Evaluation of NV556, a Novel Cyclophilin Inhibitor, as a Potential Antifibrotic Compound for Liver Fibrosis

 ,
,  and
and 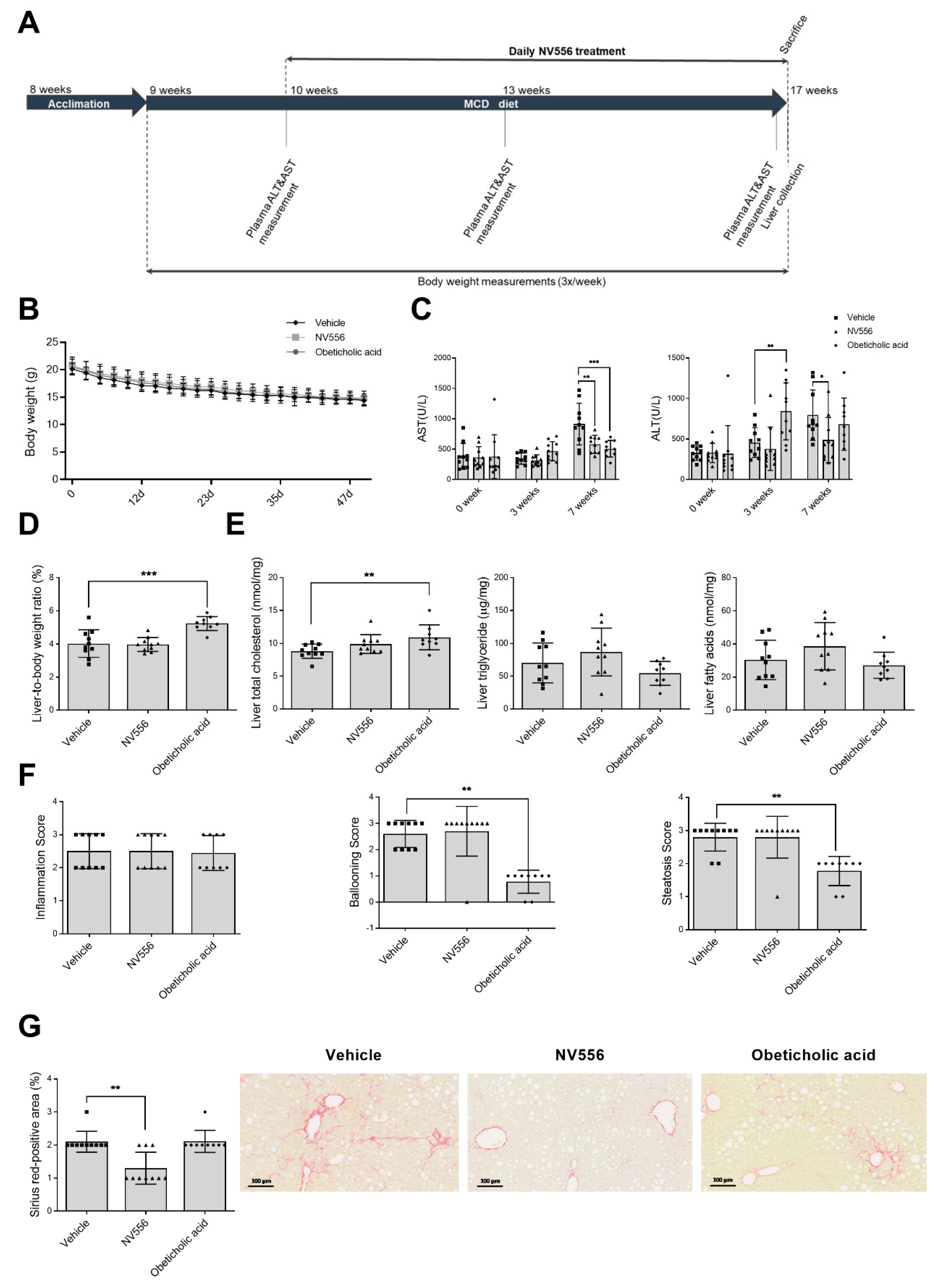{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Animal Models
2.2.1. Methionine-Choline-Deficient (MCD) Diet
2.2.2. STAM Model of Nonalcoholic Steatohepatitis
2.3. Liver Histology and Biochemistry Analysis
2.4. Cell Culture and Repopulation of 3D Human Liver Scaffolds
2.5. Gene Expression
2.5.1. LX-2 Cells
2.5.2. STAM Model
2.6. ELISA
2.7. Data and Statistical Analysis
3. Results
3.1. NV556 Decreases Fibrosis in MCD Model
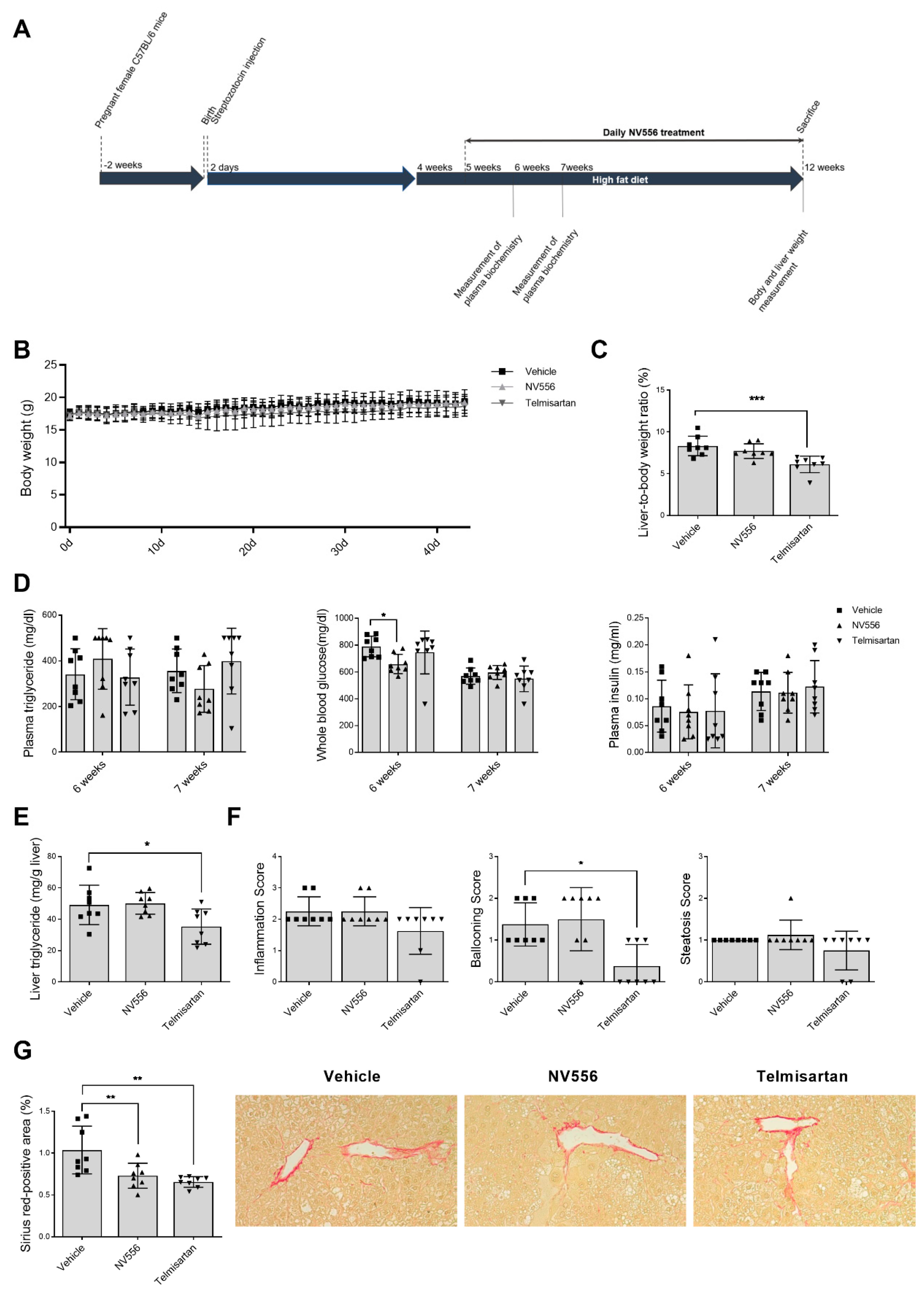
3.2. NV556 Decreases Fibrosis in STAM Model
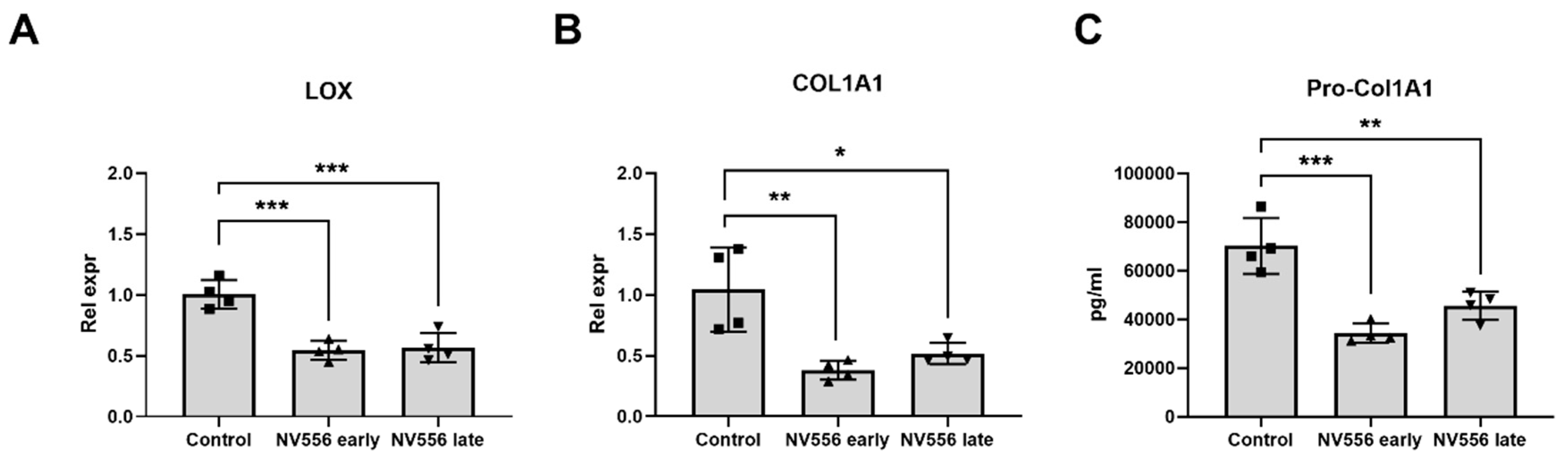
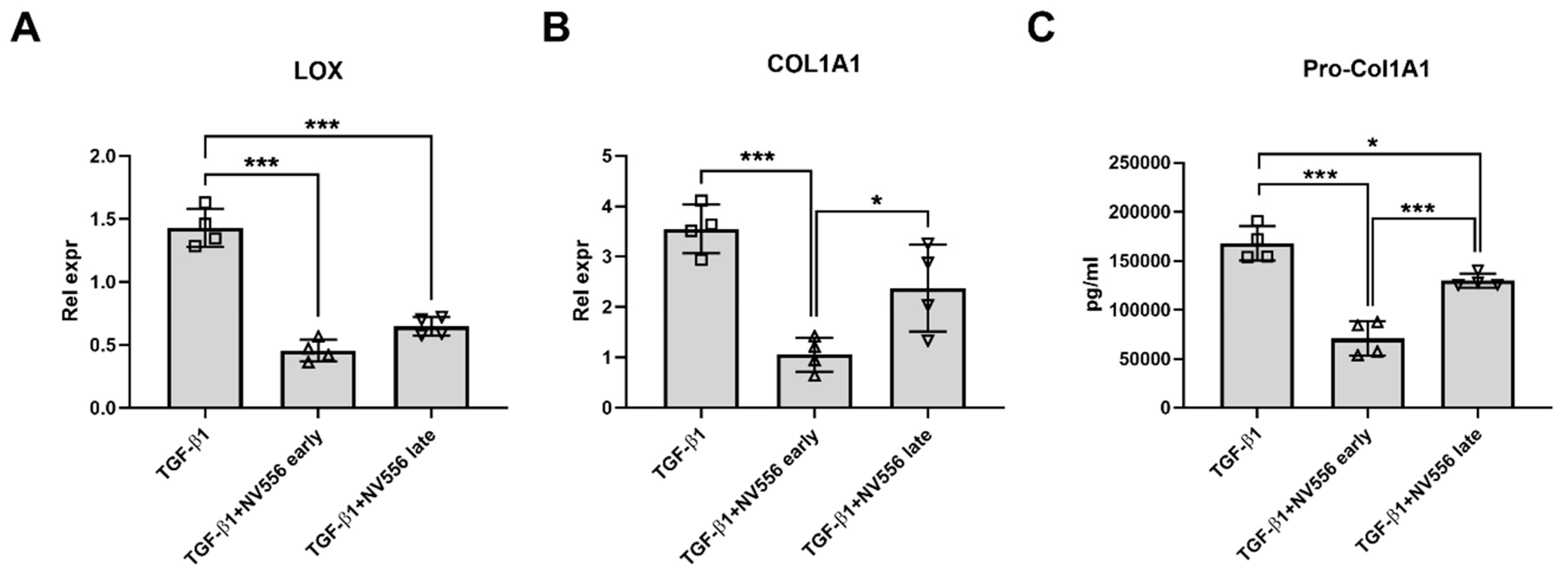
3.3. NV556 Treatment Reduces Collagen I Production in LX2 Human Hepatic Stellate Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bataller, R.; Rombouts, K.; Altamirano, J.; Marra, F. Fibrosis in alcoholic and nonalcoholic steatohepatitis. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Carloni, V.; Luong, T.V.; Rombouts, K. Hepatic stellate cells and extracellular matrix in hepatocellular carcinoma: More complicated than ever. Liver Int. 2014, 34, 834–843. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Liu, X.; Koyama, Y.; Wang, P.; Lan, T.; Kim, I.G.; Kim, I.H.; Ma, H.Y.; Kisseleva, T. The types of hepatic myofibroblasts contributing to liver fibrosis of different etiologies. Front. Pharmacol. 2014, 5, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Xu, J.; Brenner, D.A.; Kisseleva, T. Reversibility of Liver Fibrosis and Inactivation of Fibrogenic Myofibroblasts. Curr. Pathobiol. Rep. 2013, 1, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Arthur, M.J. Reversibility of liver fibrosis and cirrhosis following treatment for hepatitis C. Gastroenterology 2002, 122, 1525–1528. [Google Scholar] [CrossRef]
- Ratouis, A.; Degott, C.; Belghiti, J.; Bernades, P.; Levy, P.; Hammel, P.; Couvelard, A.; O’Toole, D.; Sauvanet, A.; Flejou, J.F.; et al. Regression of Liver Fibrosis after Biliary Drainage in Patients with Chronic Pancreatitis and Stenosis of the Common Bile Duct. N. Engl. J. Med. 2001, 344, 418–423. [Google Scholar]
- Seki, E.; Brenner, D.A. Recent advancement of molecular mechanisms of liver fibrosis. J. Hepato Biliary Pancreat. Sci. 2015, 22, 512–518. [Google Scholar] [CrossRef] [Green Version]
- Troeger, J.S.; Mederacke, I.; Gwak, G.; Dapito, D.H.; Mu, X.; Hsu, C.C.; Pradere, J.; Friedman, R.A.; Schwabe, R.F. Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology 2012, 143, 1073–1083. [Google Scholar] [CrossRef]
- Kisseleva, T.; Brenner, D.A. Hepatic stellate cells and the reversal of fibrosis. J. Gastroenterol. Hepatol. 2006, 21, 84–87. [Google Scholar] [CrossRef]
- Wake, K. “Sternzellen” in the liver: Perisinusoidal cells with special reference to storage of vitamin A. Am. J. Anat. 1971, 132, 429–461. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Mazza, G.; Al-Akkad, W.; Rombouts, K. Engineering in vitro models of hepatofibrogenesis. Adv. Drug Deliv. Rev. 2017, 121, 147–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazza, G.; Al-Akkad, W.; Telese, A.; Longato, L.; Urbani, L.; Robinson, B.; Hall, A.; Kong, K.; Frenguelli, L.; Marrone, G.; et al. Rapid production of human liver scaffolds for functional tissue engineering by high shear stress oscillation-decellularization. Sci. Rep. 2017, 7, 5534. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.L.; Walker, J.R.; Campagna-Slater, V.; Finerty, P.J.; Paramanathan, R.; Bernstein, G.; MacKenzie, F.; Tempel, W.; Ouyang, H.; Lee, W.H.; et al. Structural and biochemical characterization of the human cyclophilin family of peptidyl-prolyl isomerases. PLoS Biol. 2010, 8, e1000439. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Heitman, J. The cyclophilins. Genome Biol. 2005, 6, 226. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Liu, R.; Huang, C.; Jiang, L.; Zhou, Y.; Chen, Q. Cyclophilin A was revealed as a candidate marker for human oral submucous fibrosis by proteomic analysis. Cancer Biomark. 2017, 20, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Iordanskaia, T.; Malesevic, M.; Fischer, G.; Pushkarsky, T.; Bukrinsky, M.; Nadler, E.P. Targeting Extracellular Cyclophilins Ameliorates Disease Progression in Experimental Biliary Atresia. Mol. Med. 2015, 21, 657–664. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, Y.; Wang, T.; You, H.; Jia, J. N-methyl-4-isoleucine cyclosporine attenuates CCl -induced liver fibrosis in rats by interacting with cyclophilin B and D. J. Gastroenterol. Hepatol. 2011, 26, 558–567. [Google Scholar] [CrossRef]
- Heinzmann, D.; Bangert, A.; Müller, A.-M.; Von Ungern-Sternberg, S.N.I.; Emschermann, F.; Schönberger, T.; Chatterjee, M.; Mack, A.F.; Klingel, K.; Kandolf, R.; et al. The Novel Extracellular Cyclophilin A (CyPA) —Inhibitor MM284 Reduces Myocardial Inflammation and Remodeling in a Mouse Model of Troponin I -Induced Myocarditis. PLoS ONE 2015, 10, 0124606. [Google Scholar] [CrossRef]
- Kohjima, M.; Enjoji, M.; Higuchi, N.; Kotoh, K.; Kato, M.; Takayanagi, R.; Nakamuta, M. NIM811, a nonimmunosuppressive cyclosporine analogue, suppresses collagen production and enhances collagenase activity in hepatic stellate cells. Liver Int. 2007, 27, 1273–1281. [Google Scholar] [CrossRef] [Green Version]
- Kon, K.; Kim, J.-S.; Jaeschke, H.; Lemasters, J.J. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology 2004, 40, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Farmer, J.D.; Lane, W.S.; Friedman, J.; Weissman, I.; Schreiber, S.L. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991, 66, 807–815. [Google Scholar] [CrossRef]
- Wasilenko, S.; Bintner, J.; Mason, A.L.; Montano-Loza, A.J.; Mason, A.; Montano-Loza, A.J. Cyclosporine A Protects Against Primary Biliary Cirrhosis Recurrence After Liver Transplantation. Arab. Archaeol. Epigr. 2010, 10, 852–858. [Google Scholar]
- Rehman, H.; Ramshesh, V.K.; Theruvath, T.P.; Kim, I.; Currin, R.T.; Giri, S.; Lemasters, J.J.; Zhong, Z. NIM811 (N-methyl-4-isoleucine cyclosporine), a mitochondrial permeability transition inhibitor, attenuates cholestatic liver injury but not fibrosis in mice. J. Pharmacol. Exp. Ther. 2008, 327, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Kuo, J.; Bobardt, M.; Chatterji, U.; Mayo, P.R.; Trepanier, D.J.; Foster, R.T.; Gallay, P.; Ure, D.R. A Pan-Cyclophilin Inhibitor, CRV431, Decreases Fibrosis and Tumor Development in Chronic Liver Disease Models. J. Pharmacol. Exp. Ther. 2019, 371, 231–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansson, M.J.; Moss, S.J.; Bobardt, M.; Chatterji, U.; Coates, N.; Garcia-Rivera, J.A.; Elmér, E.; Kendrew, S.; Leyssen, P.; Neyts, J.; et al. Bioengineering and semisynthesis of an optimized cyclophilin inhibitor for treatment of chronic viral infection. Chem. Biol. 2015, 22, 285–292. [Google Scholar] [CrossRef]
- Fujii, M.; Shibazaki, Y.; Wakamatsu, K.; Honda, Y.; Kawauchi, Y.; Suzuki, K.; Arumugam, S.; Watanabe, K.; Ichida, T.; Asakura, H.; et al. A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med. Mol. Morphol. 2013, 46, 141–152. [Google Scholar] [CrossRef]
- Miao, B.; Zondlo, S.; Gibbs, S.; Cromley, D.; Hosagrahara, V.P.; Kirchgessner, T.G.; Billheimer, J.; Mukherjee, R. Raising HDL cholesterol without inducing hepatic steatosis and hypertriglyceridemia by a selective LXR modulator. J. Lipid Res. 2004, 45, 1410–1417. [Google Scholar] [CrossRef] [Green Version]
- Briand, F.; Brousseau, E.; Quinsat, M.; Burcelin, R.; Sulpice, T. Obeticholic acid raises LDL-cholesterol and reduces HDL-cholesterol in the Diet-Induced NASH (DIN) hamster model. Eur. J. Pharmacol. 2018, 818, 449–456. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.-C.; Torbenson, M.S.; Yeh, M.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Stanley, G.H.S. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)). Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2014, 385, 956–965. [Google Scholar] [CrossRef]
- Park, J.G.; Mok, J.S.; Han, Y.I.; Park, T.S.; Kang, K.W.; Choi, C.S.; Park, H.D.; Park, J. Connectivity mapping of angiotensin-PPAR interactions involved in the amelioration of non-alcoholic steatohepatitis by Telmisartan. Sci. Rep. 2019, 9, 4003. [Google Scholar] [CrossRef]
- Sherry, B.; Yarlett, N.; Strupp, A.; Cerami, A. Identification of cyclophilin as a proinflammatory secretory product of lipopolysaccharide-activated macrophages. Proc. Natl. Acad. Sci. USA 1992, 89, 3511–3515. [Google Scholar] [CrossRef]
- Hou, W.; Leong, K.G.; Ozols, E.; Tesch, G.H.; Nikolic-Paterson, D.J.; Ma, F.Y.; Nikolic-Paterson, D.J. Cyclophilin D promotes tubular cell damage and the development of interstitial fibrosis in the obstructed kidney. Clin. Exp. Pharmacol. Physiol. 2017, 45, 250–260. [Google Scholar] [CrossRef]
- Kuo, J.; Serrano, S.S.; Grönberg, A.; Massoumi, R.; Hansson, M.J.; Gallay, P. Cyclophilin Inhibitor NV556 Reduces Fibrosis and Hepatocellular Carcinoma Development in Mice With Non-Alcoholic Steatohepatitis. Front. Pharmacol. 2019, 10, 1129. [Google Scholar] [CrossRef]
- Pyott, S.M.; Schwarze, U.; Christiansen, H.E.; Pepin, M.G.; Leistritz, D.F.; Dineen, R.; Harris, C.; Burton, B.K.; Angle, B.; Kim, K.; et al. Mutations in PPIB (cyclophilin B) delay type I procollagen chain association and result in perinatal lethal to moderate osteogenesis imperfecta phenotypes. Hum. Mol. Genet. 2011, 20, 1595–1609. [Google Scholar] [CrossRef] [Green Version]
- Cabral, W.A.; Perdivara, I.; Weis, M.; Terajima, M.; Blissett, A.R.; Chang, W.; Perosky, J.E.; Makareeva, E.N.; Mertz, E.L.; Leikin, S.; et al. Abnormal Type I Collagen Post-translational Modification and Crosslinking in a Cyclophilin B KO Mouse Model of Recessive Osteogenesis Imperfecta. PLoS Genet. 2014, 10, e1004465. [Google Scholar] [CrossRef]
- Steinmann, B.; Bruckner, P.; Superti-Furga, A. Cyclosporin A slows collagen triple-helix formation in vivo: Indirect evidence for a physiologic role of peptidyl-prolyl cis-trans-isomerase. J. Biol. Chem. 1991, 266, 1299–1303. [Google Scholar]
- Liu, S.B.; Ikenaga, N.; Peng, Z.W.; Sverdlov, D.Y.; Greenstein, A.; Smith, V.; Schuppan, D. Lysyl oxidase activity contributes to collagen stabilization during liver fibrosis progression and limits spontaneous fibrosis reversal in mice. FASEB J. 2015, 30, 1599–1609. [Google Scholar] [CrossRef] [PubMed]
- Pinnell, S.R.; Martin, G.R. The cross-linking of collagen and elastin: Enzymatic conversion of lysine in peptide linkage to alpha-aminoadipic-delta-semialdehyde (allysine) by an extract from bone. Proc. Natl. Acad. Sci. USA 1968, 61, 708–716. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simón Serrano, S.; Grönberg, A.; Longato, L.; Rombouts, K.; Kuo, J.; Gregory, M.; Moss, S.; Elmér, E.; Mazza, G.; Gallay, P.; et al. Evaluation of NV556, a Novel Cyclophilin Inhibitor, as a Potential Antifibrotic Compound for Liver Fibrosis. Cells 2019, 8, 1409. https://doi.org/10.3390/cells8111409
Simón Serrano S, Grönberg A, Longato L, Rombouts K, Kuo J, Gregory M, Moss S, Elmér E, Mazza G, Gallay P, et al. Evaluation of NV556, a Novel Cyclophilin Inhibitor, as a Potential Antifibrotic Compound for Liver Fibrosis. Cells. 2019; 8(11):1409. https://doi.org/10.3390/cells8111409
Chicago/Turabian StyleSimón Serrano, Sonia, Alvar Grönberg, Lisa Longato, Krista Rombouts, Joseph Kuo, Matthew Gregory, Steven Moss, Eskil Elmér, Giuseppe Mazza, Philippe Gallay, and et al. 2019. "Evaluation of NV556, a Novel Cyclophilin Inhibitor, as a Potential Antifibrotic Compound for Liver Fibrosis" Cells 8, no. 11: 1409. https://doi.org/10.3390/cells8111409
APA StyleSimón Serrano, S., Grönberg, A., Longato, L., Rombouts, K., Kuo, J., Gregory, M., Moss, S., Elmér, E., Mazza, G., Gallay, P., Pinzani, M., Hansson, M. J., & Massoumi, R. (2019). Evaluation of NV556, a Novel Cyclophilin Inhibitor, as a Potential Antifibrotic Compound for Liver Fibrosis. Cells, 8(11), 1409. https://doi.org/10.3390/cells8111409