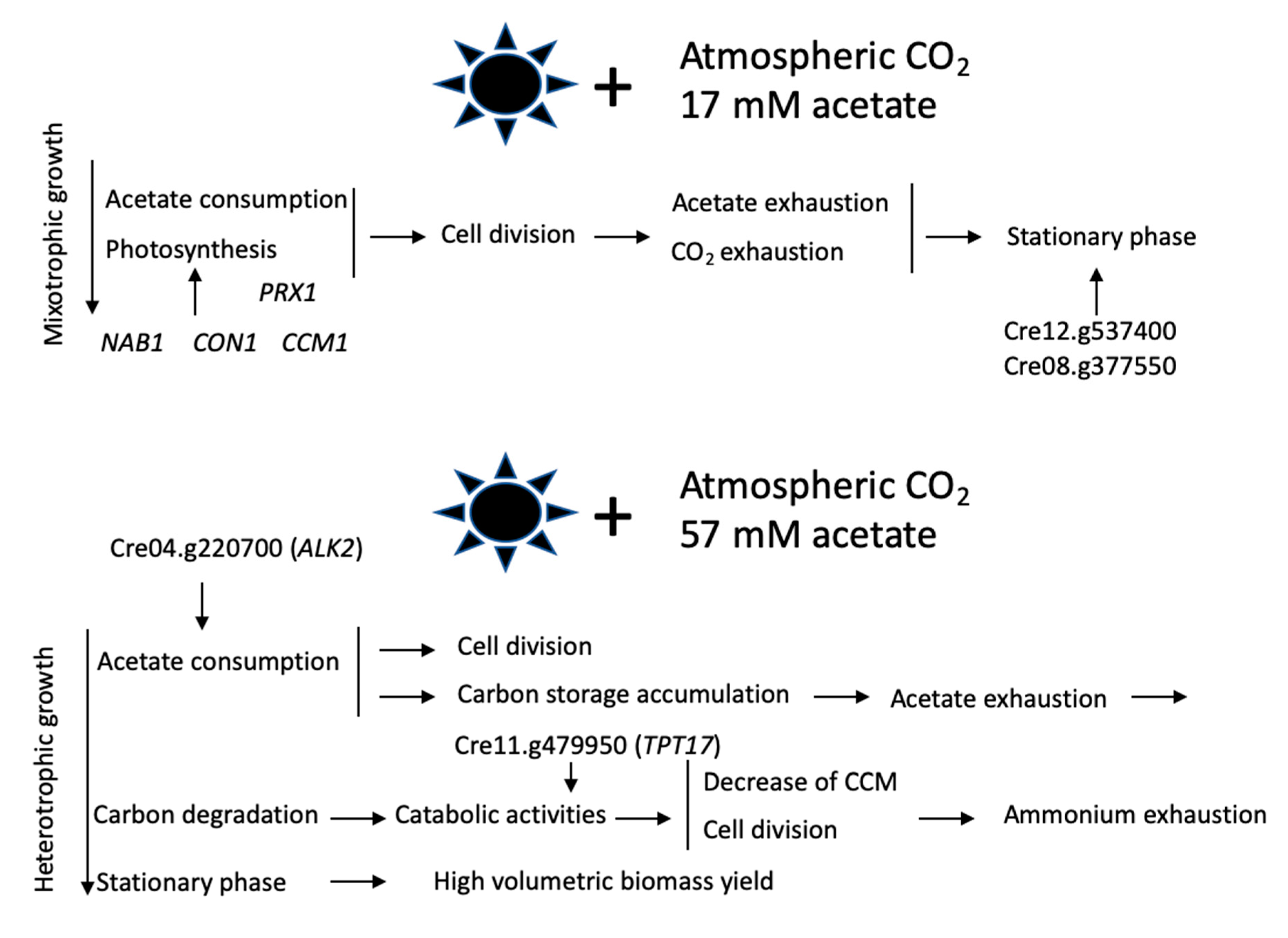
4. Discussion
5. Conclusion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | ||
|---|---|---|
| ac1t1 | 0.38910 | 40.666 |
| ac1t2 | 0.14485 | 15.139 |
| ac1t3 | 0.67040 | 70.066 |
| ac1t4 | 0.49241 | 51.463 |
| ac2t1 | 0.32035 | 33.481 |
| ac2t2 | 0.18783 | 19.631 |
| ac2t3 | 0.27087 | 28.309 |
| ac2t4 | 0.62907 | 65.746 |
| ac3t1 | 0.19546 | 20.428 |
| ac3t2 | 0.20691 | 21.625 |
| ac3t3 | 0.55731 | 58.246 |
| ac3t4 | 0.75679 | 79.094 |
| ac4t1 | 0.19919 | 20.818 |
| ac4t2 | 0.19710 | 20.600 |
| ac4t3 | 0.21022 | 21.971 |
| ac4t4 | 0.37858 | 39.567 |
References
- Fan, J.; Yan, C.; Andre, C.; Shanklin, J.; Schwender, J.; Xu, C. Oil accumulation is controlled by carbon precursor supply for fatty acid synthesis in Chlamydomonas reinhardtii. Plant Cell Physiol. 2012, 53, 1380–1390. [Google Scholar] [CrossRef] [PubMed]
- Plancke, C.; Vigeolas, H.; Höhner, R.; Roberty, S.; Emonds-Alt, B.; Larosa, V.; Willamme, R.; Duby, F.; Onga Dhali, D.; Thonart, P.; et al. Lack of isocitrate lyase in Chlamydomonas leads to changes in carbon metabolism and in the response to oxidative stress under mixotrophic growth. Plant J. 2014, 77, 404–417. [Google Scholar] [CrossRef] [PubMed]
- Salinas, T.; Larosa, V.; Cardol, P.; Maréchal-Drouard, L.; Remacle, C. Respiratory-deficient mutants of the unicellular green alga Chlamydomonas: A review. Biochimie 2014, 100, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Harris, E.H. The Chlamydomonas Sourcebook; Elsevier Inc.: Amsterdam, The Netherlands, 1989; p. 26. [Google Scholar]
- Heifetz, P.B.; Förster, B.; Osmond, C.B.; Giles, L.J.; Boynton, J.E. Effects of acetate on facultative autotrophy in Chlamydomonas reinhardtii assessed by photosynthetic measurements and stable isotope analyses. Plant Physiol. 2000, 122, 1439–1445. [Google Scholar] [CrossRef]
- Humby, P.L.; Snyder, E.C.R.; Durnford, D.G. Conditional senescence in Chlamydomonas reinhardtii (Chlorophyceae). J. Phycol. 2013, 49, 389–400. [Google Scholar] [CrossRef]
- Martinez, M.J. Genomic analysis of stationary-phase and exit in Saccharomyces cerevisiae: Gene expression and identification of novel essential genes. Mol. Biol. Cell 2004, 15, 5295–5305. [Google Scholar] [CrossRef]
- Kamei, Y.; Tamada, Y.; Nakayama, Y.; Fukusaki, E.; Mukai, Y. Changes in transcription and metabolism during the early stage of replicative cellular senescence in budding yeast. J. Biol. Chem. 2014, 289, 32081–32093. [Google Scholar] [CrossRef]
- Pérez-Pérez, M.E.; Crespo, J.L. Autophagy in the model alga Chlamydomonas reinhardtii. Autophagy 2010, 6, 562–563. [Google Scholar] [CrossRef]
- Esperanza, M.; Houde, M.; Seoane, M.; Cid, Á.; Rioboo, C. Does a short-term exposure to atrazine provoke cellular senescence in Chlamydomonas reinhardtii? Aquat. Toxicol. 2017, 189, 184–193. [Google Scholar] [CrossRef]
- Bogaert, K.A.; Manoharan-Basil, S.S.; Perez, E.; Levine, R.D.; Remacle, F.; Remacle, C. Surprisal analysis of genome-wide transcript profiling identifies differentially expressed genes and pathways associated with four growth conditions in the microalga Chlamydomonas. PLoS ONE 2018, 13, e0195142. [Google Scholar] [CrossRef]
- Remacle, F.; Kravchenko-Balasha, N.; Levitzki, A.; Levine, R.D. Information-theoretic analysis of phenotype changes in early stages of carcinogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 10324–10329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remacle, F.; Levine, R.D. Statistical thermodynamics of transcription profiles in normal development and tumorigeneses in cohorts of patients. Eur. Biophys. J. 2015, 44, 709–726. [Google Scholar] [CrossRef] [PubMed]
- Kravchenko-Balasha, N.; Remacle, F.; Gross, A.; Rotter, V.; Levitzki, A.; Levine, R.D. Convergence of logic of cellular regulation in different premalignant cells by an information theoretic approach. BMC Syst. Biol. 2011, 5, 42. [Google Scholar] [CrossRef] [PubMed]
- Kravchenko-Balasha, N.; Levitzki, A.; Goldstein, A.; Rotter, V.; Gross, A.; Remacle, F.; Levine, R.D. On a fundamental structure of gene networks in living cells. Proc. Natl. Acad. Sci. USA 2012, 109, 4702–4707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zadran, S.; Remacle, F.; Levine, R.D. miRNA and mRNA cancer signatures determined by analysis of expression levels in large cohorts of patients. Proc. Natl. Acad. Sci. USA 2013, 110, 19160–19165. [Google Scholar] [CrossRef] [Green Version]
- Fakhimi, N.; Dubini, A.; Tavakoli, O.; González-Ballester, D. Acetic acid is key for synergetic hydrogen production in Chlamydomonas-bacteria co-cultures. Bioresour. Technol. 2019, 289, 121648. [Google Scholar] [CrossRef]
- Willamme, R.; Alsafra, Z.; Arumugam, R.; Eppe, G.; Remacle, F.; Levine, R.D.D.; Remacle, C. Metabolomic analysis of the green microalga Chlamydomonas reinhardtii cultivated under day/night conditions. J. Biotechnol. 2015, 215, 20–26. [Google Scholar] [CrossRef]
- Pröschold, T.; Harris, E.H.; Coleman, A.W. Portrait of a species: Chlamydomonas reinhardtii. Genetics 2005, 170, 1601–1610. [Google Scholar] [CrossRef]
- Pulich, W.M.; Ward, C.H. Physiology and ultrastructure of an oxygen-resistant Chlorella mutant under heterotrophic conditions. Plant Physiol. 1973, 51, 337–344. [Google Scholar] [CrossRef]
- Newman, S.M.; Boynton, J.E.; Gillham, N.W.; Randolph-Anderson, B.L.; Johnson, A.M.; Harris, E.H. Transformation of chloroplast ribosomal RNA genes in Chlamydomonas: Molecular and genetic characterization of integration events. Genetics 1990, 126, 875–888. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Merchant, S.S.; Prochnik, S.E.; Vallon, O.; Harris, E.H.; Karpowicz, S.J.; Witman, G.B.; Terry, A.; Salamov, A.; Fritz-Laylin, L.K.; Maréchal-Drouard, L.; et al. The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 2007, 318, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. TopHat and Cufflinks_Protocol. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Tenenbaum, D. KEGGREST: Client-Side REST Access to KEGG. R Package Version 1.24.0; Fred Hutchinson Cancer Research Center: Seattle, WA, USA, 2019. [Google Scholar]
- Terauchi, A.M.; Peers, G.; Kobayashi, M.C.; Niyogi, K.K.; Merchant, S.S. Trophic status of Chlamydomonas reinhardtii influences the impact of iron deficiency on photosynthesis. Photosynth. Res. 2010, 105, 39–49. [Google Scholar] [CrossRef]
- Willamme, R.; Bogaert, K.A.; Remacle, F.; Remacle, C. Surprisal analysis of the transcriptomic response of the green microalga Chlamydomonas to the addition of acetate during day/night cycles. Chem. Phys. 2018, 514, 154–163. [Google Scholar] [CrossRef]
- Berger, H.; Blifernez-Klassen, O.; Ballottari, M.; Bassi, R.; Wobbe, L.; Kruse, O. Integration of carbon assimilation modes with photosynthetic light capture in the green alga Chlamydomonas reinhardtii. Mol. Plant 2014, 7, 1545–1559. [Google Scholar] [CrossRef]
- Berger, H.; De Mia, M.; Morisse, S.; Marchand, C.H.; Lemaire, S.D.; Wobbe, L.; Kruse, O. A light switch based on protein s-nitrosylation fine-tunes photosynthetic light harvesting in Chlamydomonas. Plant Physiol. 2016, 171, 821–832. [Google Scholar] [CrossRef]
- Tokutsu, R.; Fujimura-Kamada, K.; Matsuo, T.; Yamasaki, T.; Minagawa, J. The CONSTANS flowering complex controls the protective response of photosynthesis in the green alga Chlamydomonas. Nat. Commun. 2019, 10, 4099. [Google Scholar] [CrossRef]
- Gabilly, S.T.; Baker, C.R.; Wakao, S.; Crisanto, T.; Guan, K.; Bi, K.; Guiet, E.; Guadagno, C.R.; Niyogi, K.K. Regulation of photoprotection gene expression in Chlamydomonas by a putative E3 ubiquitin ligase complex and a homolog of CONSTANS. Proc. Natl. Acad. Sci. USA 2019, 116, 17556–17562. [Google Scholar] [CrossRef]
- Fukuzawa, H.; Miura, K.; Ishizaki, K.; Kucho, K.I.; Saito, T.; Kohinata, T.; Ohyama, K. Ccm1, a regulatory gene controlling the induction of a carbon-concentrating mechanism in Chlamydomonas reinhardtii by sensing CO2 availability. Proc. Natl. Acad. Sci. USA 2001, 98, 5347–5352. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Si, Y.; Douglass, S.; Casero, D.; Merchant, S.S.; Pellegrini, M.; Ladunga, I.; Liu, P.; Spalding, M.H. Transcriptome-wide changes in Chlamydomonas reinhardtii gene expression regulated by carbon dioxide and the CO2-concentrating mechanism regulator CIA5/CCM1. Plant Cell 2012, 24, 1876–1893. [Google Scholar] [CrossRef] [PubMed]
- Pollock, S.V.; Colombo, S.L.; Prout, D.L.; Godfrey, A.C.; Moroney, J.V. Rubisco activase is required for optimal photosynthesis in the green alga Chlamydomonas reinhardtii in a low-CO2 atmosphere. Plant Physiol. 2003, 133, 1854–1861. [Google Scholar] [CrossRef] [PubMed]
- Moroney, J.V.; Kitayama, M.; Togasaki, R.K.; Tolbert, N.E. Evidence for inorganic carbon transport by intact chloroplasts of Chlamydomonas reinhardtii. Plant Physiol. 1987, 83, 460–463. [Google Scholar] [CrossRef]
- Gasch, A.P.; Spellman, P.T.; Kao, C.M.; Carmel-Harel, O.; Eisen, M.B.; Storz, G.; Botstein, D.; Brown, P.O. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell 2000, 11, 4241–4257. [Google Scholar] [CrossRef]
- Hosono, K.; Sasaki, T.; Minoshima, S.; Shimizu, N. Identification and characterization of a novel gene family YPEL in a wide spectrum of eukaryotic species. Gene 2004, 340, 31–43. [Google Scholar] [CrossRef]
- Kelley, K.D.; Miller, K.R.; Todd, A.; Kelley, A.R.; Tuttle, R.; Berberich, S.J. YPEL3, a p53-regulated gene that induces cellular senescence. Cancer Res. 2010, 70, 3566–3575. [Google Scholar] [CrossRef]
- Piras, V.; Selvarajoo, K. The reduction of gene expression variability from single cells to populations follows simple statistical laws. Genomics 2015, 105, 137–144. [Google Scholar] [CrossRef]
- Gross, A.; Levine, R.D. Surprisal analysis of transcripts expression levels in the presence of noise: A reliable determination of the onset of a tumor phenotype. PLoS ONE 2013, 8, e61554. [Google Scholar] [CrossRef]



| Acetate Concentration | Doubling/Day |
|---|---|
| 17 mM | 3.0 ± 0.3 * |
| 31 mM | 2.2 ± 0.3 |
| 44 mM | 1.9 ± 0.1 |
| 57 mM | 2.0 ± 0.1 |
| KEGG Pathways | P01 | 104 × N0 2 |
|---|---|---|
| Oxidative phosphorylation | 0 | 3.55 |
| Carbon fixation in photosynthetic organisms | 0 | 3.33 |
| Phagosome | 0 | 3.16 |
| Citrate cycle (TCA cycle) | 0 | 3.05 |
| 2-Oxocarboxylic acid metabolism | 0 | 2.55 |
| Valine, leucine and isoleucine biosynthesis | 0 | 2.42 |
| alpha-Linolenic acid metabolism | 0 | 2.31 |
| Lysine biosynthesis | 0 | 2.04 |
| Pyruvate metabolism | 0 | 1.96 |
| Fatty acid biosynthesis | 0 | 1.84 |
| Ten Most Positive Pathways (Exponential Phase) | Ten Most Negative Pathways (Stationary Phase) | ||||||
|---|---|---|---|---|---|---|---|
| KEGG Pathways | 1 | 2 | 3 | KEGG Pathways | 1 | 2 | 3 |
| Porphyrin and chlorophyll metabolism | 23.50 | 9800 | 24 | Regulation of autophagy | 0 | 7.76 | 0 |
| DNA replication | 9.62 | 3930 | 24.5 | Valine, leucine and isoleucine degradation | 5.54 | 5.57 | 9.95 |
| Carotenoid biosynthesis | 4.82 | 1790 | 26.9 | Endocytosis | 1.88 | 1.37 | 13.7 |
| N-Glycan biosynthesis | 10.4 | 3700 | 28.2 | Plant hormone signal transduction | 1.92 | 1.36 | 14.1 |
| RNA polymerase | 5.85 | 1780 | 32.9 | Peroxisome | 8.78 | 3.63 | 24.2 |
| Ubiquinone and other terpenoid-quinone biosynthesis | 18.4 | 5050 | 36.4 | Propanoate metabolism | 14.5 | 4.61 | 31.4 |
| Lysine biosynthesis | 14.4 | 3690 | 38.9 | beta-Alanine metabolism | 20.9 | 5.70 | 36.7 |
| Histidine metabolism | 14.6 | 3330 | 43.9 | SNARE interactions in vesicular transport | 6.5 | 1.47 | 44.3 |
| Photosynthesis—antenna proteins | 94.2 | 222 | 4240 | Selenocompound metabolism | 20.4 | 3.74 | 54.5 |
| Photosynthesis | 48.4 | 9.37 | 51700 | Pentose and glucuronate interconversions | 7.65 | 1.24 | 61.6 |
| Ten Most Positive Pathways—High-Acetate-Grown Samples | Ten Most Negative Pathways—Low-Acetate-Grown Samples | ||||||
|---|---|---|---|---|---|---|---|
| KEGG pathways | 1 | 107 × 2 | 3 | KEGG pathways | 1 | 105 × 2 | 3 |
| Nucleotide excision repair | 8.52 | 23.30 | 36.6 | alpha-Linolenic acid metabolism | 0 | 3.72 | 0 |
| Sphingolipid metabolism | 6.58 | 17.80 | 36.9 | Photosynthesis—antenna proteins | 1.30 | 22.30 | 5.83 |
| Ubiquitin mediated proteolysis | 5.30 | 11.80 | 45 | Fatty acid biosynthesis | 2.54 | 8.82 | 28.80 |
| N-Glycan biosynthesis | 10.30 | 21.90 | 47.3 | Carotenoid biosynthesis | 2.07 | 4.22 | 49.10 |
| Base excision repair | 9.49 | 8.16 | 116.0 | Photosynthesis | 14.9 | 17.40 | 85.60 |
| beta-Alanine metabolism | 16.90 | 9.19 | 184.0 | Ribosome | 4.46 | 3.90 | 114.00 |
| Homologous recombination | 9.31 | 2.06 | 452.0 | Pyruvate metabolism | 4.53 | 3.15 | 144.00 |
| DNA replication | 29.90 | 0 | Inf | Lysine biosynthesis | 5.84 | 3.25 | 179.00 |
| Proteasome | 7.32 | 0 | Inf | Porphyrin and chlorophyll metabolism | 10.3 | 4.88 | 211.00 |
| SNARE interactions in vesicular transport | 3.90 | 0 | Inf | Carbon fixation in photosynthetic organisms | 11.7 | 5.41 | 216.00 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bogaert, K.A.; Perez, E.; Rumin, J.; Giltay, A.; Carone, M.; Coosemans, N.; Radoux, M.; Eppe, G.; Levine, R.D.; Remacle, F.; et al. Metabolic, Physiological, and Transcriptomics Analysis of Batch Cultures of the Green Microalga Chlamydomonas Grown on Different Acetate Concentrations. Cells 2019, 8, 1367. https://doi.org/10.3390/cells8111367
Bogaert KA, Perez E, Rumin J, Giltay A, Carone M, Coosemans N, Radoux M, Eppe G, Levine RD, Remacle F, et al. Metabolic, Physiological, and Transcriptomics Analysis of Batch Cultures of the Green Microalga Chlamydomonas Grown on Different Acetate Concentrations. Cells. 2019; 8(11):1367. https://doi.org/10.3390/cells8111367
Chicago/Turabian StyleBogaert, Kenny A., Emilie Perez, Judith Rumin, Axel Giltay, Michele Carone, Nadine Coosemans, Michele Radoux, Gauthier Eppe, Raphael D. Levine, Francoise Remacle, and et al. 2019. "Metabolic, Physiological, and Transcriptomics Analysis of Batch Cultures of the Green Microalga Chlamydomonas Grown on Different Acetate Concentrations" Cells 8, no. 11: 1367. https://doi.org/10.3390/cells8111367