Tauroursodeoxycholate—Bile Acid with Chaperoning Activity: Molecular and Cellular Effects and Therapeutic Perspectives

Abstract
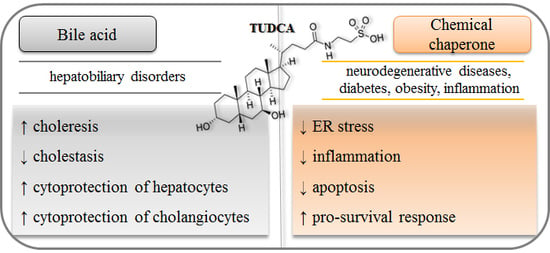
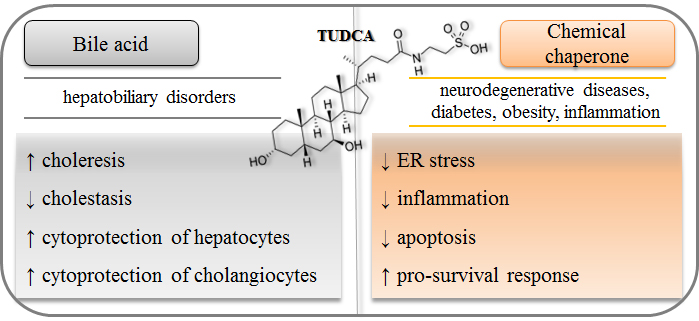
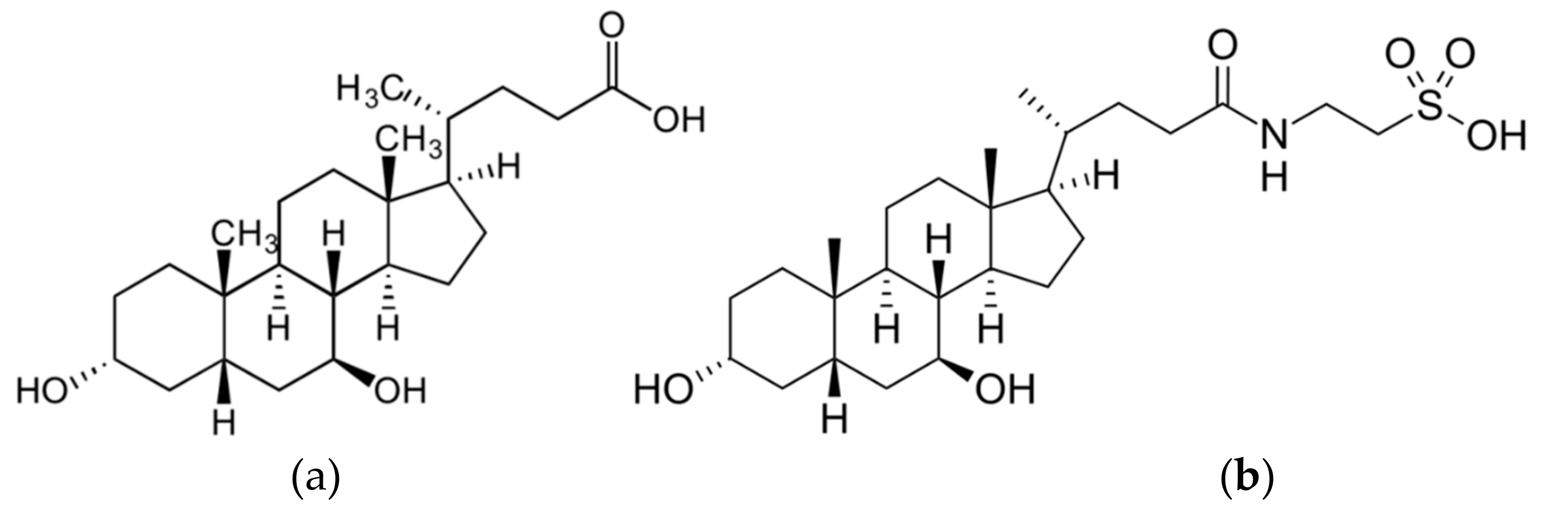
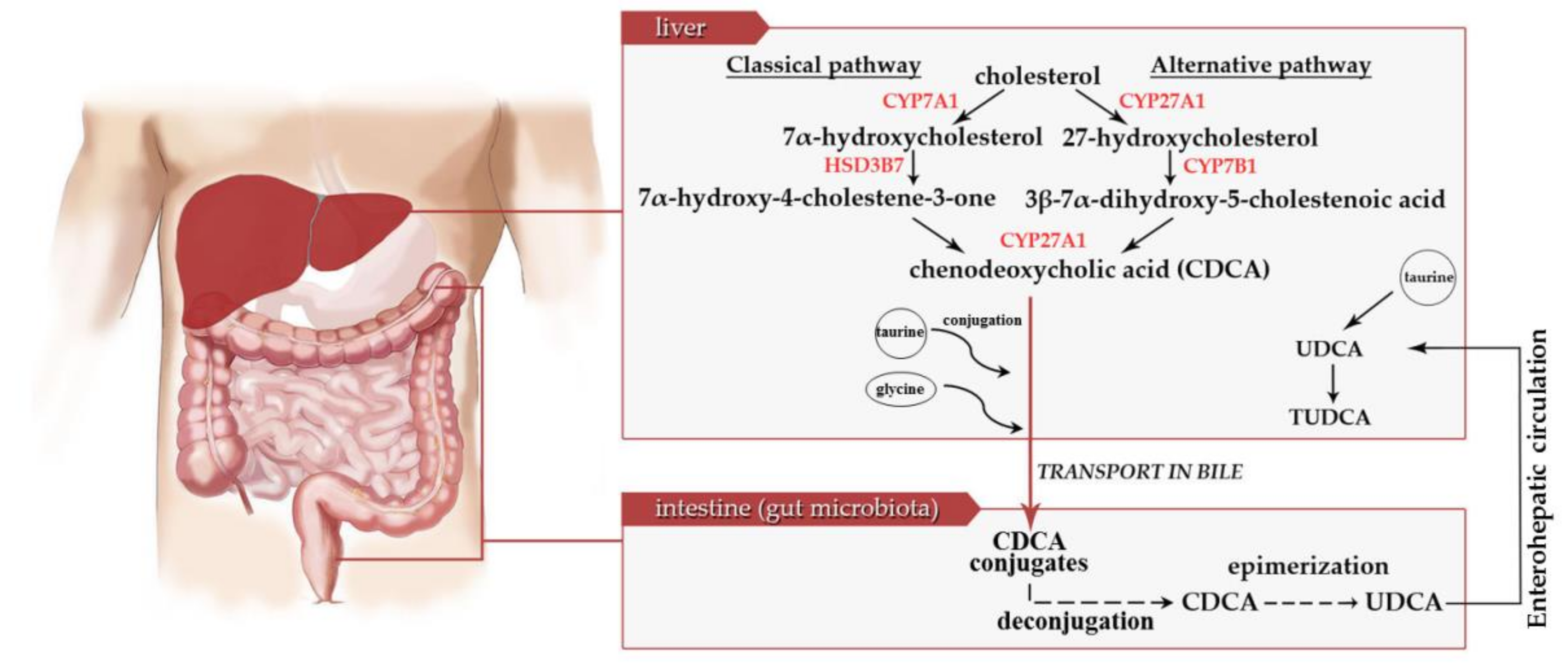
:
{kind=link}
{kind=link}
{kind=link}
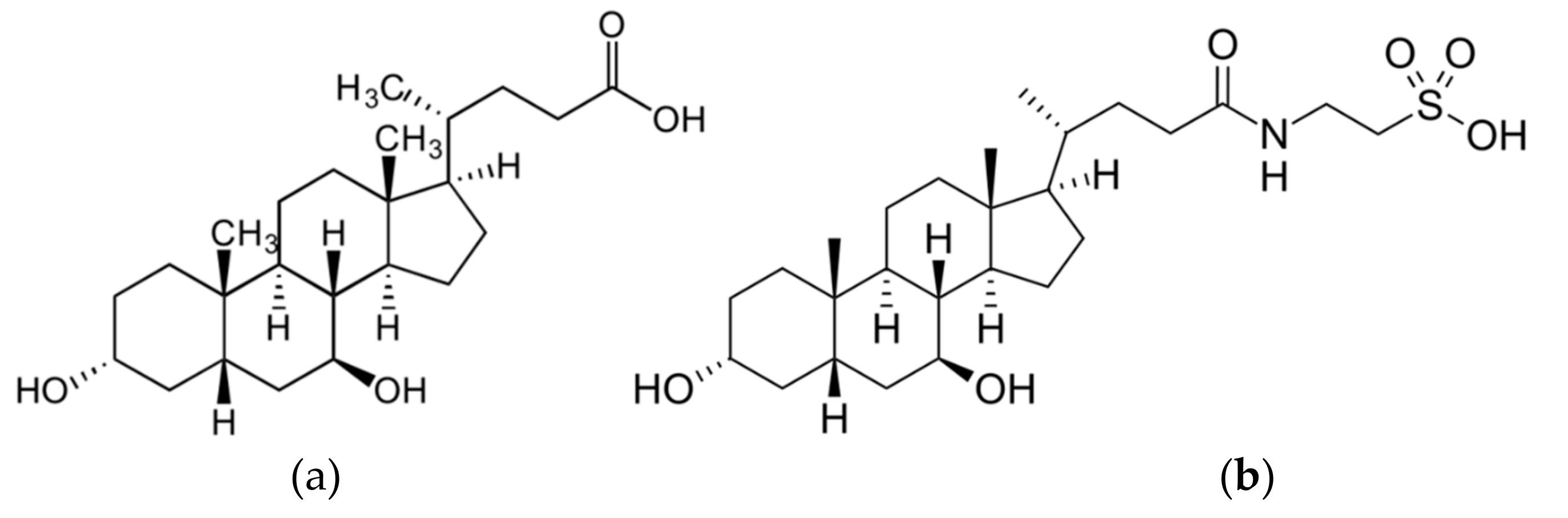
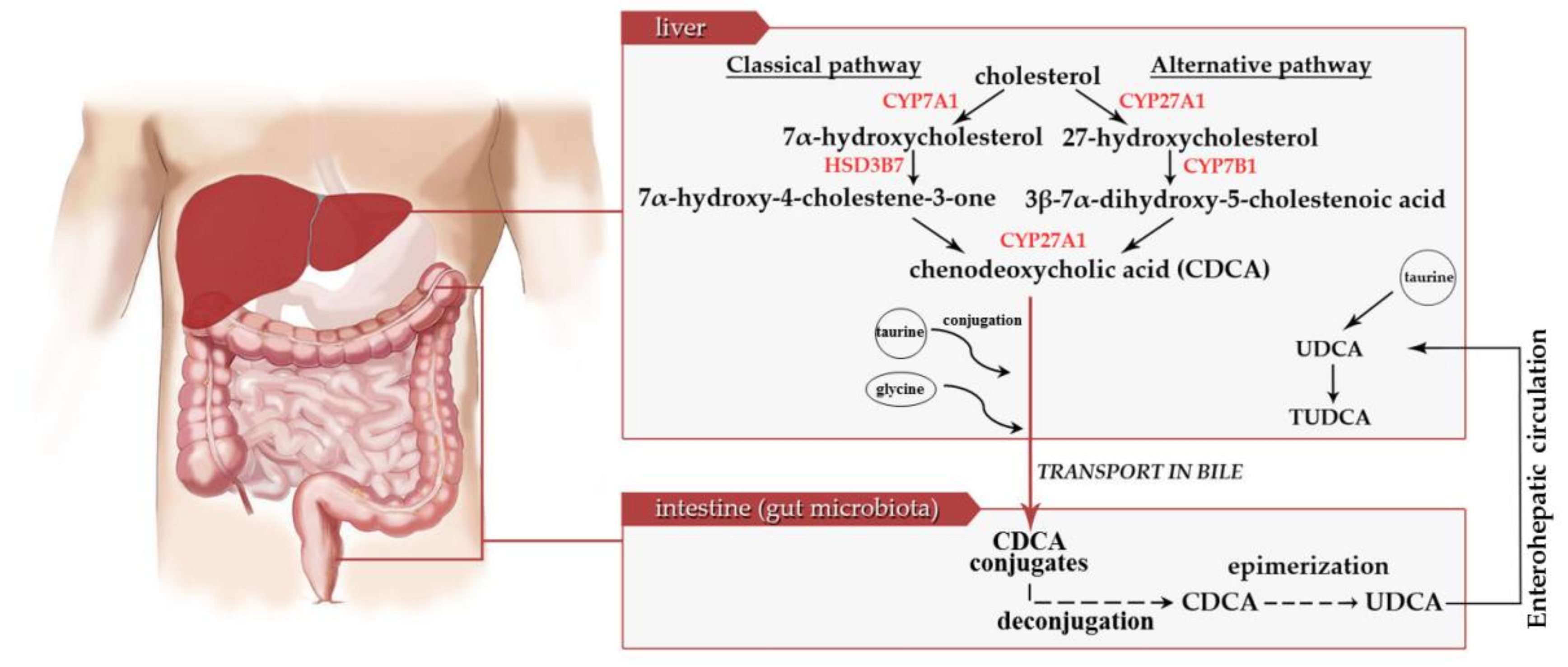
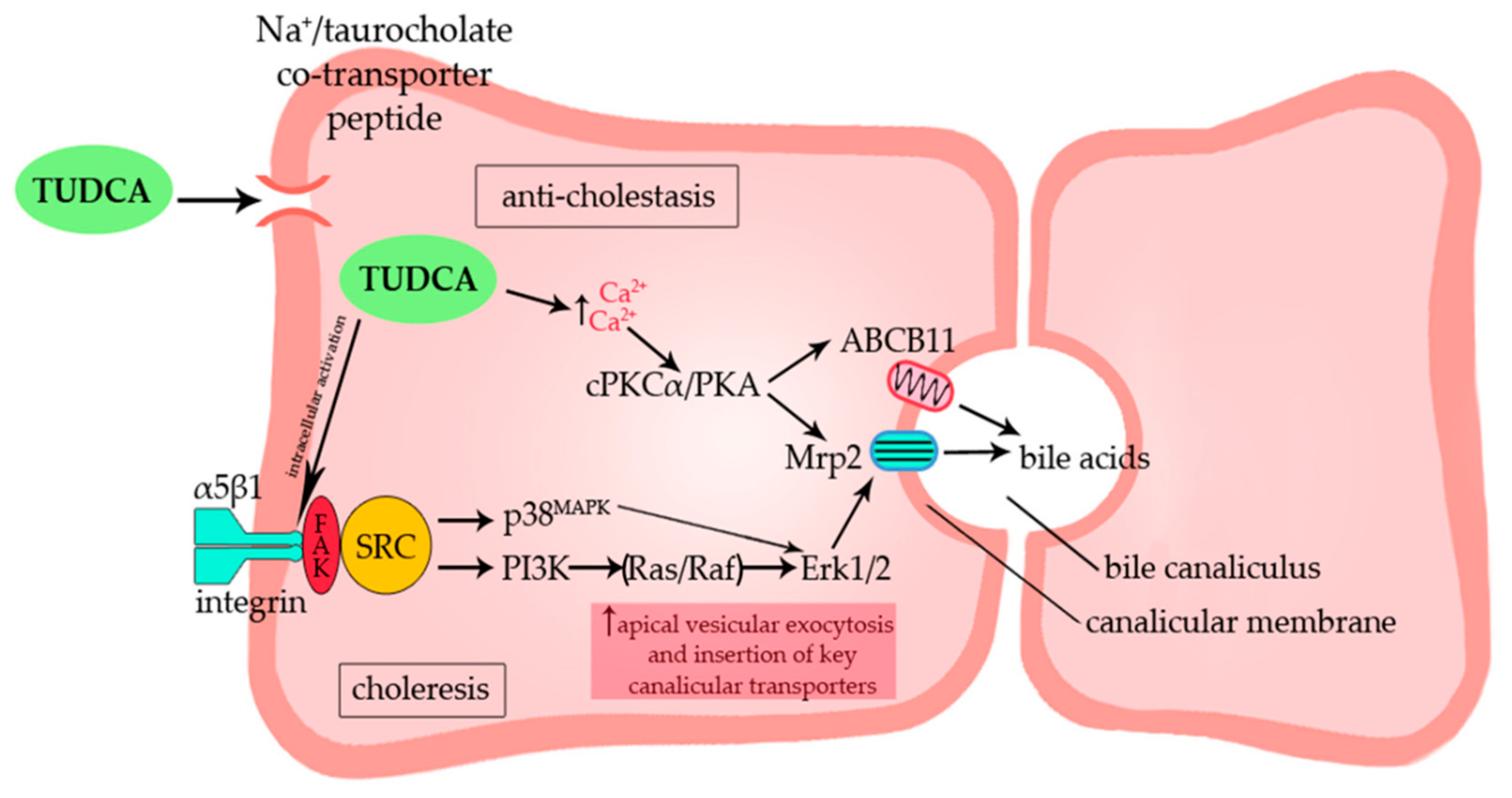{kind=link}
1. Introduction
2. TUDCA as Bile Acid in Hepatobiliary Disorders
3. TUDCA as Chemical Chaperone in ER Stress-Related Diseases
3.1. TUDCA in Diabetes and Obesity
3.2. TUDCA in Neurodegenerative Diseases
3.3. TUDCA in Inflammation
3.4. TUDCA in Cancer
4. Therapeutic Perspectives and Limitations
5. Conclusions
Funding
Conflicts of Interest
References
- Wang, D.Q.; Carey, M.C. Therapeutic uses of animal biles in traditional Chinese medicine: An ethnopharmacological, biophysical chemical and medicinal review. World J. Gastroenterol. 2014, 20, 9952–9975. [Google Scholar] [CrossRef] [PubMed]
- Lepercq, P.; Gérard, P.; Béguet, F.; Raibaud, P.; Grill, J.P.; Relano, P.; Cayuela, C.; Juste, C. Epimerization of chenodeoxycholic acid to ursodeoxycholic acid by Clostridium baratii isolated from human feces. FEMS Microbiol. Lett. 2004, 235, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, H.D.; Gerhard, G.S. Bile acids in neurodegenerative disorders. Front. Aging Neurosci. 2016, 8, 263. [Google Scholar] [CrossRef] [PubMed]
- Vang, S.; Longley, K.; Steer, C.J.; Low, W.C. The Unexpected uses of urso- and tauroursodeoxycholic acid in the treatment of non-liver diseases. Global Adv. Health Med. 2014, 3, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chiang, J.Y. Bile acid signaling in metabolic disease and drug therapy. Pharmacol. Rev. 2014, 66, 948–983. [Google Scholar] [CrossRef]
- McMillin, M.; DeMorrow, S. Effects of bile acids on neurological function and disease. FASEB J. 2016, 30, 3658–3668. [Google Scholar] [CrossRef]
- Kiriyama, Y.; Nochi, H. The biosynthesis, signaling, and neurological functions of bile acids. Biomolecules 2019, 9, 232. [Google Scholar] [CrossRef]
- Beuers, U. β1 integrin is a long-sought sensor for tauroursodeoxycholic acid. Hepatology 2013, 57, 867–869. [Google Scholar] [CrossRef]
- Gohlke, H.; Schmitz, B.; Sommerfeld, A.; Reinehr, R.; Häussinger, D. α5β1-integrins are sensors for tauroursodeoxycholic acid in hepatocytes. Hepatology 2013, 57, 1117–1129. [Google Scholar] [CrossRef]
- Studer, E.; Zhou, X.; Zhao, R.; Wang, Y.; Takabe, K.; Nagahashi, M.; Pandak, W.M.; Dent, P.; Spiegel, S.; Shi, R.; et al. Conjugated bile acids activate the sphingosine-1-phosphate receptor 2 in primary rodent hepatocytes. Hepatology 2012, 55, 267–276. [Google Scholar] [CrossRef]
- Carrera-Bastos, P.; Fontes O’Keefe, J.; Lindeberg, S.; Cordain, L. The western diet and lifestyle and diseases of civilization. Res. Rep. Clin. Cardiol. 2011, 2, 15–35. [Google Scholar] [CrossRef]
- Kusaczuk, M.; Bartoszewicz, M.; Cechowska-Pasko, M. Phenylbutyric acid: Simple structure- multiple effects. Curr. Pharm. Des. 2015, 21, 2147–2166. [Google Scholar] [CrossRef] [PubMed]
- Ringe, D.; Petsko, G.A. Q&A: What are pharmacological chaperones and why are they interesting? J. Biol. 2009, 8, 80. [Google Scholar] [PubMed]
- Cortez, L.; Sim, V. The therapeutic potential of chemical chaperones in protein folding diseases. Prion 2014, 8, 197–202. [Google Scholar] [CrossRef]
- Hartl, F.U.; Hayer-Hartl, M. Converging concepts of protein folding in vitro and in vivo. Nat. Struct. Mol. Biol. 2009, 16, 574–581. [Google Scholar] [CrossRef]
- Leandro, P.; Gomes, C.M. Protein misfolding in conformational disorders: Rescue of folding defects and chemical chaperoning. Mini Rev. Med. Chem. 2008, 8, 901–911. [Google Scholar] [CrossRef]
- Arakawa, T.; Ejima, D.; Kita, Y.; Tsumoto, K. Small molecule pharmacological chaperones: From thermodynamic stabilization to pharmaceutical drugs. Biochim. Biophys. Acta 2006, 1764, 1677–1687. [Google Scholar] [CrossRef]
- Lenin, R.; Maria, M.S.; Agrawal, M.; Balasubramanyam, J.; Mohan, V.; Balasubramanyam, M. Amelioration of glucolipotoxicity-induced endoplasmic reticulum stress by a “chemical chaperone” in human THP-1 monocytes. Exp. Diabetes Res. 2012, 28, 1–10. [Google Scholar] [CrossRef]
- Lee, Y.Y.; Hong, S.H.; Lee, Y.J.; Chung, S.S.; Jung, H.S.; Park, S.G.; Park, K.S. Tauroursodeoxycholate (TUDCA), chemical chaperone, enhances function of islets by reducing ER stress. Biochem. Biophys. Res. Commun. 2010, 397, 735–739. [Google Scholar] [CrossRef]
- Da-Silva, W.S.; Ribich, S.; Arrojo e Drigo, R.; Castillo, M.; Patti, M.E.; Bianco, A.C. The chemical chaperones tauroursodeoxycholic and 4-phenylbutyric acid accelerate thyroid hormone activation and energy expenditure. FEBS Lett. 2011, 585, 539–544. [Google Scholar] [CrossRef]
- Paridaens, A.; Raevens, S.; Devisscher, L.; Bogaerts, E.; Verhelst, X.; Hoorens, A.; Van Vlierberghe, H.; van Grunsven, L.A.; Geerts, A.; Colle, I. Modulation of the unfolded protein response by tauroursodeoxycholic acid counteracts apoptotic cell death and fibrosis in a mouse model for secondary biliary liver fibrosis. Int. J. Mol. Sci. 2017, 18, 214. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, X.; Zheng, G.; Shan, Q.; Lu, J.; Fan, S.; Sun, C.; Wu, D.; Zhang, C.; Su, W.; et al. Troxerutin attenuates enhancement of hepatic gluconeogenesis by inhibiting NOD activation-mediated inflammation in high-fat diet-treated mice. Int. J. Mol. Sci. 2016, 18, 31. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Ghaffar, A.; Elhossary, G.G.; Mahmoud, A.M.; Elshazly, A.H.M.; Hassanin, O.A.; Saleh, A. Potential prophylactic effect of chemical chaperones for alleviation of endoplasmic reticulum stress in experimental diabetic cataract. Bull. Natl. Res. Cent. 2019, 43, 71. [Google Scholar] [CrossRef]
- Rodrigues, C.M.; Stieers, C.L.; Keene, C.D.; Ma, X.; Kren, B.T.; Low, W.C.; Steer, C.J. Tauroursodeoxycholic acid partially prevents apoptosis induced by 3-nitropropionic acid: Evidence for a mitochondrial pathway independent of the permeability transition. J. Neurochem. 2000, 75, 2368–2379. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.M.P.; Fan, G.; Wong, P.Y.; Kren, B.T.; Steer, C.J. Ursodeoxycholic acid may inhibit deoxycholic acid-induced apoptosis by modulating mitochondrial transmembrane potential and reactive oxygen species production. Mol. Med. 1998, 4, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Azzaroli, F.; Soroka, C.J.; Wang, L.; Lee, J.; Crispe, I.N.; Boyer, J.L. Ursodeoxycholic acid diminishes Fas-ligand-induced apoptosis in mouse hepatocytes. Hepatology 2002, 36, 49–54. [Google Scholar] [CrossRef]
- Castro-Caldas, M.; Carvalho, A.N.; Carvalho, E.; Rodrigues, C.J.; Henderson, C.R.; Wolf, C.M.P.; Rodrigues, M.J.; Gama, M.J. Tauroursodeoxycholic acid prevents MPTP-induced dopaminergic cell death in a mouse model of Parkinson’s disease. Mol. Neurobiol. 2012, 46, 475–486. [Google Scholar]
- Rodrigues, C.M.P.; Fan, G.; Ma, X.; Kren, B.T.; Steer, C.J. A novel role for ursodeoxycholic acid in inhibiting apoptosis by modulating mitochondrial membrane perturbation. J. Clin. Investig. 1998, 101, 2790–2799. [Google Scholar] [CrossRef]
- Schoemaker, M.H.; Conde de la Rosa, L.; Buist-Homan, M.; Vrenken, T.E.; Havinga, R.; Poelstra, K.; Haisma, H.J.; Jansen, P.L.; Moshage, H. Tauroursodeoxycholic acid protects rat hepatocytes from bile acid-induced apoptosis via activation of survival pathways. Hepatology 2004, 39, 1563–1573. [Google Scholar] [CrossRef]
- Rodrigues, C.M.P.; Ma, X.; Linehan-Stieers, C.; Fan, G.; Kren, B.T.; Steer, C.J. Ursodeoxycholic acid prevents cytochrome c release in apoptosis by inhibiting mitochondrial membrane depolarization and channel formation. Cell Death Differ. 1999, 6, 842–854. [Google Scholar] [CrossRef]
- Castro, R.E.; Solá, S.; Ma, X.; Ramalho, R.M.; Kren, B.T.; Steer, C.J.; Rodrigues, C.M. A distinct microarray gene expression profile in primary rat hepatocytes incubated with ursodeoxycholic acid. J. Hepatol. 2005, 42, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Castro, R.E.; Amaral, J.D.; Solá, S.; Kren, B.T.; Steer, C.J.; Rodrigues, C.M. Differential regulation of cyclin D1 and cell death by bile acids in primary rat hepatocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G327–G334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solá, S.; Ma, X.; Castro, R.E.; Kren, B.T.; Steer, C.J.; Rodrigues, C.M. Ursodeoxycholic acid modulates E2F-1 and p53 expression through a caspase-independent mechanism in transforming growth factor b1-induced apoptosis of rat hepatocytes. J. Biol. Chem. 2003, 278, 48831–48838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral, J.D.; Castro, R.E.; Solá, S.; Steer, C.J.; Rodrigues, C.M. p53 is a key molecular target of ursodeoxycholic acid in regulating apoptosis. J. Biol. Chem. 2007, 282, 34250–34259. [Google Scholar] [CrossRef] [Green Version]
- Ramalho, R.M.; Ribeiro, P.S.; Solá, S.; Castro, R.E.; Steer, C.J.; Rodrigues, C.M. Inhibition of the E2F-1/p53/Bax pathway by tauroursodeoxycholic acid in amyloid beta-peptide-induced apoptosis of PC12 cells. J. Neurochem. 2004, 90, 567–575. [Google Scholar] [CrossRef]
- Solá, S.; Amaral, J.D.; Castro, R.E.; Ramalho, R.M.; Borralho, P.M.; Kren, B.T.; Tanaka, H.; Steer, C.J.; Rodrigues, C.M. Nuclear translocation of UDCA by the glucocorticoid receptor is required to reduce TGF-β1-induced apoptosis in rat hepatocytes. Hepatology 2005, 42, 925–934. [Google Scholar] [CrossRef]
- Solá, S.; Amaral, J.D.; Borralho, P.M.; Ramalho, R.M.; Castro, R.E.; Aranha, M.M.; Steer, C.J.; Rodrigues, C.M. Functional modulation of nuclear steroid receptors by tauroursodeoxycholic acid reduces amyloid beta-peptide-induced apoptosis. Mol. Endocrinol. 2006, 20, 2292–2303. [Google Scholar] [CrossRef] [Green Version]
- Castro, R.E.; Solá, S.; Ramalho, R.M.; Steer, C.J.; Rodrigues, C.M. The bile acid tauroursodeoxycholic acid modulates phosphorylation and translocation of Bad via phosphatidylinositol 3-kinase in glutamate-induced apoptosis of rat cortical neurons. J. Pharmacol. Exp. Ther. 2004, 311, 845–852. [Google Scholar] [CrossRef]
- Cash, J.G.; Kuhel, D.G.; Basford, J.E.; Jaeschke, A.; Chatterjee, T.K.; Weintraub, N.L.; Hui, D.Y. Apolipoprotein E4 impairs macrophage efferocytosis and potentiates apoptosis by accelerating endoplasmic reticulum stress. J. Biol. Chem. 2012, 287, 27876–27884. [Google Scholar] [CrossRef] [Green Version]
- Bradham, C.A.; Plümpe, J.; Manns, M.P.; Brenner, D.A.; Trautwein, C. Mechanisms of hepatic toxicity. I. TNF-induced liver injury. Am. J. Physiol. 1998, 275, G387–G392. [Google Scholar]
- Solá, S.; Castro, R.E.; Laires, P.A.; Steer, C.J.; Rodrigues, C.M. Tauroursodeoxycholic acid prevents amyloid β-peptide-induced neuronal death through a phosphatidylinositol 3-kinase-dependent signaling pathway. Mol Med. 2003, 9, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Gu, G.; Wang, J.; Chai, Y.; Fan, Y.; Yang, M.; Xu, X.; Gao, W.; Li, F.; Yin, D.; et al. Administration of tauroursodeoxycholic acid attenuates early brain injury via Akt pathway activation. Front. Cell Neurosci. 2017, 11, 193. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Tan, H.Y.; Wang, N.; Hong, M.; Li, L.; Cheung, F.; Feng, Y. Substitutes for bear bile for the treatment of liver diseases: Research progress and future perspective. Evid. Based Complementary Altern. Med. 2016, 430507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Festi, D.; Montagnani, M.; Azzaroli, F.; Lodato, F.; Mazzella, G.; Roda, A.; Di Biase, A.R.; Roda, E.; Simoni, P.; Colecchia, A. Clinical efficacy and effectiveness of ursodeoxycholic acid in cholestatic liver diseases. Curr. Clin. Pharmacol. 2007, 2, 155–177. [Google Scholar] [CrossRef]
- Poupon, R. Ursodeoxycholic acid and bile-acid mimetics as therapeutic agents for cholestatic liver diseases: An overview of their mechanisms of action. Clin. Res. Hepatol. Gas. 2012, 36, S3–S12. [Google Scholar] [CrossRef]
- Feng, Y.; Siu, K.; Wang, N.; Ng, K.M.; Tsao, S.W.; Nagamatsu, T.; Tong, Y. Bear bile: Dilemma of traditional medicinal use and animal protection. J. Ethnobiol. Ethnomed. 2009, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Rust, C.; Beuers, U. Medical treatment of primary biliary cirrhosis and primary sclerosing cholangitis. Clin. Rev. Allergy Immunol. 2005, 28, 135–145. [Google Scholar] [CrossRef]
- Caestecker, J.S.; Jazrawi, R.P.; Petroni, M.L.; Northfield, T.C. Ursodeoxycholic acid in chronic liver disease. Gut 1991, 32, 1061–1065. [Google Scholar] [CrossRef]
- Cullen, S.N.; Rust, C.; Fleming, K.; Edwards, C.; Beuers, U.; Chapman, R.W. High dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis is safe and effective. Hepatology 2008, 48, 792–800. [Google Scholar] [CrossRef]
- Pusl, T.; Beuers, U. Ursodeoxycholic acid treatment of vanishing bile duct syndromes. World J. Gastroenterol. 2006, 12, 3487–3495. [Google Scholar]
- Paumgartner, G.; Beuers, U. Ursodeoxycholic acid in cholestatic liver disease: Mechanisms of action and therapeutic use revisited. Hepatology 2002, 36, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Baiocchi, L.; Zhou, T.; Liangpunsakul, S.; Lenci, I.; Santopaolo, F.; Meng, F. Dual role of bile acids on the biliary epithelium: Friend or foe? Int. J. Mol. Sci. 2019, 20, 1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratziu, V.; Ledinghen, V. A randomized controlled trial of high-dose ursodesoxycholic acid for nonalcoholic steatohepatitis. Hepatology 2011, 54, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.D.; Li, L.; Wang, J.Y. Ursodeoxycholic acid for nonalcoholic steatohepatitis. Eur. J. Gastroenterol. Hepatol. 2012, 24, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Were, A.; Dechêne, A.; Herzer, K.; Hilgard, P.; Syn, W.K.; Gerken, G.; Canbay, A. Steroid and ursodesoxycholic acid combination therapy in severe drug-induced liver injury. Digestion 2011, 84, 54–59. [Google Scholar]
- Suraweera, D.; Rahal, H.; Jimenez, M.; Viramontes, M.; Choi, G.; Saab, S. Treatment of primary biliary cholangitis ursodeoxycholic acid non-responders: A systematic review. Liver Int. 2017, 37, 1877–1886. [Google Scholar] [CrossRef] [PubMed]
- Setchell, K.D.; Rodrigues, C.M.; Podda, M.; Crosignani, A. Metabolism of orally administered tauroursodeoxycholic acid in patients with primary biliary cirrhosis. Gut 1996, 38, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Pavlović, N.; Goločorbin-Kon, S.; Ðanić, M.; Stanimirov, B.; Al-Salami, H.; Stankov, K.; Mikov, M. Bile acids and their derivatives as potential modifiers of drug release and pharmacokinetic profiles. Front. Pharmacol. 2018, 9, 1283. [Google Scholar] [CrossRef]
- Zhou, Y.; Doyen, R.; Lichtenberger, L.M. The role of membrane cholesterol in determining bile acid cytotoxicity and cytoprotection of ursodeoxycholic acid. Biochim. Biophys. Acta 2009, 1788, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.L.; Zhao, L.; Li, L.; Li, A.H.; Ye, J.; Yang, L.; Xu, K.S.; Hou, X.H. Efficacy and safety of tauroursodeoxycholic acid in the treatment of liver cirrhosis: A double-blind randomized controlled trial. J. Huazhong Univ. Sci. Technolog. Med. Sci. 2013, 33, 189–194. [Google Scholar] [CrossRef]
- Ma, H.; Zeng, M.; Han, Y.; Yan, H.; Tang, H.; Sheng, J.; Hu, H.; Cheng, L.; Xie, Q.; Zhu, Y.; et al. A multicenter, randomized, double-blind trial comparing the efficacy and safety of TUDCA and UDCA in Chinese patients with primary biliary cholangitis. Medicine 2016, 95, 5391. [Google Scholar] [CrossRef] [PubMed]
- Colell, A.; Coll, O.; García-Ruiz, C.; París, R.; Tiribelli, C.; Kaplowitz, N.; Fernández-Checa, J.C. Tauroursodeoxycholic acid protects hepatocytes from ethanol-fed rats against tumor necrosis factor-induced cell death by replenishing mitochondrial glutathione. Hepatology 2001, 34, 964–971. [Google Scholar] [CrossRef] [PubMed]
- Santiago, P.; Scheinberg, A.R.; Levy, C. Cholestatic liver diseases: New targets, new therapies. Therap. Adv. Gastroenterol. 2018, 11, 17562848–18787400. [Google Scholar] [CrossRef] [PubMed]
- Beuers, U. Drug insight: Mechanisms and sites of action of ursodeoxycholic acid in cholestasis. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 318–328. [Google Scholar] [CrossRef]
- Marschall, H.U.; Wagner, M.; Zollner, G.; Fickert, P.; Diczfalusy, U.; Gumhold, J.; Silbert, D.; Fuchsbichler, A.; Benthin, L.; Grundström, R.; et al. Complementary stimulation of hepatobiliary transport and detoxification systems by rifampicin and ursodeoxycholic acid in humans. Gastroenterology 2005, 129, 476–485. [Google Scholar] [CrossRef]
- Beuers, U.; Nathanson, M.H.; Isales, C.M.; Boyer, J.L. Tauroursodeoxycholic acid stimulates hepatocellular exocytosis and mobilizes extracellular Ca++ mechanisms defective in cholestasis. J. Clin. Investig. 1993, 92, 2984–3293. [Google Scholar] [CrossRef]
- Bouscarel, B.; Fromm, H.; Nussbaum, R. Ursodeoxycholate mobilizes intracellular Ca++ and activates phosphorylase a in isolated hepatocytes. Am. J. Physiol. 1993, 264, G243–G251. [Google Scholar]
- Beuers, U.; Thiel, M.; Bardenheuer, H.; Paumgartner, G. Tauroursodeoxycholic acid inhibits the cytosolic Ca++ increase in human neutrophils stimulated by formyl-methionyl-leucyl-phenylalanine. Biochem. Biophys. Res. Commun. 1990, 171, 1115–1121. [Google Scholar] [CrossRef]
- Beuers, U.; Nathanson, M.H.; Boyer, J.L. Effects of tauroursodeoxycholic acid on cytosolic Ca++ signals in isolated rat hepatocytes. Gastroenterology 1993, 104, 604–612. [Google Scholar] [CrossRef]
- Beuers, U.; Throckmorton, D.C.; Anderson, M.S.; Isales, C.M.; Thasler, W.; Kullak-Ublick, G.A. Tauroursodeoxycholic acid activates protein kinase C in isolated rat hepatocytes. Gastroenterology 1996, 110, 1553–1563. [Google Scholar] [CrossRef]
- Bouscarel, B.; Gettys, T.W.; Fromm, H.; Dubner, H. Ursodeoxycholic acid inhibits glucagon-induced cAMP formation in hamster hepatocytes: A role for PKC. Am. J. Physiol. 1995, 268, G300–G310. [Google Scholar] [CrossRef] [PubMed]
- Stravitz, R.T.; Rao, Y.P.; Vlahcevic, Z.R.; Gurley, E.C.; Jarvis, W.D.; Hylemon, P.B. Hepatocellular protein kinase C activation by bile acids: Implications for regulation of cholesterol 7 alpha-hydroxylase. Am. J. Physiol. 1996, 271, G293–G303. [Google Scholar] [CrossRef] [PubMed]
- Kurz, A.K.; Graf, D.; Schmitt, M.; vom Dahl, S.; Haussinger, D. Tauroursodesoxycholate-induced choleresis involves p38(MAPK) activation and translocation of the bile salt export pump in rats. Gastroenterology 2001, 121, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Schliess, F.; Kurz, A.K.; vom Dahl, S.; Haussinger, D. Mitogen-activated protein kinases mediate the stimulation of bile acid secretion by tauroursodeoxycholate in rat liver. Gastroenterology 1997, 113, 1306–1314. [Google Scholar] [CrossRef]
- Marzioni, M.; Francis, H.; Benedetti, A.; Ueno, Y.; Fava, G.; Venter, J.; Reichenbach, R.; Mancino, M.G.; Summers, R.; Alpini, G.; et al. Ca2+ -dependent cytoprotective effects of ursodeoxycholic and tauroursodeoxycholic acid on the biliary epithelium in a rat model of cholestasis and loss of bile ducts. Am. J. Pathol. 2006, 168, 398–409. [Google Scholar] [CrossRef] [Green Version]
- Häussinger, D.; Kurz, A.K.; Wettstein, M.; Graf, D.; vom Dahl, S.; Schliess, F. Involvement of integrins and Src in tauroursodeoxycholate-induced and swelling-induced choleresis. Gastroenterology 2003, 124, 1476–1487. [Google Scholar] [CrossRef]
- Li, Q.; Dutta, A.; Kresge, C.; Bugde, A.; Feranchak, A.P. Bile acids stimulate cholangiocyte fluid secretion by activation of transmembrane member 16A Cl- channels. Hepatology 2018, 68, 187–199. [Google Scholar] [CrossRef] [Green Version]
- Beuers, U.; Bilzer, M.; Chittattu, A.; Kullak-Ublick, G.A.; Keppler, D.; Paumgartner, G.; Dombrowski, F. Tauroursodeoxycholic acid inserts the apical conjugate export pump, Mrp2, into canalicular membranes and stimulates organic anion secretion by protein kinase C-dependent mechanisms in cholestatic rat liver. Hepatology 2001, 33, 1206–1216. [Google Scholar] [CrossRef]
- Milkiewicz, P.; Roma, M.G.; Elias, E.; Coleman, R. Hepatoprotection with tauroursodeoxycholate and beta muricholate against taurolithocholate induced cholestasis: Involvement of signal transduction pathways. Gut 2002, 51, 113–119. [Google Scholar] [CrossRef]
- Wimmer, R.; Hohenester, S.; Pusl, T.; Denk, G.U.; Rust, C.; Beuers, U. Tauroursodeoxycholic acid exerts anticholestatic effects by a cooperative cPKC alpha-/PKA-dependent mechanism in rat liver. Gut 2008, 57, 1448–1454. [Google Scholar] [CrossRef] [Green Version]
- Dombrowski, F.; Stieger, B.; Beuers, U. Tauroursodeoxycholic acid inserts the bile salt export pump into canalicular membranes of cholestatic rat liver. Lab. Investig. 2006, 86, 166–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glasova, H.; Berghaus, T.M.; Kullak-Ublick, G.A.; Paumgartner, G.; Beuers, U. Tauroursodeoxycholic acid mobilizes alpha-PKC after uptake in human HepG2 hepatoma cells. Eur. J. Clin. Investig. 2002, 32, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Chipurupalli, S.; Kannan, E.; Tergaonkar, V.; D’Andrea, R.; Robinson, N. Hypoxia Induced ER Stress Response as an Adaptive Mechanism in Cancer. Int. J. Mol. Sci. 2019, 20, 749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusaczuk, M.; Cechowska-Pasko, M. Molecular chaperone ORP150 in ER stress-related diseases. Curr. Pharm. Des. 2013, 19, 2807–2818. [Google Scholar] [CrossRef]
- Uppala, J.K.; Gani, A.R.; Ramaiah, K.V.A. Chemical chaperone, TUDCA unlike PBA, mitigates protein aggregation efficiently and resists ER and non-ER stress induced HepG2 cell death. Sci. Rep. 2017, 7, 3831. [Google Scholar] [CrossRef]
- Gani, A.R.; Uppala, J.K.; Ramaiah, K.V. Tauroursodeoxycholic acid prevents stress induced aggregation of proteins in vitro and promotes PERK activation in HepG2 cells. Arch. Biochem. Biophys. 2015, 568, 8–15. [Google Scholar] [CrossRef]
- Papp, E.; Csermely, P. Chemical chaperones: Mechanisms of action and potential use. Handb. Exp. Pharmacol. 2006, 172, 405–416. [Google Scholar]
- Perlmutter, D.H. Chemical chaperones: A pharmacological strategy for disorders of protein folding and trafficking. Pediatr. Res. 2002, 52, 832–836. [Google Scholar] [CrossRef]
- Dandage, R.; Bandyopadhyay, A.; Jayaraj, G.G.; Saxena, K.; Dalal, V.; Das, A.; Chakraborty, K. Classification of chemical chaperones based on their effect on protein folding landscapes. ACS Chem. Biol. 2015, 10, 813–820. [Google Scholar] [CrossRef]
- Majtan, T.; Liu, L.; Carpenter, J.F.; Kraus, J.P. Rescue of cystathionine beta-synthase (CBS) mutants with chemical chaperones: Purification and characterization of eight CBS mutant enzymes. J. Biol. Chem. 2010, 285, 15866–15873. [Google Scholar] [CrossRef] [Green Version]
- Cohen, F.E.; Kelly, J.W. Therapeutic approaches to protein-misfolding diseases. Nature 2003, 426, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Kwon, Y.T. Protein quality control by molecular chaperones in neurodegeneration. Front. Neurosci. 2017, 11, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omura, T.; Asari, M.; Yamamoto, J.; Oka, K.; Hoshina, C.; Maseda, C.; Awaya, T.; Tasaki, Y.; Shiono, H.; Yonezawa, A.; et al. Sodium tauroursodeoxycholate prevents paraquat-induced cell death by suppressing endoplasmic reticulum stress responses in human lung epithelial A549 cells. Biochem. Biophys. Res. Commun. 2013, 432, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yoon, Y.M.; Lee, S.H. TUDCA-treated mesenchymal stem cells protect against ER Stress in the hippocampus of a murine chronic kidney disease model. Int. J. Mol. Sci. 2019, 20, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, H.; Zhou, C.; Chi, J.; Pan, S.; Lin, H.; Gao, F.; Ni, T.; Meng, L.; Zhang, J.; Jiang, C.; et al. The role of tauroursodeoxycholic acid on dedifferentiation of vascular smooth muscle cells by modulation of endoplasmic reticulum stress and as an oral drug inhibiting in-stent restenosis. Cardiovasc. Drugs Ther. 2019, 33, 25–33. [Google Scholar] [CrossRef]
- Bernstein, C.; Payne, C.M.; Bernstein, H.; Garewal, H. Activation of the metallothionein IIA promoter and other key stress response elements by ursodeoxycholate in HepG2 cells: Relevance to the cytoprotective function of ursodeoxycholate. Pharmacology 2002, 65, 2–9. [Google Scholar] [CrossRef]
- Mitsuyoshi, H.; Nakashima, T.; Sumida, Y.; Yoh, T.; Nakajima, Y.; Ishikawa, H.; Inaba, K.; Sakamoto, Y.; Okanoue, T.; Kashima, K. Ursodeoxycholic acid protects hepatocytes against oxidative injury via induction of antioxidants. Biochem. Biophys. Res. Commun. 1999, 263, 537–542. [Google Scholar] [CrossRef]
- Galán, M.; Kassan, M.; Choi, S.K.; Partyka, M.; Trebak, M.; Henrion, D.; Matrougui, K. A novel role for epidermal growth factor receptor tyrosine kinase and its downstream endoplasmic reticulum stress in cardiac damage and microvascular dysfunction in type 1 diabetes mellitus. Hypertension 2012, 60, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Kars, M.; Yang, L.; Gregor, M.F.; Mohammed, B.S.; Pietka, T.A.; Finck, B.N.; Patterson, B.W.; Horton, J.D.; Mittendorfer, B.; Hotamisligil, G.S.; et al. Tauroursodeoxycholic Acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes 2010, 59, 1899–1905. [Google Scholar] [CrossRef] [Green Version]
- Gaspar, J.M.; Martins, A.; Cruz, R.; Rodrigues, C.M.; Ambrósio, A.F.; Santiago, A.R. Tauroursodeoxycholic acid protects retinal neural cells from cell death induced by prolonged exposure to elevated glucose. Neuroscience 2013, 253, 380–388. [Google Scholar] [CrossRef]
- Marquardt, A.; Al-Dabet, M.M.; Ghosh, S.; Kohli, S.; Manoharan, J.; ElWakiel, A.; Gadi, I.; Bock, F.; Nazir, S.; Wang, H.; et al. Farnesoid X receptor agonism protects against diabetic tubulopathy: Potential add-on therapy for diabetic nephropathy. J. Am. Soc. Nephrol. 2017, 28, 3182–3189. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Zhou, Y.; Cao, H.; Wen, P.; Jiang, L.; He, W.; Dai, C.; Yang, J. Autophagy attenuates diabetic glomerular damage through protection of hyperglycemia-induced podocyte injury. PLoS ONE 2013, 8, e60546. [Google Scholar] [CrossRef] [PubMed]
- Bronczek, G.A.; Vettorazzi, J.F.; Soares, G.M.; Kurauti, M.A.; Santos, C.; Bonfim, M.F.; Carneiro, E.M.; Balbo, S.L.; Boschero, A.C.; Costa Júnior, J.M. The bile acid TUDCA improves beta-cell mass and reduces insulin degradation in mice with early-stage of type-1 diabetes. Front. Physiol. 2019, 10, 561. [Google Scholar] [CrossRef] [PubMed]
- Turdi, S.; Hu, N.; Ren, J. Tauroursodeoxycholic acid mitigates high fat diet-induced cardiomyocyte contractile and intracellular Ca2+ anomalies [published correction appears in PLoS One. 2016;11(4):e0154907]. PLoS ONE 2013, 8, e63615. [Google Scholar]
- Boden, G.; Duan, X.; Homko, C.; Molina, E.J.; Song, W.; Perez, O.; Cheung, P.; Merali, S. Increase in endoplasmic reticulum stress-related proteins and genes in adipose tissue of obese, insulin-resistant individuals. Diabetes 2008, 57, 2468–2544. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.K.; Das, S.K.; Mondal, A.K.; Hackney, O.G.; Chu, W.S.; Kern, P.A.; Rasouli, N.; Spencer, H.J.; Yao-Borengasser, A.; Elbein, S.C. Endoplasmic reticulum stress markers are associated with obesity in nondiabetic subjects. J. Clin. Endocrinol. Metab. 2008, 93, 4532–4541. [Google Scholar] [CrossRef]
- Vettorazzi, J.F.; Kurauti, M.A.; Soares, G.M.; Borck, P.C.; Ferreira, S.M.; Branco, R.C.S.; Michelone, L.S.L.; Boschero, A.C.; Junior, J.M.C.; Carneiro, E.M. Bile acid TUDCA improves insulin clearance by increasing the expression of insulin-degrading enzyme in the liver of obese mice. Sci. Rep. 2017, 7, 14876. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Zhong, J.J.; Jin, J.F.; Yin, X.M.; Miao, H. Tauroursodeoxycholate, a chemical chaperone, prevents palmitate-induced apoptosis in pancreatic β-cells by reducing ER stress. Exp. Clin. Endocrinol. Diabetes 2013, 121, 43–47. [Google Scholar] [CrossRef]
- Seyhun, E.; Malo, A.; Schäfer, C.; Moskaluk, C.A.; Hoffmann, R.T.; Göke, B.; Kubisch, C.H. Tauroursodeoxycholic acid reduces endoplasmic reticulum stress, acinar cell damage, and systemic inflammation in acute pancreatitis. Am. J. Physiol. Gastrointest. Live Physiol. 2011, 301, G773–G782. [Google Scholar] [CrossRef] [Green Version]
- Panzhinskiy, E.; Hua, Y.; Culver, B.; Ren, J.; Nair, S. Endoplasmic reticulum stress upregulates protein tyrosine phosphatase 1B and impairs glucose uptake in cultured myotubes. Diabetologia 2013, 56, 598–607. [Google Scholar] [CrossRef]
- Battson, M.L.; Lee, D.M.; Jarrell, D.K.; Hou, S.; Ecton, K.E.; Phan, A.B.; Gentile, C.L. Tauroursodeoxycholic acid reduces arterial stiffness and improves endothelial dysfunction in type 2 diabetic mice. J. Vasc. Res. 2017, 54, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Engin, F.; Yermalovich, A.; Ngyuen, T.; Hummasti, S.; Fu, W.; Eizirik, D.L.; Mathis, D.; Hotamisligil, G.S. Restoration of the unfolded protein response in pancreatic β cells protects mice against type 1 diabetes. Sci. Transl. Med. 2013, 5, ra156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vettorazzi, J.F.; Ribeiro, R.A.; Borck, P.C.; Branco, R.C.; Soriano, S.; Merino, B.; Boschero, A.C.; Nadal, A.; Quesada, I.; Carneiro, E.M. The bile acid TUDCA increases glucose-induced insulin secretion via the cAMP/PKA pathway in pancreatic beta cells. Metabolism 2016, 65, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Düfer, M.; Hörth, K.; Wagner, R.; Schittenhelm, B.; Prowald, S.; Wagner, T.F.; Oberwinkler, J.; Lukowski, R.; Gonzalez, F.J.; Krippeit-Drews, P.; et al. Bile acids acutely stimulate insulin secretion of mouse β-cells via farnesoid X receptor activation and K(ATP) channel inhibition. Diabetes 2012, 61, 1479–1489. [Google Scholar] [CrossRef] [Green Version]
- Xia, W.; Zhou, Y.; Wang, L.; Wang, L.; Liu, X.; Lin, Y.; Liu, L. Tauroursodeoxycholic acid inhibits TNF-induced lipolysis in 3T3-L1 adipocytes via the IRE-JNK-perilipin-A signaling pathway. Mol. Med. 2017, 15, 1753–1758. [Google Scholar] [CrossRef]
- Guo, Q.; Shi, Q.; Li, H.; Liu, J.; Wu, S.; Sun, H.; Zhou, B. Glycolipid metabolism disorder in the liver of obese mice is improved by TUDCA via the restoration of defective hepatic autophagy. Int. J. Endocrinol. 2015, 2015, 687938. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Wang, D.; Xu, J.; Chi, B. Effect of tauroursodeoxycholic acid and 4-phenylbutyric acid on metabolism of copper and zinc in type 1 diabetic mice model. Biol. Trace Elem. Res. 2016, 170, 348–356. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Guo, W.; Jia, Y.; Xu, J. Effect of 4-phenylbutyric acid and tauroursodeoxycholic acid on magnesium and calcium metabolism in streptozocin-induced type 1 diabetic mice. Biol. Trace Elem. Res. 2019, 189, 501–510. [Google Scholar] [CrossRef] [Green Version]
- Viktorinova, A.; Toserova, E.; Krizko, M.; Durackova, Z. Altered metabolism of copper, zinc, and magnesium is associated with increased levels of glycated hemoglobin in patients with diabetes mellitus. Metabolism 2009, 58, 1477–1482. [Google Scholar] [CrossRef]
- Arruda, A.P.; Hotamisligil, G.S. Calcium homeostasis and organelle function in the pathogenesis of obesity and diabetes. Cell Metab. 2015, 22, 381–397. [Google Scholar] [CrossRef] [Green Version]
- Ceylan-Isik, A.F.; Sreejayan, N.; Ren, J. Endoplasmic reticulum chaperon tauroursodeoxycholic acid alleviates obesity-induced myocardial contractile dysfunction. J. Mol. Cell Cardiol. 2011, 50, 107–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, C.; Wang, Y.; Zhang, H.; Han, F. The role of endoplasmic reticulum stress in neurodegenerative disease. Apoptosis 2017, 22, 1–26. [Google Scholar] [CrossRef] [PubMed]
- MahmoudianDehkordi, S.; Arnold, M.; Nho, K.; Ahmad, S.; Jia, W.; Xie, G.; Louie, G.; Kueider-Paisley, A.; Moseley, M.; Thompson, J.W.; et al. Altered bile acid profile associates with cognitive impairment in Alzheimer’s disease-An emerging role for gut microbiome. Alzheimers Dement. 2019, 15, 76–92. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.C.; Callaerts-Vegh, Z.; Nunes, A.F.; Rodrigues, C.M.; D’Hooge, R. Tauroursodeoxycholic acid (TUDCA) supplementation prevents cognitive impairment and amyloid deposition in APP/PS1 mice. Neurobiol. Dis. 2013, 50, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Nunes, A.F.; Amaral, J.D.; Lo, A.C.; Fonseca, M.B.; Viana, R.J.; Callaerts-Vegh, Z.; D’Hooge, R.; Rodrigues, C.M. TUDCA, a bile acid, attenuates amyloid precursor protein processing and amyloid-β deposition in APP/PS1 mice. Mol. Neurobiol. 2012, 45, 440–454. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.M.; Rodrigues, C.M.; Zhao, L.R.; Steer, C.J.; Low, W.C. Tauroursodeoxycholic acid improves the survival and function of nigral transplants in a rat model of Parkinson’s disease. Cell Transplant. 2002, 11, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Rosa, A.I.; Fonseca, I.; Nunes, M.J.; Moreira, S.; Rodrigues, E.; Carvalho, A.N.; Rodrigues, C.M.P.; Gama, M.J.; Castro-Caldas, M. Novel insights into the antioxidant role of tauroursodeoxycholic acid in experimental models of Parkinson’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2171–2181. [Google Scholar] [CrossRef]
- Moreira, S.; Fonseca, I.; Nunes, M.J.; Rosa, A.; Lemos, L.; Rodrigues, E.; Carvalho, A.N.; Outeiro, T.F.; Rodrigues, C.M.P.; Gama, M.J.; et al. Nrf2 activation by tauroursodeoxycholic acid in experimental models of Parkinson’s disease. Exp. Neurol. 2017, 295, 77–87. [Google Scholar] [CrossRef]
- Graham, S.F.; Rey, N.L.; Ugur, Z.; Yilmaz, A.; Sherman, E.; Maddens, M.; Bahado-Singh, R.O.; Becker, K.; Schulz, E.; Meyerdirk, L.K.; et al. Metabolomic profiling of bile acids in an experimental model of prodromal Parkinson’s disease. Metabolites 2018, 8, 71. [Google Scholar] [CrossRef] [Green Version]
- Rosa, A.I.; Duarte-Silva, S.; Silva-Fernandes, A.; Nunes, M.J.; Carvalho, A.N.; Rodrigues, E.; Gama, M.J.; Rodrigues, C.M.P.; Maciel, P.; Castro-Caldas, M. Tauroursodeoxycholic acid improves motor symptoms in a mouse model of Parkinson’s disease. Mol. Neurobiol. 2018, 55, 9139–9155. [Google Scholar] [CrossRef]
- Keene, C.D.; Rodrigues, C.M.; Eich, T.; Linehan-Stieers, C.; Abt, A.; Kren, B.T.; Steer, C.J.; Low, W.C. A bile acid protects against motor and cognitive deficits and reduces striatal degeneration in the 3-nitropropionic acid model of Huntington’s disease. Exp. Neurol. 2001, 171, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Elia, A.E.; Lalli, S.; Monsurrò, M.R.; Sagnelli, A.; Taiello, A.C.; Reggiori, B.; La Bella, V.; Tedeschi, G.; Albanese, A. Tauroursodeoxycholic acid in the treatment of patients with amyotrophic lateral sclerosis. Eur. J. Neurol. 2016, 23, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Parry, G.J.; Rodrigues, C.M.; Aranha, M.M.; Hilbert, S.J.; Davey, C.; Kelkar, P.; Low, W.C.; Steer, C.J. Safety, tolerability, and cerebrospinal fluid penetration of ursodeoxycholic acid in patients with amyotrophic lateral sclerosis. Clin. Neuropharmacol. 2010, 33, 17–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, J.H.; Hong, Y.H.; Sung, J.J.; Kim, S.M.; Lee, J.B.; Lee, K.W. Oral solubilized ursodeoxycholic acid therapy in amyotrophic lateral sclerosis: A randomized cross-over trial. J. Korean Med. Sci. 2012, 27, 200–206. [Google Scholar] [CrossRef] [Green Version]
- Cortez, L.M.; Campeau, J.; Norman, G.; Kalayil, M.; Van der Merwe, J.; McKenzie, D.; Sim, V.L. Bile acids reduce prion conversion, reduce neuronal loss, and prolong male survival in models of Prion disease. J. Virol. 2015, 89, 7660–7672. [Google Scholar] [CrossRef] [Green Version]
- Cai, F.; Liu, J.; Li, C.; Wang, J. Critical role of endoplasmic reticulum stress in cognitive impairment induced by microcystin-LR. Int. J. Mol. Sci. 2015, 16, 28077–28086. [Google Scholar] [CrossRef] [Green Version]
- Launay, N.; Ruiz, M.; Grau, L.; Ortega, F.J.; Ilieva, E.V.; Martínez, J.J.; Galea, E.; Ferrer, I.; Knecht, E.; Pujol, A.; et al. Tauroursodeoxycholic bile acid arrests axonal degeneration by inhibiting the unfolded protein response in X-linked adrenoleukodystrophy. Acta Neuropathol. 2017, 133, 283–301. [Google Scholar] [CrossRef] [Green Version]
- Ramalho, R.M.; Viana, R.J.; Low, W.C.; Steer, C.J.; Rodrigues, C.M. Bile acids and apoptosis modulation: An emerging role in experimental Alzheimer’s disease. Trends Mol. Med. 2008, 14, 54–62. [Google Scholar] [CrossRef]
- Keene, C.D.; Rodrigues, C.M.; Eich, T.; Chhabra, M.S.; Steer, C.J.; Low, W.C. Tauroursodeoxycholic acid, a bile acid, is neuroprotective in a transgenic animal model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2002, 99, 10671–10676. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, C.M.P.; Solá, S.; Silva, R.; Brites, D. Bilirubin and amyloid-beta peptide induce cytochrome c release through mitochondrial membrane permeabilization. Mol. Med. 2000, 6, 936–946. [Google Scholar] [CrossRef] [Green Version]
- Ramalho, R.M.; Borralho, P.M.; Castro, R.E.; Solá, S.; Steer, C.J.; Rodrigues, C.M. Tauroursodeoxycholic acid modulates p53-mediated apoptosis in Alzheimer’s disease mutant neuroblastoma cells. J. Neurochem. 2006, 98, 1610–1618. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, G.; Chakrabarti, S.; Chatterjee, U.; Saso, L. Proteinopathy, oxidative stress and mitochondrial dysfunction: Cross talk in Alzheimer’s disease and Parkinson’s disease. Drug Des. Devel. Ther. 2017, 11, 797–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oveson, B.C.; Iwase, T.; Hackett, S.F.; Lee, S.Y.; Usui, S.; Sedlak, T.W.; Snyder, S.H.; Campochiaro, P.A.; Sung, J.U. Constituents of bile, bilirubin and TUDCA, protect against oxidative stress-induced retinal degeneration. J. Neurochem. 2011, 116, 144–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, H.S.; Low, W.C. Ursodeoxycholic acid suppresses mitochondria-dependent programmed cell death induced by sodium nitroprusside in SH-SY5Y cells. Toxicology 2012, 292, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Keane, P.C.; Kurzawa, M.; Blain, P.G.; Morris, C.M. Mitochondrial dysfunction in Parkinson’s disease. Parkinson’s Dis. 2011, 2011, 716871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, J.; Basso, V.; Ziviani, E. Post translational modification of parkin. Biol. Direct. 2017, 12, 6. [Google Scholar] [CrossRef] [Green Version]
- Shiba-Fukushima, K.; Imai, Y.; Yoshida, S.; Ishihama, Y.; Kanao, T.; Sato, S.; Hattori, N. PINK1-mediated phosphorylation of the parkin ubiquitin-like domain primes mitochondrial translocation of parkin and regulates mitophagy. Sci. Rep. 2012, 2, 1002. [Google Scholar] [CrossRef]
- Graham, S.F.; Rey, N.L.; Yilmaz, A.; Kumar, P.; Madaj, Z.; Maddens, M.; Bahado-Singh, R.O.; Becker, K.; Schulz, E.; Meyerdirk, L.K.; et al. Biochemical profiling of the brain and blood metabolome in a mouse model of prodromal Parkinson’s disease reveals distinct metabolic profiles. J. Proteom. Res. 2018, 17, 2460–2469. [Google Scholar] [CrossRef]
- Noailles, A.; Fernández-Sánchez, L.; Lax, P.; Cuenca, N. Microglia activation in a model of retinal degeneration and TUDCA neuroprotective effects. J. Neuroinflammation 2014, 11, 186. [Google Scholar] [CrossRef] [Green Version]
- Mantopoulos, D.; Murakami, Y.; Comander, J.; Thanos, A.; Roh, M.; Miller, J.W.; Vavvas, D.G. Tauroursodeoxycholic acid (TUDCA) protects photoreceptors from cell death after experimental retinal detachment. PLoS ONE 2011, 6, e24245. [Google Scholar] [CrossRef]
- Boatright, J.H.; Nickerson, J.M.; Moring, A.G.; Pardue, M.T. Bile acids in treatment of ocular disease. J. Ocul. Biol. Dis. Infor. 2009, 2, 149–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Sanchez, L.; Lax, P.; Pinilla, I.; Martín-Nieto, J.; Cuenca, N. Tauroursodeoxycholic acid prevents retinal degeneration in transgenic P23H rats. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4998–5008. [Google Scholar] [CrossRef] [PubMed]
- Boatright, J.H.; Moring, A.G.; McElroy, C.; Phillips, M.J.; Do, V.T.; Chang, B.; Hawes, N.L.; Boyd, A.P.; Sidney, S.S.; Stewart, R.E.; et al. Tool from ancient pharmacopoeia prevents vision loss. Mol. Vis. 2006, 12, 1706–1714. [Google Scholar] [PubMed]
- Phillips, M.J.; Walker, T.A.; Choi, H.Y.; Faulkner, A.E.; Kim, M.K.; Sidney, S.S.; Boyd, A.P.; Nickerson, J.M.; Boatright, J.H.; Pardue, M.T. Tauroursodeoxycholic acid preservation of photoreceptor structure and function in the rd10 mouse through postnatal day 30. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2148–2155. [Google Scholar] [CrossRef] [Green Version]
- Drack, A.V.; Dumitrescu, A.V.; Bhattarai, S.; Gratie, D.; Stone, E.M.; Mullins, R.; Sheffield, V.C. TUDCA slows retinal degeneration in two different mouse models of retinitis pigmentosa and prevents obesity in Bardet-Biedl syndrome type 1 mice. Investig. Ophthalmol. Vis. Sci. 2012, 53, 100–106. [Google Scholar] [CrossRef]
- Zhang, T.; Baehr, W.; Fu, Y. Chemical chaperone TUDCA preserves cone photoreceptors in a mouse model of Leber congenital amaurosis. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3349–3356. [Google Scholar] [CrossRef] [Green Version]
- Xia, H.; Nan, Y.; Huang, X.; Gao, J.; Pu, M. Effects of tauroursodeoxycholic acid and alpha-lipoic-acid on the visual response properties of cat retinal ganglion cells: An in vitro study. Investig. Ophthalmol. Vis. Sci. 2015, 56, 6638–6645. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Vicente, V.; Lax, P.; Fernandez-Sanchez, L.; Rondon, N.; Esquiva, G.; Germain, F.; de la Villa, P.; Cuenca, P. Neuroprotective effect of tauroursodeoxycholic acid on N-methyl-D-aspartate-induced retinal ganglion cell degeneration. PLoS ONE 2015, 10, e0137826. [Google Scholar] [CrossRef] [Green Version]
- Murase, H.; Tsuruma, K.; Shimazawa, M.; Hara, H. TUDCA promotes phagocytosis by retinal pigment epithelium via MerTK activation. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2511–2518. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Wang, M.; Li, J.Z.; Wei, S.D.; Wu, H.; Lai, X.; Cao, D.; Ou, Z.B.; Gong, J. Tauroursodeoxycholic acid alleviates hepatic ischemia reperfusion injury by suppressing the function of Kupffer cells in mice. Biomed. Pharmacother. 2018, 106, 1271–1281. [Google Scholar] [CrossRef]
- Nakada, E.M.; Bhakta, N.R.; Korwin-Mihavics, B.R.; Kumar, A.; Chamberlain, N.; Bruno, S.R.; Chapman, D.G.; Hoffman, S.M.; Daphtary, N.; Aliyeva, M.; et al. Conjugated bile acids attenuate allergen-induced airway inflammation and hyperresponsiveness by inhibiting UPR transducers. JCI Insight. 2019, 4, e98101. [Google Scholar] [CrossRef] [PubMed]
- Yanguas-Casás, N.; Barreda-Manso, M.A.; Nieto-Sampedro, M.; Romero-Ramírez, L. Tauroursodeoxycholic acid reduces glial cell activation in an animal model of acute neuroinflammation. J. Neuroinflamm. 2014, 11, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanguas-Casás, N.; Barreda-Manso, M.A.; Perez-Rial, S.; Nieto-Sampedro, M.; Romero-Ramírez, L. TGFβ Contributes to the anti-inflammatory effects of tauroursodeoxycholic acid on an animal model of acute neuroinflammation. Mol. Neurobiol. 2017, 54, 6737–6749. [Google Scholar] [CrossRef] [PubMed]
- Castro, I.; Albornoz, N.; Aguilera, S.; Barrera, M.J.; González, S.; Núñez, M.; Carvajal, P.; Jara, D.; Lagos, C.; Molina, C.; et al. Aberrant MUC1 accumulation in salivary glands of Sjögren’s syndrome patients is reversed by TUDCA in vitro. Rheumatology 2019. [Google Scholar] [CrossRef]
- Yun, S.P.; Yoon, Y.M.; Lee, J.H.; Kook, M.; Han, Y.S.; Jung, S.K.; Lee, S.H. Tauroursodeoxycholic acid protects against the effects of P-Cresol-induced reactive oxygen species via the expression of cellular Prion protein. Int. J. Mol. Sci. 2018, 19, 352. [Google Scholar] [CrossRef] [Green Version]
- Cho, E.J.; Yoon, J.H.; Kwak, M.S.; Jang, E.S.; Lee, J.H.; Yu, S.J.; Kim, Y.J.; Kim, C.Y.; Lee, H.S. Tauroursodeoxycholic acid attenuates progression of steatohepatitis in mice fed a methionine–choline-deficient diet. Dig. Dis. Sci. 2014, 59, 1461–1474. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, J.; Gui, W.; Sun, D.; Dai, H.; Xiao, L.; Chu, H.; Du, F.; Zhu, Q.; Schnabl, B.; et al. Tauroursodeoxycholic acid inhibits intestinal inflammation and barrier disruption in mice with non-alcoholic fatty liver disease. Br. J. Pharmacol. 2018, 175, 469–484. [Google Scholar] [CrossRef] [Green Version]
- Aslan, M.; Kıraç, E.; Yılmaz, Ö.; Ünal, B.; Konuk, E.K.; Özcan, F.; Tuzcu, H. Effect of tauroursodeoxycholic acid on PUFA levels and inflammation in an animal and cell model of hepatic endoplasmic reticulum stress. Hum. Exp. Toxicol. 2018, 37, 803–816. [Google Scholar] [CrossRef]
- Siddesha, J.M.; Nakada, E.M.; Mihavics, B.R.; Hoffman, S.M.; Rattu, G.K.; Chamberlain, N.; Cahoon, J.M.; Lahue, K.G.; Daphtary, N.; Aliyeva, M.; et al. Effect of a chemical chaperone, tauroursodeoxycholic acid, on HDM-induced allergic airway disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L1243–L1259. [Google Scholar] [CrossRef] [Green Version]
- Seong, J.K.; Wan-Kyu, K.; Min-Jae, J.; Yoshie, A.; Hyemin, C.; Hemant, K.; In-Bo, H.; Seil, S. Anti-inflammatory effect of Tauroursodeoxycholic acid in RAW 264.7 macrophages, Bone marrow-derived macrophages, BV2 microglial cells, and spinal cord injury. Sci. Rep. 2018, 8, 3176. [Google Scholar]
- Miguel, C.; Sedaka, R.; Kasztan, M.; Lever, J.M.; Sonnenberger, M.; Abad, A.; Jin, C.; Carmines, P.K.; Pollock, D.M.; Pollock, J.S. Tauroursodeoxycholic acid (TUDCA) abolishes chronic high salt-induced renal injury and inflammation. Acta Physiol. 2019, 226, e13227. [Google Scholar] [CrossRef] [PubMed]
- Walczak, A.; Gradzik, K.; Kabzinski, J.; Przybylowska-Sygut, K.; Majsterek, I. The role of the ER-induced UPR pathway and the efficacy of its inhibitors and inducers in the inhibition of tumor progression. Oxid. Med. Cell Longev. 2019, 2019, 5729710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Kusaczuk, M.; Krętowski, R.; Naumowicz, M.; Stypułkowska, A.; Cechowska-Pasko, M. Silica nanoparticle-induced oxidative stress and mitochondrial damage is followed by activation of intrinsic apoptosis pathway in glioblastoma cells. Int. J. Nanomed. 2018, 13, 2279–2294. [Google Scholar] [CrossRef] [Green Version]
- Alpini, G.; Kanno, N.; Glaser, S.; Francis, H.; Taffetani, S.; LeSage, G. Tauroursodeoxycholate inhibits human cholangiocarcinoma growth via Ca2+-, PKC-, and MAPK-dependent pathways. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G973–G982. [Google Scholar] [CrossRef] [Green Version]
- Vandewynckel, Y.P.; Laukens, D.; Devisscher, L.; Paridaens, A.; Bogaerts, E.; Verhelst, X.; van den Bussche, A.; Raevens, S.; van Steenkiste, C.; van Troys, M.; et al. Tauroursodeoxycholic acid dampens oncogenic apoptosis induced by endoplasmic reticulum stress during hepatocarcinogen exposure. Oncotarget 2015, 6, 28011–28025. [Google Scholar] [CrossRef] [Green Version]
- Park, G.; Han, Y.K.; Han, J.Y.; Lee, C.G. Tauroursodeoxycholic acid reduces the invasion of MDA-MB-231 cells by modulating matrix metalloproteinases 7 and 13. Oncol. Lett. 2013, 12, 2227–2231. [Google Scholar] [CrossRef]
- Kim, S.H.; Chun, H.J.; Choi, H.S.; Kim, E.S.; Keum, B.; Seo, Y.S.; Jeen, Y.T.; Lee, H.S.; Um, S.H.; Kim, C.D. Ursodeoxycholic acid attenuates 5-fluorouracil-induced mucositis in a rat model. Oncol. Lett. 2018, 16, 2585–2590. [Google Scholar] [CrossRef]
- Yu, H.; Fu, Q.R.; Huang, Z.J.; Lin, J.Y.; Chen, Q.X.; Wang, Q.; Shen, D.Y. Apoptosis induced by ursodeoxycholic acid in human melanoma cells through the mitochondrial pathway. Oncol. Rep. 2019, 41, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Dongfeng, D.; An, C.; Shujia, P.; Jikai, Y.; Tao, Y.; Rui, D.; Kai, T.; Yafeng, C.; Jianguo, L.; Xilin, D. Explanation of colon cancer pathophysiology through analyzing the disrupted homeostasis of bile acids. Afr. Health Sci. 2014, 14, 925–928. [Google Scholar]
- Kim, Y.H.; Kim, J.H.; Kim, B.G.; Lee, K.L.; Kim, J.W.; Koh, S.J. Tauroursodeoxycholic acid attenuates colitis-associated colon cancer by inhibiting nuclear factor kappaB signaling. J. Gastroenterol. Hepatol. 2019, 34, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Ludolph, A.C. The TUDCA trial--innovative trial designs for amyotrophic lateral sclerosis drugs? Eur. J. Neurol. 2016, 23, 11–12. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.M.; Kim, S.; Han, Y.S.; Yun, C.W.; Lee, J.H.; Noh, H.; Lee, S.H. TUDCA-treated chronic kidney disease-derived hMSCs improve therapeutic efficacy in ischemic disease via PrPC. Redox Biol. 2019, 22, 101144. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qu, P.; Ma, X.; Qiao, F.; Ma, Y.; Qing, S.; Zhang, Y.; Wang, Y.; Cui, W. Tauroursodeoxycholic acid (TUDCA) alleviates endoplasmic reticulum stress of nuclear donor cells under serum starvation. PLoS ONE 2018, 13, e0196785. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Zhao, B.; Qi, M.; Yao, Y.; Xu, L.; Ji, R.; Chen, W.; Wang, J.; Huang, S.; Ma, L.; et al. Tudca Ameliorates Liver Injury Via Activation of SIRT1-FXR Signaling in a Rat Hemorrhagic Shock Model. Shock 2019. [Google Scholar] [CrossRef]
- Li, P.; Fu, D.; Sheng, Q.; Yu, S.; Bao, X.; Lv, Z. TUDCA attenuates intestinal injury and inhibits endoplasmic reticulum stress-mediated intestinal cell apoptosis in necrotizing enterocolitis. Int. Immunopharmacol. 2019, 74, 105665. [Google Scholar] [CrossRef]
- Arai, Y.; Choi, B.; Kim, B.J.; Rim, W.; Park, S.; Park, H.; Ahn, J.; Lee, S.H. Tauroursodeoxycholic acid (TUDCA) counters osteoarthritis by regulating intracellular cholesterol levels and membrane fluidity of degenerated chondrocytes. Biomater. Sci. 2019, 7, 3178–3189. [Google Scholar] [CrossRef]
- Liu, C.; Cao, Y.; Yang, X.; Shan, P.; Liu, H. Tauroursodeoxycholic acid suppresses endoplasmic reticulum stress in the chondrocytes of patients with osteoarthritis. Int. J. Mol. Med. 2015, 36, 1081–1087. [Google Scholar] [CrossRef]
- Yao, X.H.; Nguyen, K.H.; Nyomba, B.L. Reversal of glucose intolerance in rat offspring exposed to ethanol before birth through reduction of nuclear skeletal muscle HDAC expression by the bile acid TUDCA. Physiol. Rep. 2014, 2, e12195. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.H.; Nguyen, H.K.; Nyomba, B.L. Prenatal ethanol exposure causes glucose intolerance with increased hepatic gluconeogenesis and histone deacetylases in adult rat offspring: Reversal by tauroursodeoxycholic acid. PLoS ONE 2013, 8, e59680. [Google Scholar] [CrossRef]
- Villota-Salazar, N.A.; Mendoza-Mendoza, A.; González-Prieto, J.M. Epigenetics: From the past to the present. Front. Life Sci. 2016, 9, 347–370. [Google Scholar] [CrossRef]
- Wawryka, J.; Barg, E. Impact of SIRT1 gene expression on the development and treatment of the metabolic syndrome in oncological patients. Pediatr. Endocrinol. Diabetes Metab. 2016, 22, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Kotb, M.A. Molecular mechanisms of ursodeoxycholic acid toxicity & side effects: Ursodeoxycholic acid freezes regeneration & induces hibernation mode. Int. J. Mol. Sci. 2012, 13, 8882–8914. [Google Scholar] [PubMed] [Green Version]
- Rudolph, G.; Gotthardt, D.N.; Kloeters-Plachky, P.; Kulaksiz, H.; Schirmacher, P.; Stiehl, A. In PSC with colitis treated with UDCA, most colonic carcinomas develop in the first years after the start of treatment. Dig. Dis. Sci. 2011, 56, 3624–3630. [Google Scholar] [CrossRef] [PubMed]
- Hylemon, P.B.; Zhou, H.; Pandak, W.M.; Ren, S.; Gil, G.; Dent, P. Bile acids as regulatory molecules. J. Lipid Res. 2009, 50, 1509–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dosa, P.I.; Ward, T.; Castro, R.E.; Rodrigues, C.M.P.; Steer, C.J. Synthesis and evaluation of water-soluble prodrugs of ursodeoxycholic acid (UDCA), an anti-apoptotic bile acid. Chem. Med. Chem. 2013, 8, 1002–1011. [Google Scholar] [CrossRef]
- Xu, Y.; Yang, L.; Zhao, S.; Wang, Z. Large-scale production of tauroursodeoxycholic acid products through fermentation optimization of engineered Escherichia coli cell factory. Microbial Cell Factories 2019, 18, 34. [Google Scholar] [CrossRef]
- Feng, R.; Lia, J.; Chen, J.; Duan, L.; Liu, X.; Di, D.; Deng, Y.; Song, Y. Preparation and toxicity evaluation of a novel nattokinase- tauroursodeoxycholate complex. Asian J. Pharm. Sci. 2018, 13, 173–182. [Google Scholar] [CrossRef]

© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kusaczuk, M. Tauroursodeoxycholate—Bile Acid with Chaperoning Activity: Molecular and Cellular Effects and Therapeutic Perspectives. Cells 2019, 8, 1471. https://doi.org/10.3390/cells8121471
Kusaczuk M. Tauroursodeoxycholate—Bile Acid with Chaperoning Activity: Molecular and Cellular Effects and Therapeutic Perspectives. Cells. 2019; 8(12):1471. https://doi.org/10.3390/cells8121471
Chicago/Turabian StyleKusaczuk, Magdalena. 2019. "Tauroursodeoxycholate—Bile Acid with Chaperoning Activity: Molecular and Cellular Effects and Therapeutic Perspectives" Cells 8, no. 12: 1471. https://doi.org/10.3390/cells8121471
APA StyleKusaczuk, M. (2019). Tauroursodeoxycholate—Bile Acid with Chaperoning Activity: Molecular and Cellular Effects and Therapeutic Perspectives. Cells, 8(12), 1471. https://doi.org/10.3390/cells8121471