Adjuvant Epigenetic Therapy of Decitabine and Suberoylanilide Hydroxamic Acid Exerts Anti-Neoplastic Effects in Acute Myeloid Leukemia Cells

 ,
,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Drugs
2.2. Cell Lines and Culture Conditions
2.3. In vitro Proliferation Assays
2.4. Cell Cycle Analysis
2.5. Apoptosis Assay
2.6. Protein Extraction and Quantification
2.7. Western Blotting
2.8. Statistical Analysis
3. Results
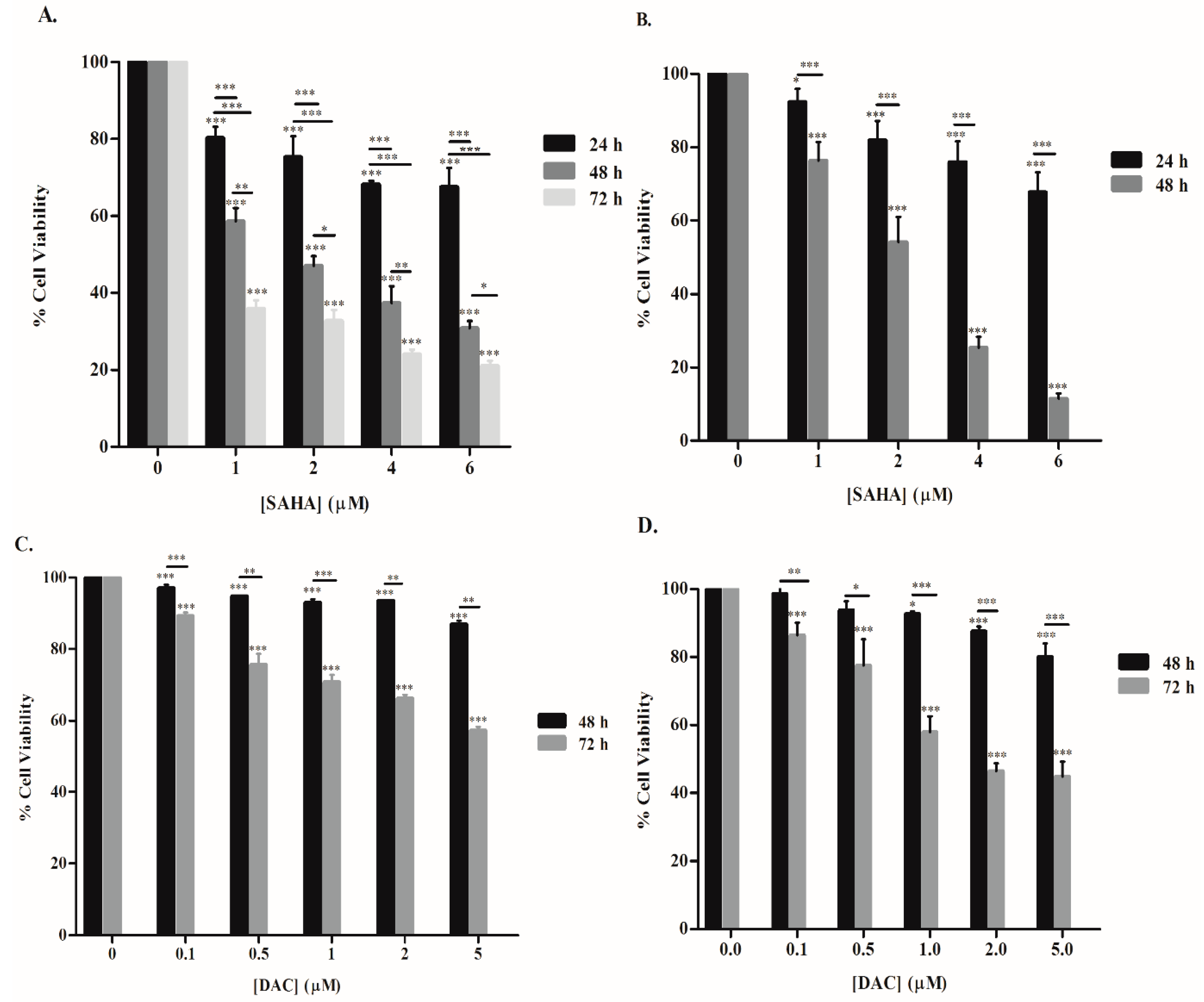
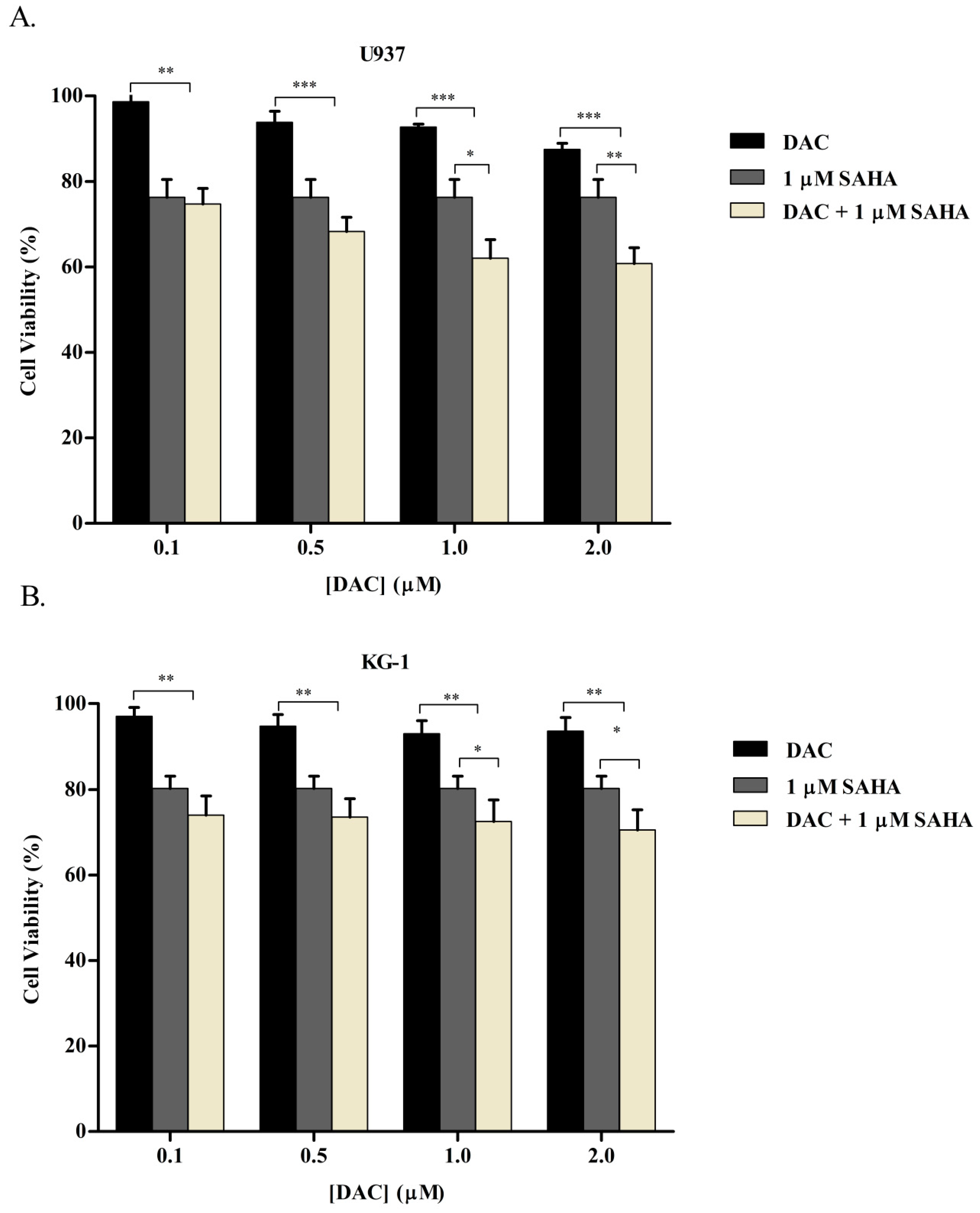
3.1. SAHA and DAC Reduce the Viability of KG-1 and U937 Cells
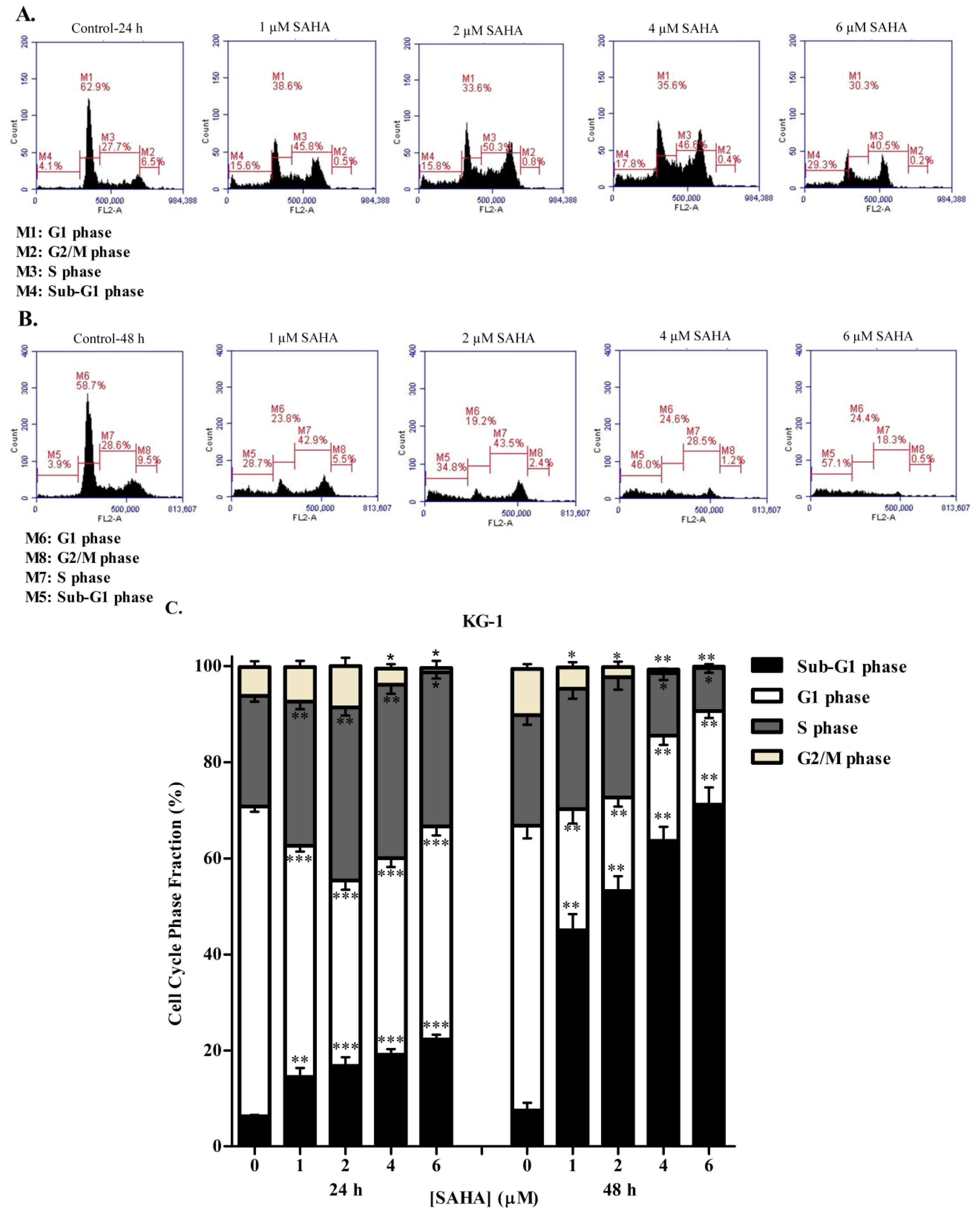
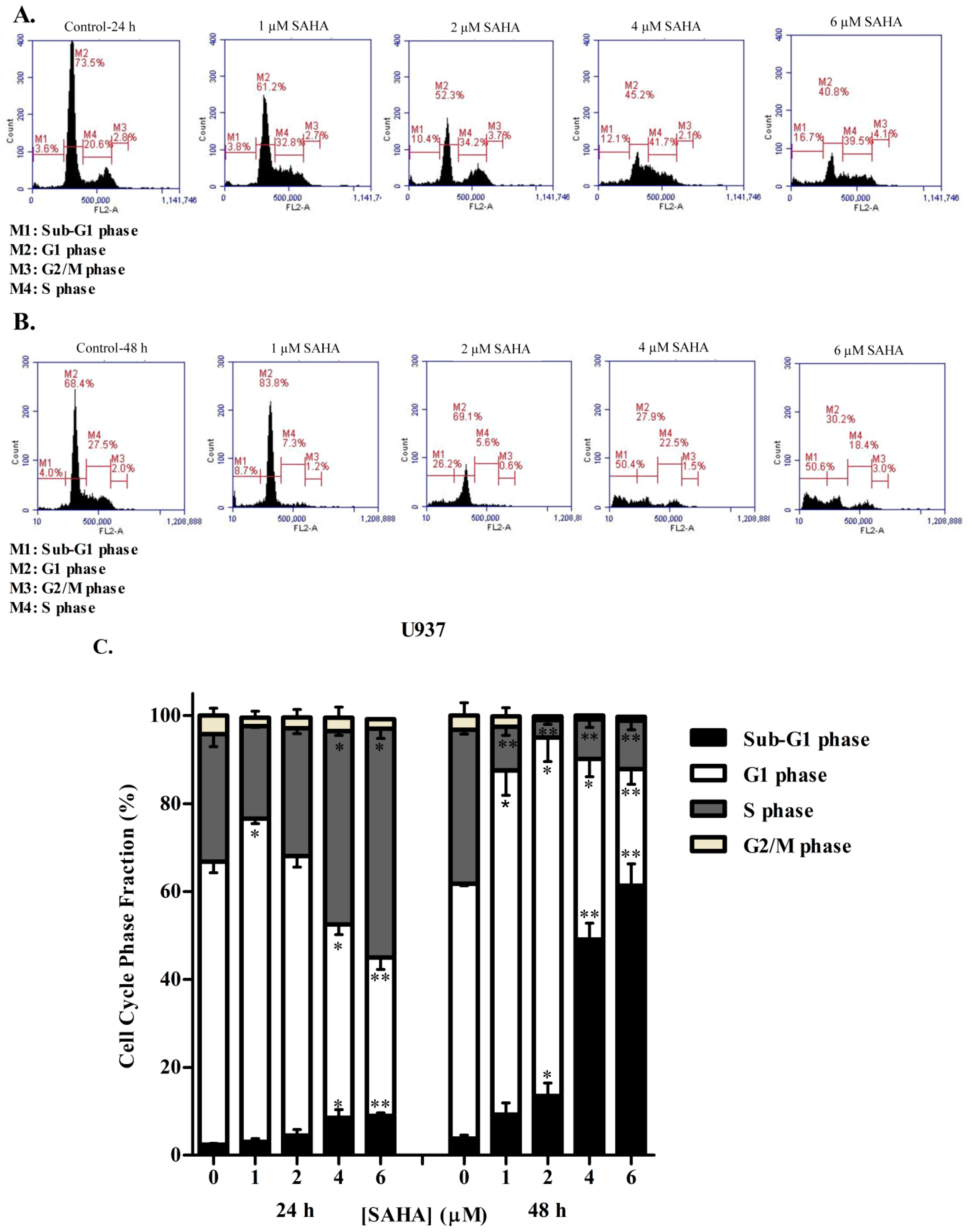
3.2. SAHA Induces Cell Cycle Arrest in the S/G2 Phase of KG-1 and U937 Cells
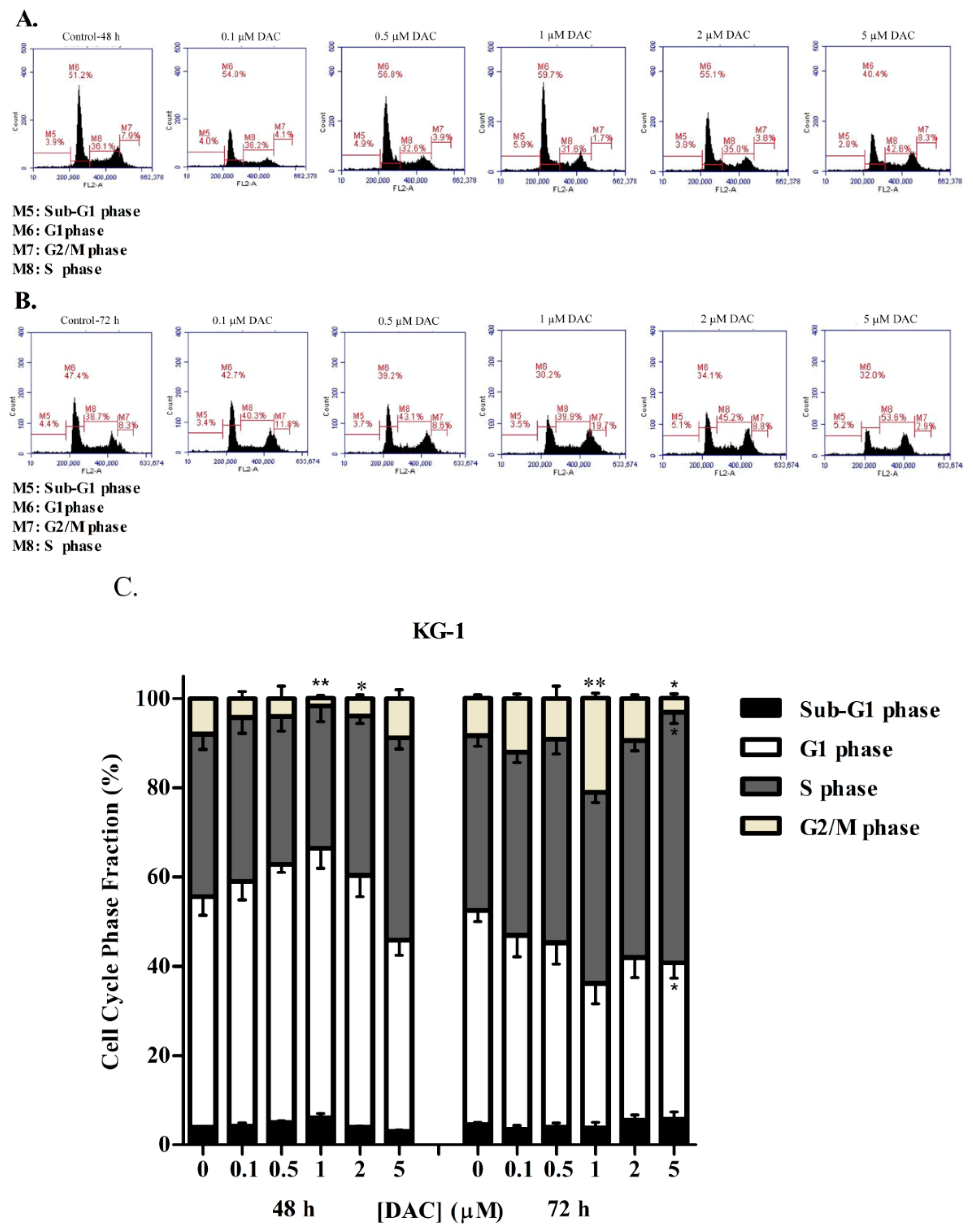
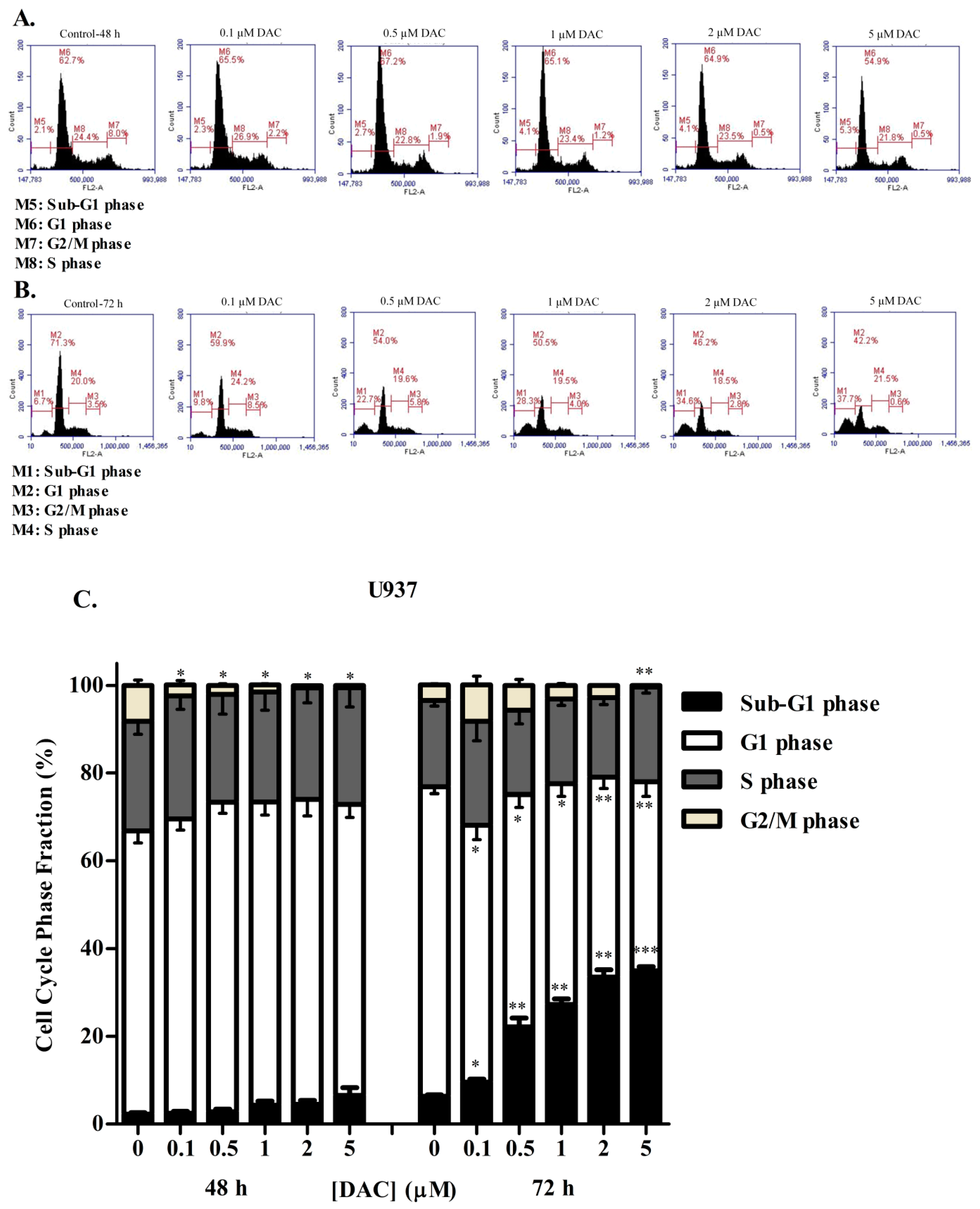
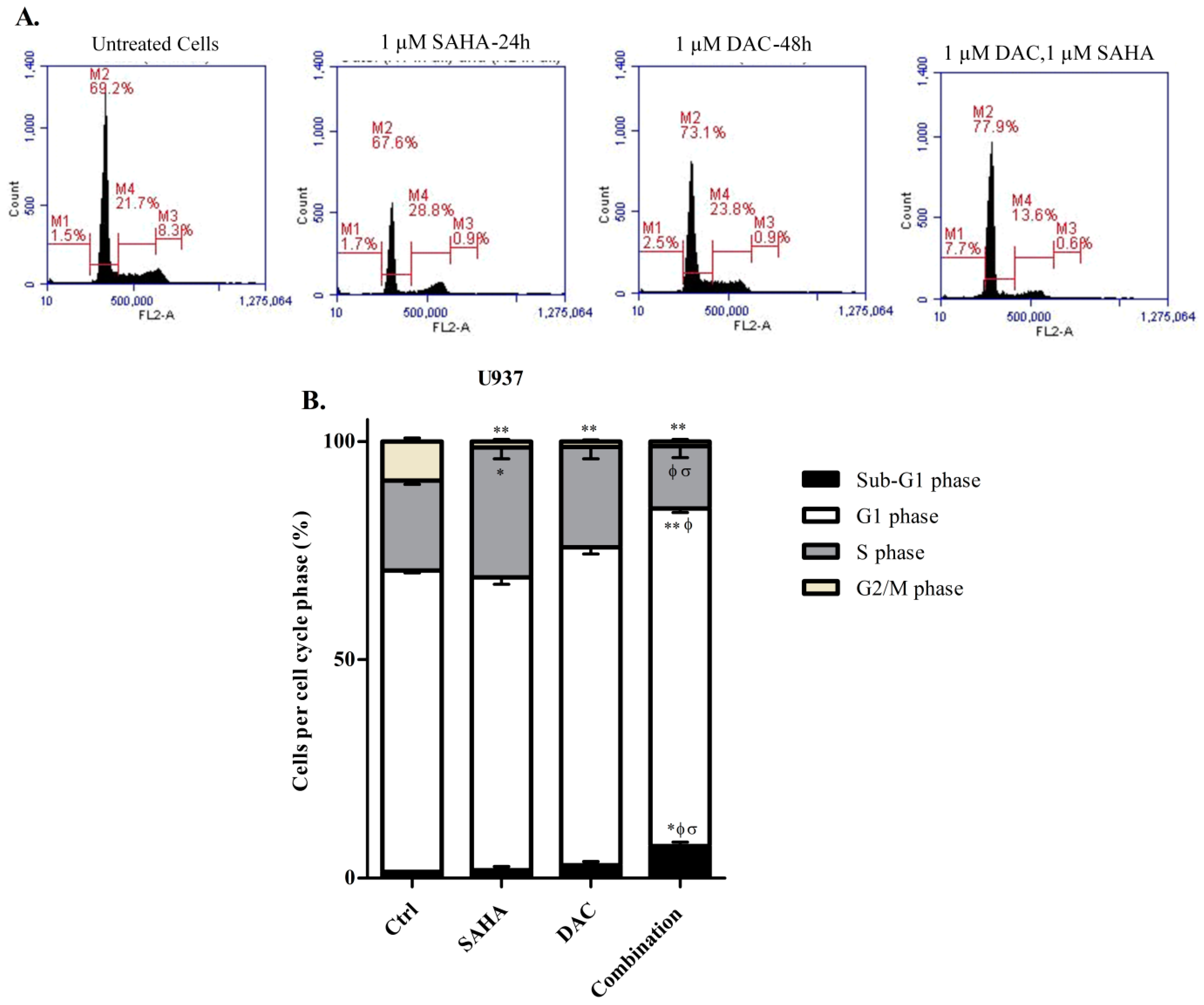
3.3. DAC Induces Cell Cycle Arrest in KG-1 and U937 Cells
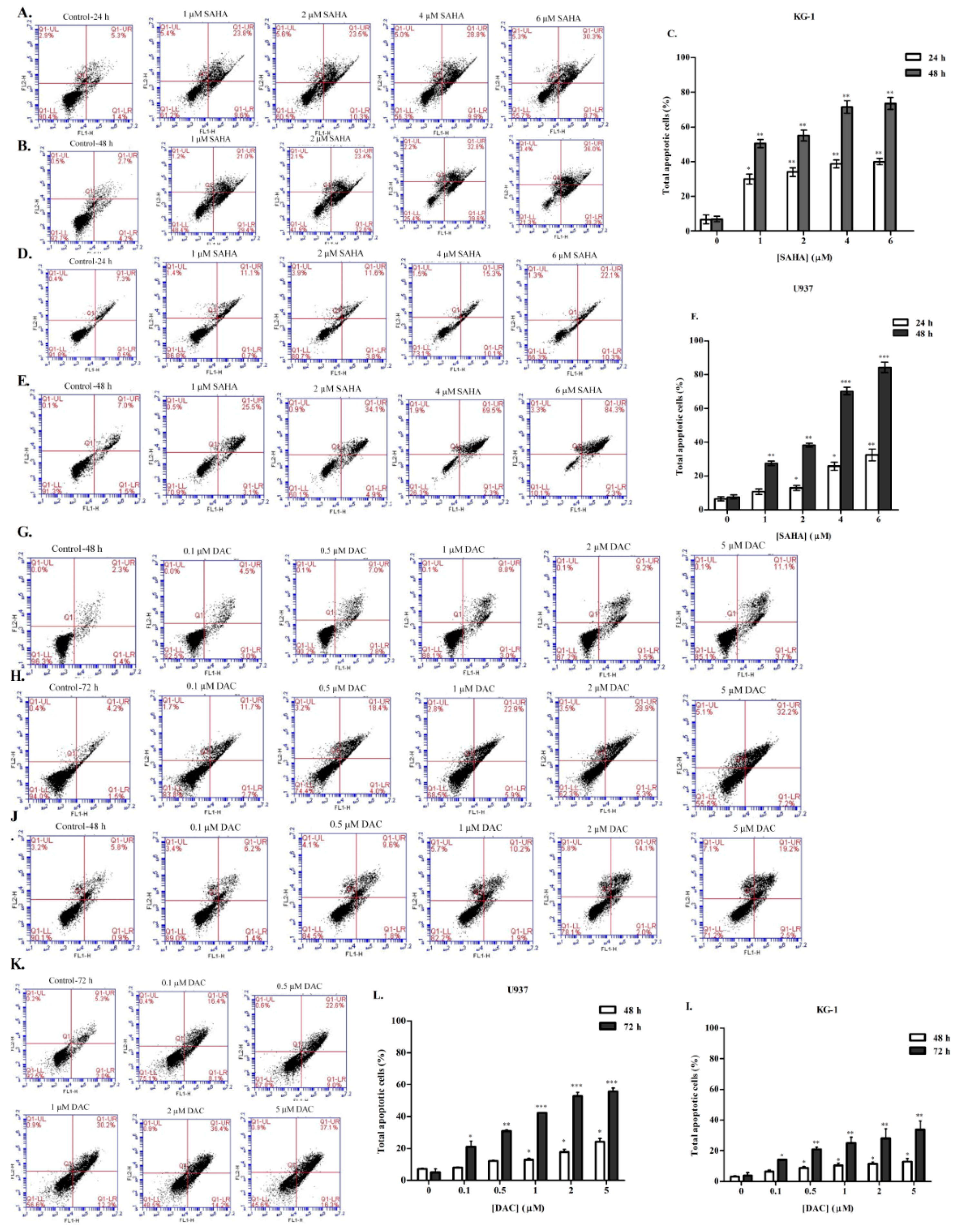
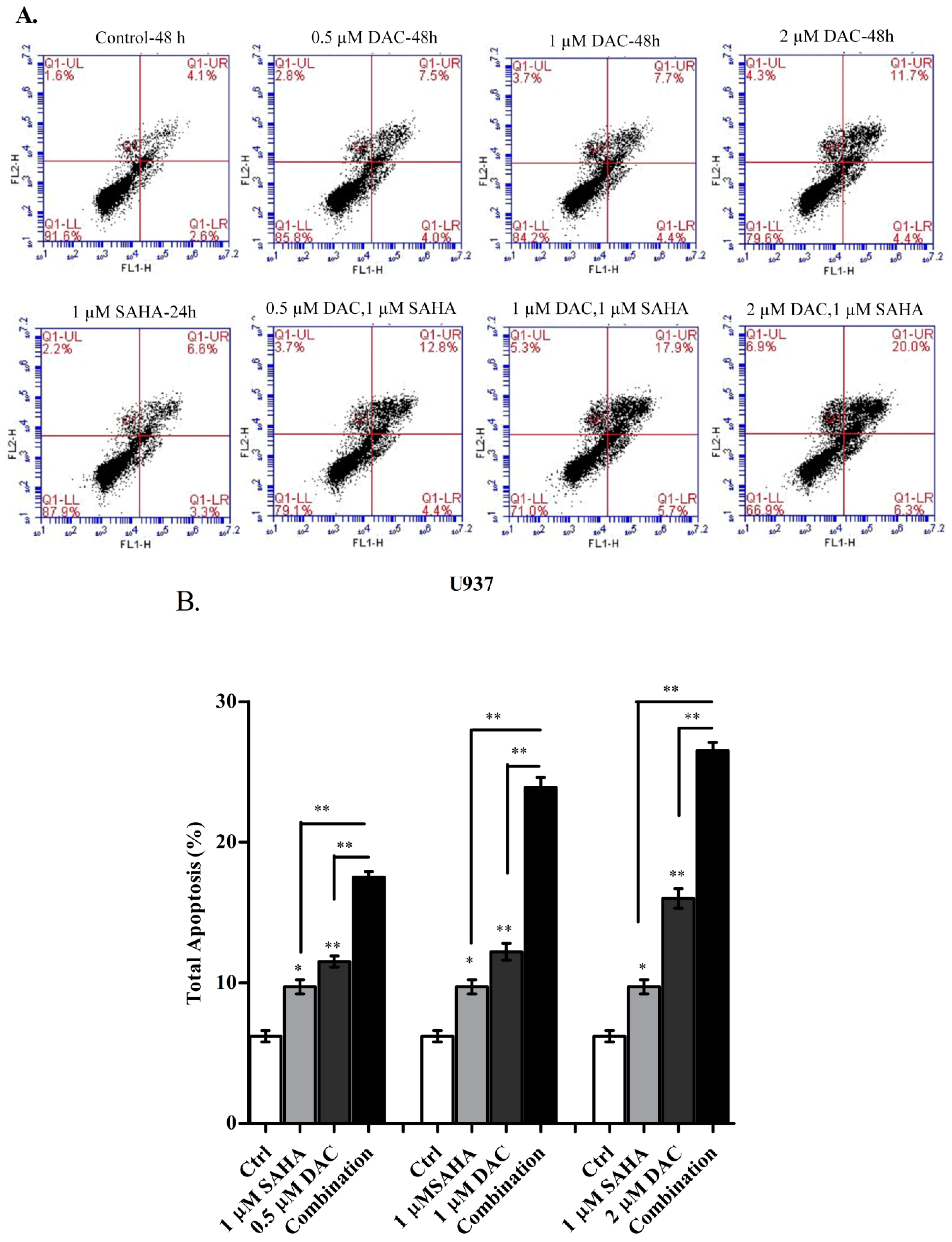
3.4. SAHA and DAC Induce Apoptosis of KG-1 and U937 Cells
3.5. Simultaneous Combination of SAHA and DAC Resulted in an Additive Cytotoxic Effect on KG-1 and U937 Cell Lines
3.6. Sequential Combination of SAHA and DAC Enhances Growth Inhibition of U937 Cell Line
3.7. The Sequential Addition of SAHA to DAC-Primed U937 Cells Expands the Apoptotic Population
3.8. Sequential Treatment with DAC and SAHA Diminished the Number of Replicating Cells and Escalated the Percentage of Dead U937 Cells
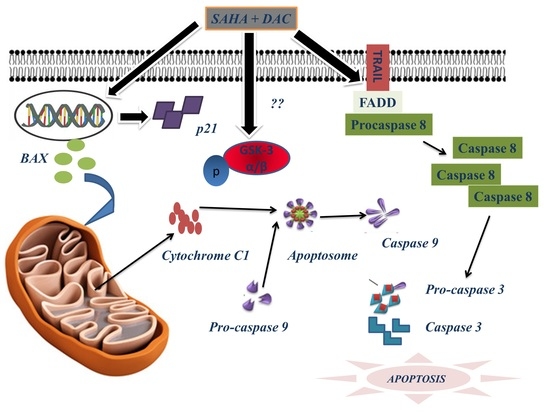
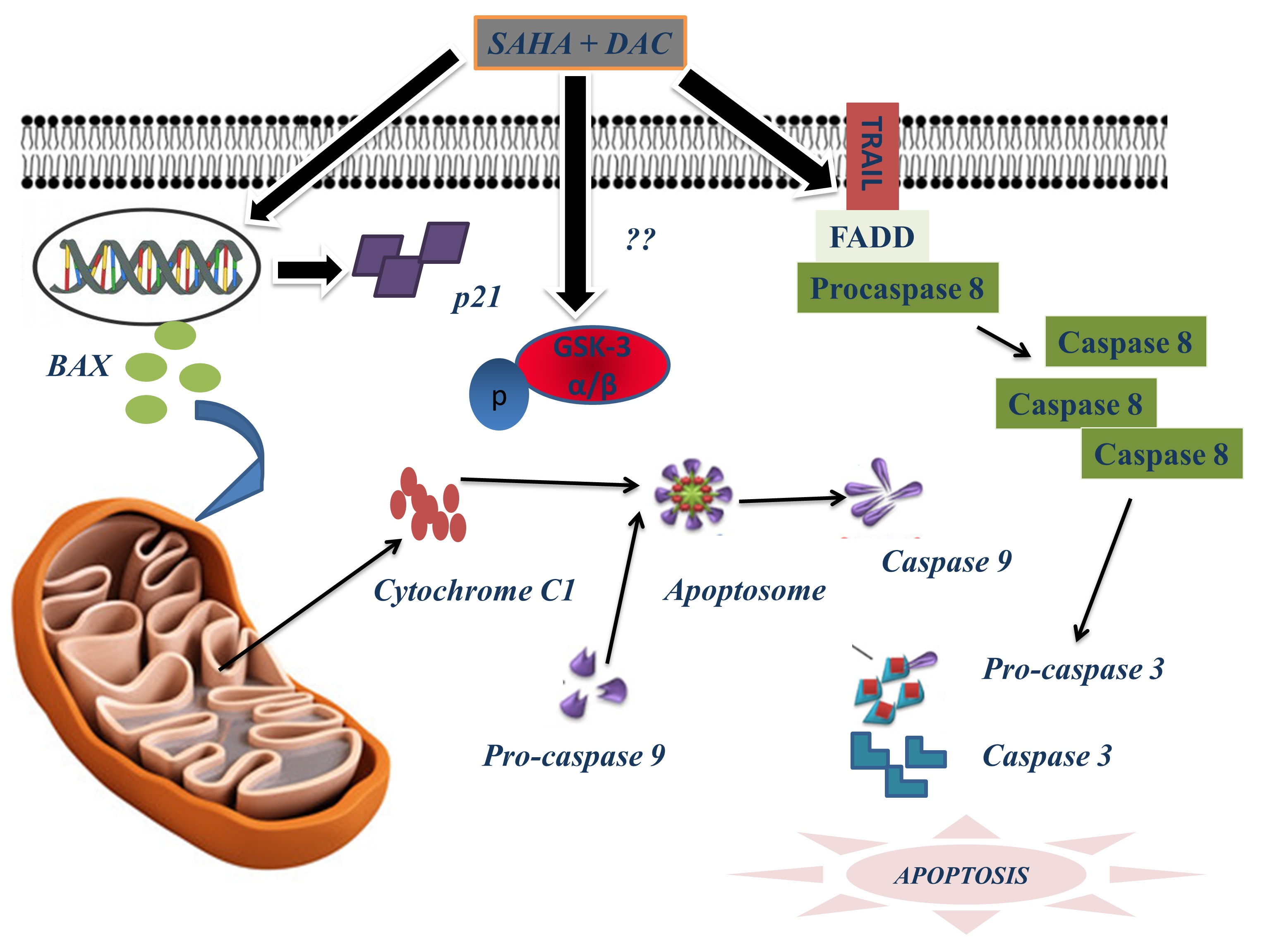
3.9. Sequential Combination of SAHA and DAC Upregulates Protein Expression of Bax, Cytochrome c1, and p21 and Increases the Cleavage of Procaspases 8, 9, and 3 in the U937 Cell Line
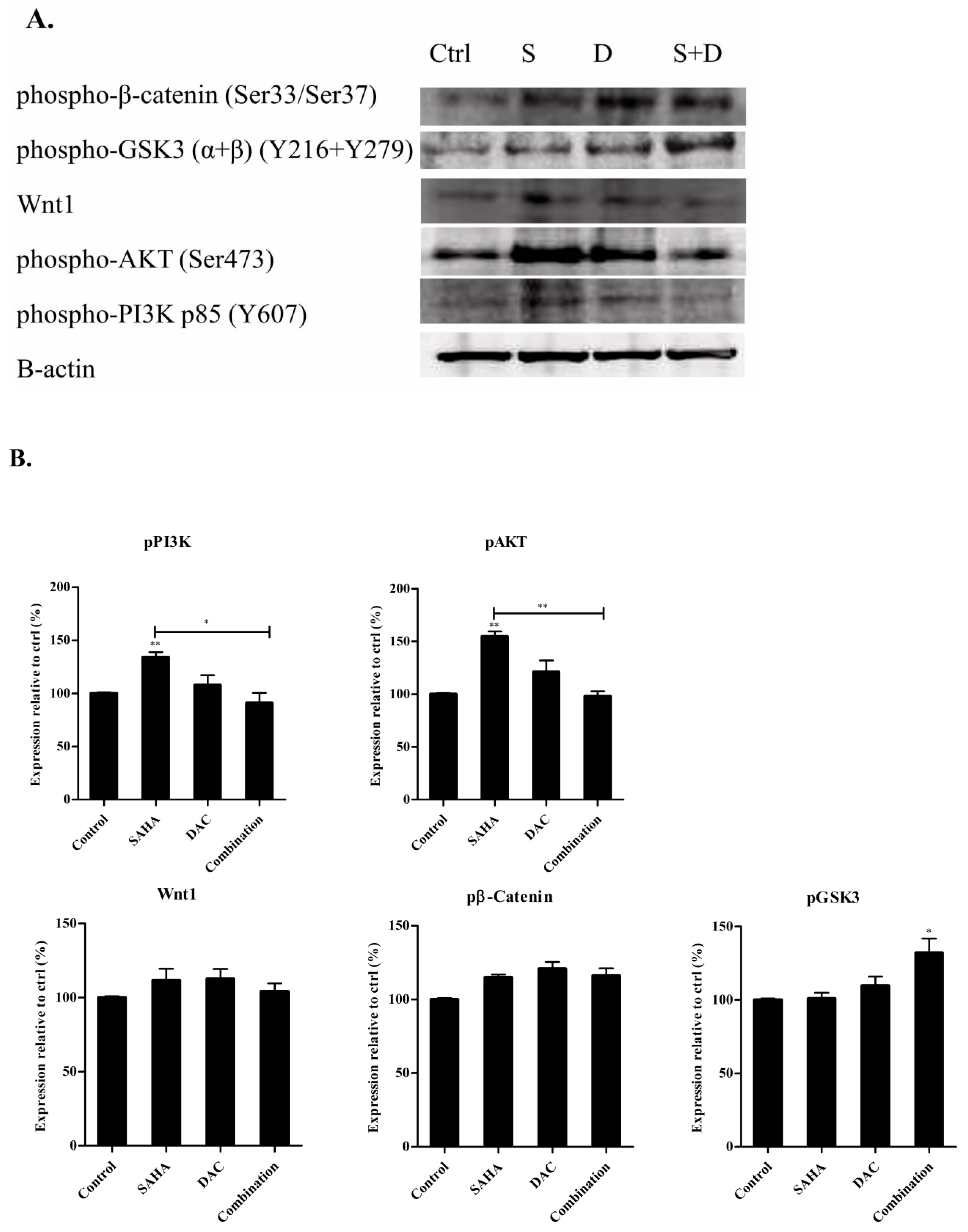
3.10. Sequential Combination of SAHA and DAC Upregulates Protein Expression of p-GSK3 in the U937 Cell Line
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Llinas-Arias, P.; Esteller, M. Epigenetic inactivation of tumour suppressor coding and non-coding genes in human cancer: An update. Open Biol. 2017, 7, 170152. [Google Scholar] [CrossRef] [PubMed]
- Meissner, A. Epigenetic modifications in pluripotent and differentiated cells. Nat. Biotechnol. 2010, 28, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suner, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef] [PubMed]
- Ropero, S.; Esteller, M. The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Glozak, M.A.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar] [CrossRef]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef]
- Di Cerbo, V.; Schneider, R. Cancers with wrong HATs: The impact of acetylation. Brief. Funct. Genom. 2013, 12, 231–243. [Google Scholar] [CrossRef]
- Baylin, S.B. DNA methylation and gene silencing in cancer. Nat. Clin. Pract. Oncol. 2005, 2, S4. [Google Scholar] [CrossRef]
- Silverman, L.R.; Mufti, G.J. Methylation inhibitor therapy in the treatment of myelodysplastic syndrome. Nat. Clin. Pract. Oncol. 2005, 2, S12. [Google Scholar] [CrossRef] [PubMed]
- Harikrishnan, K.N.; Chow, M.Z.; Baker, E.K.; Pal, S.; Bassal, S.; Brasacchio, D.; Wang, L.; Craig, J.M.; Jones, P.L.; Sif, S.; et al. Brahma links the SWI/SNF chromatin-remodeling complex with MeCP2-dependent transcriptional silencing. Nat. Genet. 2005, 37, 254–264. [Google Scholar] [PubMed]
- El-Osta, A.; Wolffe, A.P. DNA methylation and histone deacetylation in the control of gene expression: Basic biochemistry to human development and disease. Gene Expr. 2000, 9, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Ng, H.H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998, 393, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.N.; Torrisani, J.; Unterberger, A.; Provencal, N.; Shikimi, K.; Karimi, M.; Ekstrom, T.J.; Szyf, M. Histone deacetylase inhibitor Trichostatin A induces global and gene-specific DNA demethylation in human cancer cell lines. Biochem. Pharmacol. 2007, 73, 1297–1307. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhang, L.; Li, J.; Huang, J.; Wen, R.; Ma, L.; Zhou, D.; Li, L. Trichostatin A and 5-azacytidine both cause an increase in global histone H4 acetylation and a decrease in global DNA and H3K9 methylation during mitosis in maize. BMC Plant. Biol. 2010, 10, 178. [Google Scholar] [CrossRef]
- Wouters, B.J.; Delwel, R. Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood 2016, 127, 42–52. [Google Scholar] [CrossRef]
- Ferrara, F.; Schiffer, C.A. Acute myeloid leukaemia in adults. Lancet (Lond. Engl.) 2013, 381, 484–495. [Google Scholar] [CrossRef]
- Malik, P.; Cashen, A.F. Decitabine in the treatment of acute myeloid leukemia in elderly patients. Cancer Manag. Res. 2014, 6, 53–61. [Google Scholar]
- Plass, C.; Oakes, C.; Blum, W.; Marcucci, G. Epigenetics in Acute Myeloid Leukemia. Semin. Oncol. 2008, 35, 378–387. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Lugthart, S.; Li, Y.; Erpelinck-Verschueren, C.; Deng, X.; Christos, P.J.; Schifano, E.; Booth, J.; van Putten, W.; Skrabanek, L.; et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 2010, 17, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Rush, L.J.; Dai, Z.; Smiraglia, D.J.; Gao, X.; Wright, F.A.; Frühwald, M.; Costello, J.F.; Held, W.A.; Yu, L.; Krahe, R.; et al. Novel methylation targets in de novo acute myeloid leukemia with prevalence of chromosome 11 loci. Blood 2001, 97, 3226–3233. [Google Scholar] [CrossRef] [PubMed]
- Agrawal-Singh, S.; Isken, F.; Agelopoulos, K.; Klein, H.U.; Thoennissen, N.H.; Koehler, G.; Hascher, A.; Baumer, N.; Berdel, W.E.; Thiede, C.; et al. Genome-wide analysis of histone H3 acetylation patterns in AML identifies PRDX2 as an epigenetically silenced tumor suppressor gene. Blood 2012, 119, 2346–2357. [Google Scholar] [CrossRef] [PubMed]
- Hollenbach, P.W.; Nguyen, A.N.; Brady, H.; Williams, M.; Ning, Y.; Richard, N.; Krushel, L.; Aukerman, S.L.; Heise, C.; MacBeth, K.J. A comparison of azacitidine and decitabine activities in acute myeloid leukemia cell lines. PLoS ONE 2010, 5, e9001. [Google Scholar] [CrossRef] [PubMed]
- Flotho, C.; Claus, R.; Batz, C.; Schneider, M.; Sandrock, I.; Ihde, S.; Plass, C.; Niemeyer, C.M.; Lubbert, M. The DNA methyltransferase inhibitors azacitidine, decitabine and zebularine exert differential effects on cancer gene expression in acute myeloid leukemia cells. Leukemia 2009, 23, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Fredly, H.; Gjertsen, B.T.; Bruserud, Ø. Histone deacetylase inhibition in the treatment of acute myeloid leukemia: The effects of valproic acid on leukemic cells, and the clinical and experimental evidence for combining valproic acid with other antileukemic agents. Clin. Epigenetics 2013, 5, 12. [Google Scholar] [CrossRef]
- Liu, S.; Klisovic, R.B.; Vukosavljevic, T.; Yu, J.; Paschka, P.; Huynh, L.; Pang, J.; Neviani, P.; Liu, Z.; Blum, W.; et al. Targeting AML1/ETO-histone deacetylase repressor complex: A novel mechanism for valproic acid-mediated gene expression and cellular differentiation in AML1/ETO-positive acute myeloid leukemia cells. J. Pharmacol. Exp. Ther. 2007, 321, 953–960. [Google Scholar] [CrossRef]
- Zhu, X.; Liu, X.; Cheng, Z.; Zhu, J.; Xu, L.; Wang, F.; Qi, W.; Yan, J.; Liu, N.; Sun, Z.; et al. Quantitative Analysis of Global Proteome and Lysine Acetylome Reveal the Differential Impacts of VPA and SAHA on HL60 Cells. Sci. Rep. 2016, 6, 19926. [Google Scholar] [CrossRef]
- Silva, G.; Cardoso, B.A.; Belo, H.; Almeida, A.M. Vorinostat induces apoptosis and differentiation in myeloid malignancies: Genetic and molecular mechanisms. PLoS ONE 2013, 8, e53766. [Google Scholar] [CrossRef]
- Olsen, E.A.; Kim, Y.H.; Kuzel, T.M.; Pacheco, T.R.; Foss, F.M.; Parker, S.; Frankel, S.R.; Chen, C.; Ricker, J.L.; Arduino, J.M.; et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 2007, 25, 3109–3115. [Google Scholar] [CrossRef]
- Petruccelli, L.A.; Dupere-Richer, D.; Pettersson, F.; Retrouvey, H.; Skoulikas, S.; Miller, W.H., Jr. Vorinostat induces reactive oxygen species and DNA damage in acute myeloid leukemia cells. PLoS ONE 2011, 6, e20987. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- Young, C.S.; Clarke, K.M.; Kettyle, L.M.; Thompson, A.; Mills, K.I. Decitabine-Vorinostat combination treatment in acute myeloid leukemia activates pathways with potential for novel triple therapy. Oncotarget 2017, 8, 51429–51446. [Google Scholar] [CrossRef] [PubMed]
- Kentsis, A.; Look, A.T. Distinct and dynamic requirements for mTOR signaling in hematopoiesis and leukemogenesis. Cell Stem Cell 2012, 11, 281–282. [Google Scholar] [CrossRef][Green Version]
- Kharas, M.G.; Okabe, R.; Ganis, J.J.; Gozo, M.; Khandan, T.; Paktinat, M.; Gilliland, D.G.; Gritsman, K. Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood 2010, 115, 1406–1415. [Google Scholar] [CrossRef]
- Tamburini, J.; Elie, C.; Bardet, V.; Chapuis, N.; Park, S.; Broet, P.; Cornillet-Lefebvre, P.; Lioure, B.; Ugo, V.; Blanchet, O.; et al. Constitutive phosphoinositide 3-kinase/Akt activation represents a favorable prognostic factor in de novo acute myelogenous leukemia patients. Blood 2007, 110, 1025–1028. [Google Scholar] [CrossRef]
- Chapuis, N.; Park, S.; Leotoing, L.; Tamburini, J.; Verdier, F.; Bardet, V.; Green, A.S.; Willems, L.; Agou, F.; Ifrah, N.; et al. IκB kinase overcomes PI3K/Akt and ERK/MAPK to control FOXO3a activity in acute myeloid leukemia. Blood 2010, 116, 4240–4250. [Google Scholar] [CrossRef]
- Morgan, R.G.; Pearn, L.; Liddiard, K.; Pumford, S.L.; Burnett, A.K.; Tonks, A.; Darley, R.L. gamma-Catenin is overexpressed in acute myeloid leukemia and promotes the stabilization and nuclear localization of beta-catenin. Leukemia 2013, 27, 336–343. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA: A Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Sun, Y.; Chen, B.R.; Deshpande, A. Epigenetic Regulators in the Development, Maintenance, and Therapeutic Targeting of Acute Myeloid Leukemia. Front. Oncol. 2018, 8, 41. [Google Scholar] [CrossRef]
- Mizuno, S.; Chijiwa, T.; Okamura, T.; Akashi, K.; Fukumaki, Y.; Niho, Y.; Sasaki, H. Expression of DNA methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemia. Blood 2001, 97, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.P.; Baylin, S.B.; Herman, J.G. DNA methylation changes in hematologic malignancies: Biologic and clinical implications. Leukemia 1997, 11 (Suppl. 1), S7–S11. [Google Scholar]
- Fahrner, J.A.; Eguchi, S.; Herman, J.G.; Baylin, S.B. Dependence of histone modifications and gene expression on DNA hypermethylation in cancer. Cancer Res. 2002, 62, 7213–7218. [Google Scholar] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Stapnes, C.; Ryningen, A.; Hatfield, K.; Oyan, A.M.; Eide, G.E.; Corbascio, M.; Kalland, K.H.; Gjertsen, B.T.; Bruserud, O. Functional characteristics and gene expression profiles of primary acute myeloid leukaemia cells identify patient subgroups that differ in susceptibility to histone deacetylase inhibitors. Int. J. Oncol. 2007, 31, 1529–1538. [Google Scholar] [CrossRef]
- Brodska, B.; Holoubek, A.; Otevrelova, P.; Kuzelova, K. Combined treatment with low concentrations of decitabine and SAHA causes cell death in leukemic cell lines but not in normal peripheral blood lymphocytes. BioMed Res. Int. 2013, 2013, 659254. [Google Scholar] [CrossRef][Green Version]
- Jiemjit, A.; Fandy, T.E.; Carraway, H.; Bailey, K.A.; Baylin, S.; Herman, J.G.; Gore, S.D. p21(WAF1/CIP1) induction by 5-azacytosine nucleosides requires DNA damage. Oncogene 2008, 27, 3615–3623. [Google Scholar] [CrossRef]
- Jones, P.A.; Taylor, S.M. Cellular differentiation, cytidine analogs and DNA methylation. Cell 1980, 20, 85–93. [Google Scholar] [CrossRef]
- Momparler, R.L. Pharmacology of 5-Aza-2’-deoxycytidine (decitabine). Semin. Hematol. 2005, 42, S9–S16. [Google Scholar] [CrossRef]
- Soncini, M.; Santoro, F.; Gutierrez, A.; Frige, G.; Romanenghi, M.; Botrugno, O.A.; Pallavicini, I.; Pelicci, P.; Di Croce, L.; Minucci, S. The DNA demethylating agent decitabine activates the TRAIL pathway and induces apoptosis in acute myeloid leukemia. Biochim. Et Biophys. Acta 2013, 1832, 114–120. [Google Scholar] [CrossRef]
- Steinhart, L.; Belz, K.; Fulda, S. Smac mimetic and demethylating agents synergistically trigger cell death in acute myeloid leukemia cells and overcome apoptosis resistance by inducing necroptosis. Cell Death Dis. 2013, 4, e802. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Cheng, Y.; You, L.; Qian, J.; Qian, W. Homoharringtonine and SAHA synergistically enhance apoptosis in human acute myeloid leukemia cells through upregulation of TRAIL and death receptors. Mol. Med. Rep. 2013, 7, 1838–1844. [Google Scholar] [CrossRef] [PubMed]
- Bewersdorf, J.P.; Shallis, R.; Stahl, M.; Zeidan, A.M. Epigenetic therapy combinations in acute myeloid leukemia: What are the options? Ther. Adv. Hematol. 2019, 10, 2040620718816698. [Google Scholar] [CrossRef] [PubMed]
- Abou Najem, S.; Khawaja, G.; Hodroj, M.H.; Rizk, S. Synergistic Effect of Epigenetic Inhibitors Decitabine and Suberoylanilide Hydroxamic Acid on Colorectal Cancer in vitro. Curr. Mol. Pharmacol. 2019, 12, 281–300. [Google Scholar] [CrossRef]
- Chen, M.Y.; Liao, W.S.; Lu, Z.; Bornmann, W.G.; Hennessey, V.; Washington, M.N.; Rosner, G.L.; Yu, Y.; Ahmed, A.A.; Bast, R.C., Jr. Decitabine and suberoylanilide hydroxamic acid (SAHA) inhibit growth of ovarian cancer cell lines and xenografts while inducing expression of imprinted tumor suppressor genes, apoptosis, G2/M arrest, and autophagy. Cancer 2011, 117, 4424–4438. [Google Scholar] [CrossRef]
- Teodoridis, J.M.; Hall, J.; Marsh, S.; Kannall, H.D.; Smyth, C.; Curto, J.; Siddiqui, N.; Gabra, H.; McLeod, H.L.; Strathdee, G.; et al. CpG island methylation of DNA damage response genes in advanced ovarian cancer. Cancer Res. 2005, 65, 8961–8967. [Google Scholar] [CrossRef]
- Steele, N.; Finn, P.; Brown, R.; Plumb, J.A. Combined inhibition of DNA methylation and histone acetylation enhances gene re-expression and drug sensitivity in vivo. Br. J. Cancer 2009, 100, 758–763. [Google Scholar] [CrossRef]
- Appleton, K.; Mackay, H.J.; Judson, I.; Plumb, J.A.; McCormick, C.; Strathdee, G.; Lee, C.; Barrett, S.; Reade, S.; Jadayel, D.; et al. Phase I and pharmacodynamic trial of the DNA methyltransferase inhibitor decitabine and carboplatin in solid tumors. J. Clin. Oncol. 2007, 25, 4603–4609. [Google Scholar] [CrossRef]
- Yang, H.; Hoshino, K.; Sanchez-Gonzalez, B.; Kantarjian, H.; Garcia-Manero, G. Antileukemia activity of the combination of 5-aza-2’-deoxycytidine with valproic acid. Leuk. Res. 2005, 29, 739–748. [Google Scholar] [CrossRef]
- Takam Kamga, P.; Cassaro, A.; Dal Collo, G.; Adamo, A.; Gatti, A.; Carusone, R.; Midolo, M.; Di Trapani, M.; Resci, F.; Bonifacio, M.; et al. Role of Wnt/β-Catenin Signalling in Acute Myeloid Leukemia (AML) Cell Response to Chemotherapy. Blood 2016, 128, 2753. [Google Scholar] [CrossRef]
- Gandillet, A.; Park, S.; Lassailly, F.; Griessinger, E.; Vargaftig, J.; Filby, A.; Lister, T.A.; Bonnet, D. Heterogeneous sensitivity of human acute myeloid leukemia to β-catenin down-modulation. Leukemia 2011, 25, 770–780. [Google Scholar] [CrossRef] [PubMed]












© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abou Najem, S.; Khawaja, G.; Hodroj, M.H.; Babikian, P.; Rizk, S. Adjuvant Epigenetic Therapy of Decitabine and Suberoylanilide Hydroxamic Acid Exerts Anti-Neoplastic Effects in Acute Myeloid Leukemia Cells. Cells 2019, 8, 1480. https://doi.org/10.3390/cells8121480
Abou Najem S, Khawaja G, Hodroj MH, Babikian P, Rizk S. Adjuvant Epigenetic Therapy of Decitabine and Suberoylanilide Hydroxamic Acid Exerts Anti-Neoplastic Effects in Acute Myeloid Leukemia Cells. Cells. 2019; 8(12):1480. https://doi.org/10.3390/cells8121480
Chicago/Turabian StyleAbou Najem, Sonia, Ghada Khawaja, Mohammad Hassan Hodroj, Patil Babikian, and Sandra Rizk. 2019. "Adjuvant Epigenetic Therapy of Decitabine and Suberoylanilide Hydroxamic Acid Exerts Anti-Neoplastic Effects in Acute Myeloid Leukemia Cells" Cells 8, no. 12: 1480. https://doi.org/10.3390/cells8121480
APA StyleAbou Najem, S., Khawaja, G., Hodroj, M. H., Babikian, P., & Rizk, S. (2019). Adjuvant Epigenetic Therapy of Decitabine and Suberoylanilide Hydroxamic Acid Exerts Anti-Neoplastic Effects in Acute Myeloid Leukemia Cells. Cells, 8(12), 1480. https://doi.org/10.3390/cells8121480