Molecular Insights into the Mechanism of Necroptosis: The Necrosome as a Potential Therapeutic Target


Abstract
1. Introduction
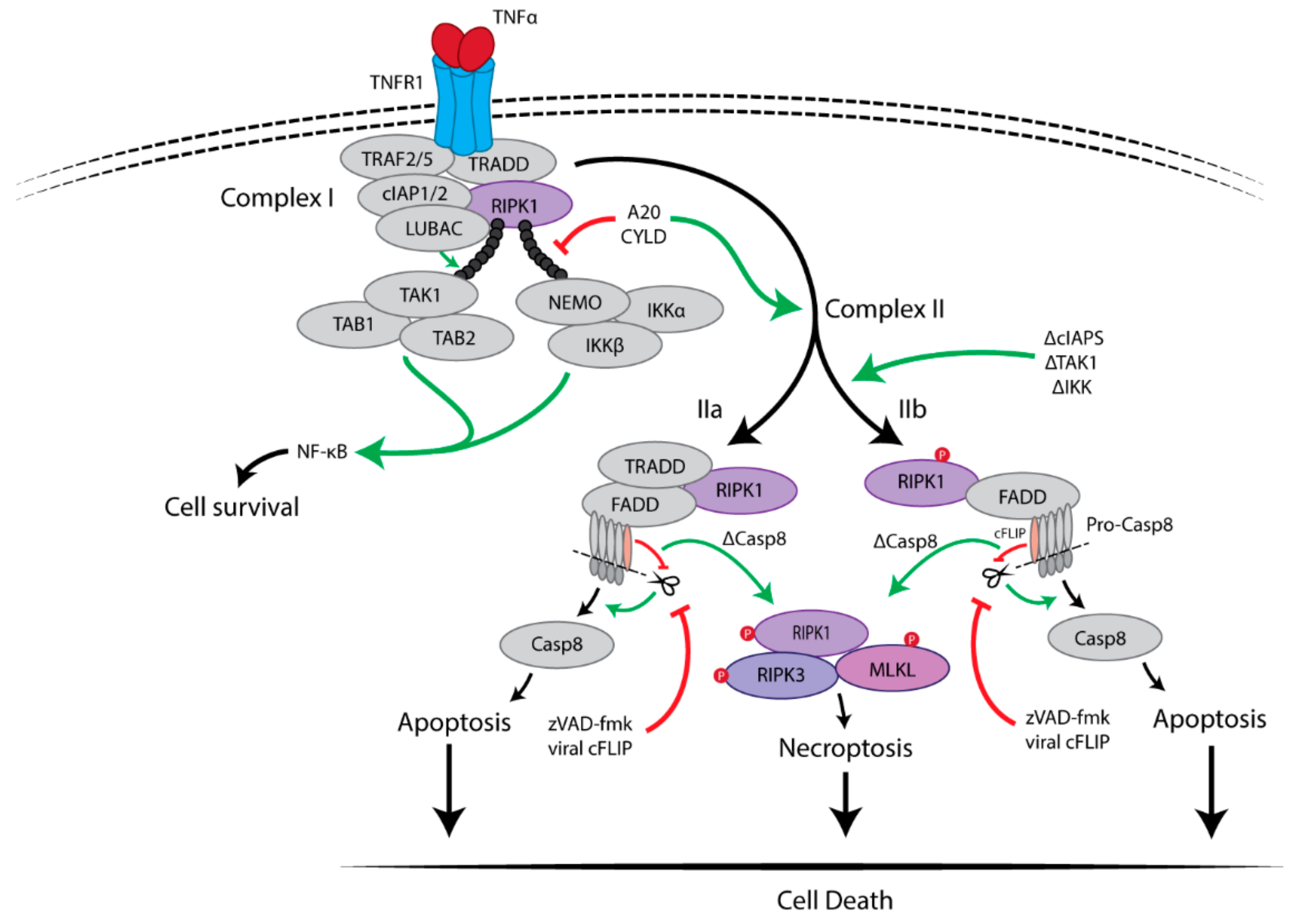
2. Necroptosis Induction
2.1. Pro-Survival Signalling Complex I
2.2. Cell Death-Inducing Signalling Complex II
Inhibition of Caspase-8 Activity Is Essential for Necroptosis
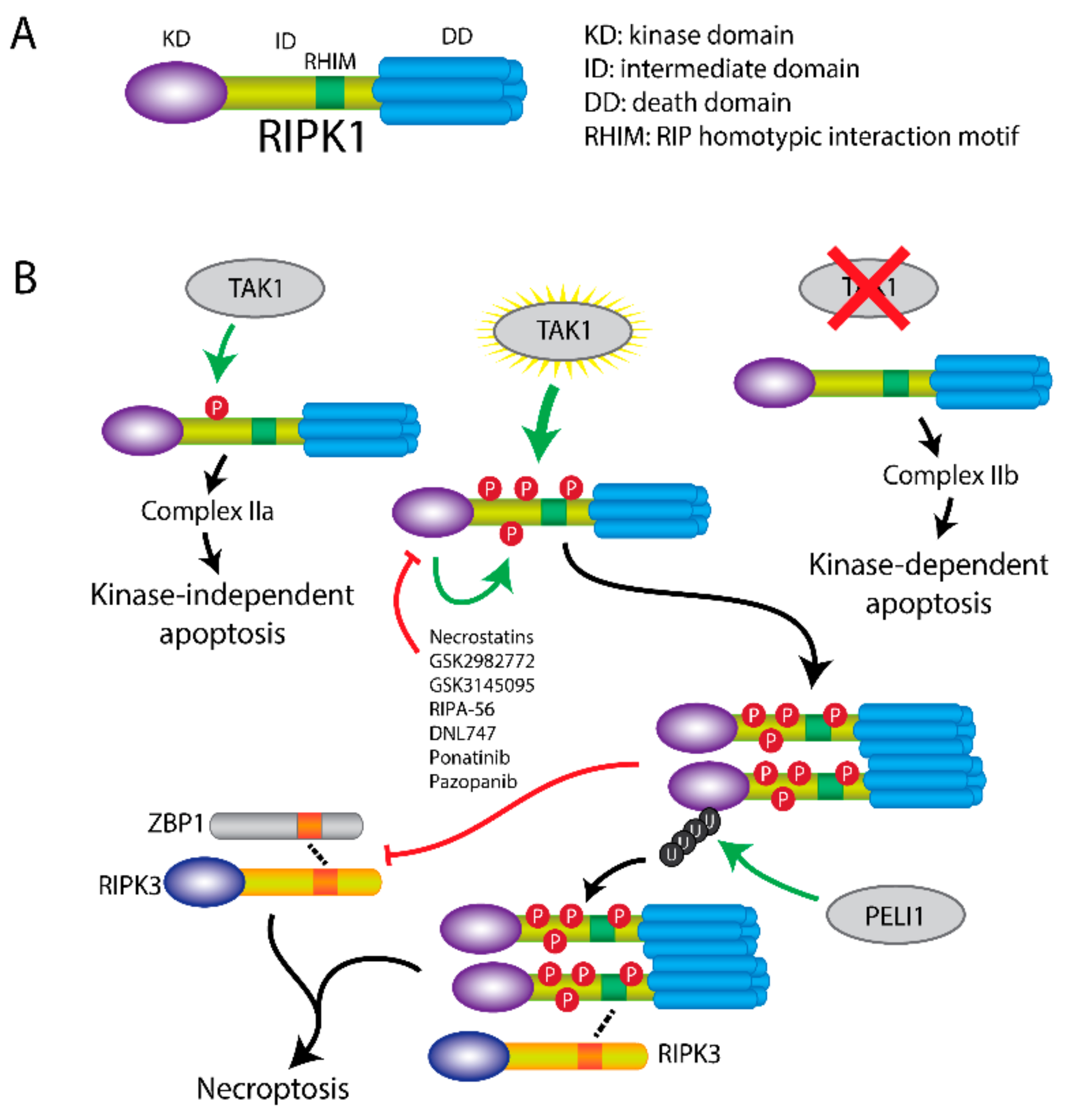
3. RIPK1 Has Multiple Functions
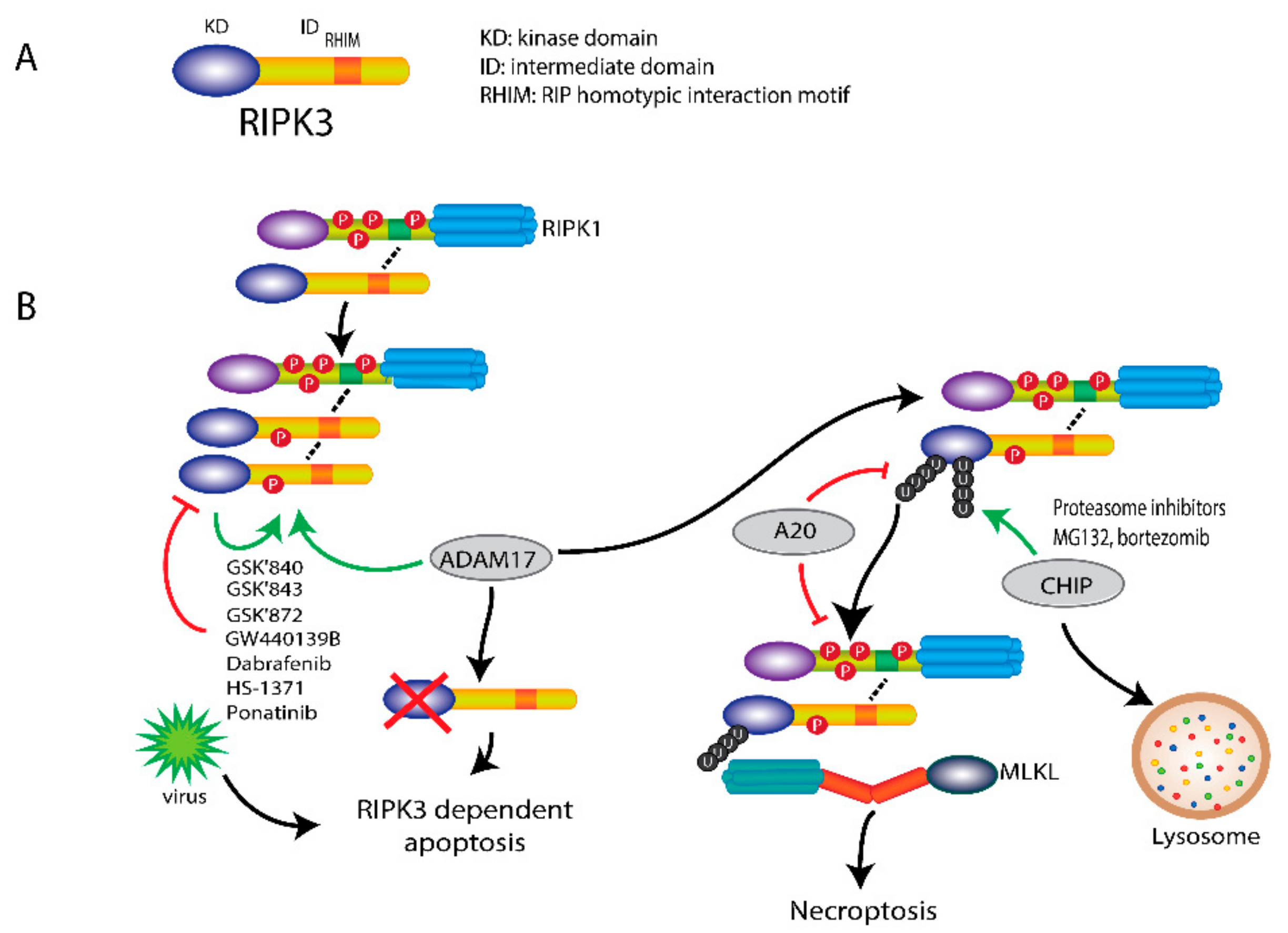
4. RIPK3 Plays a Key Role in Necroptosis
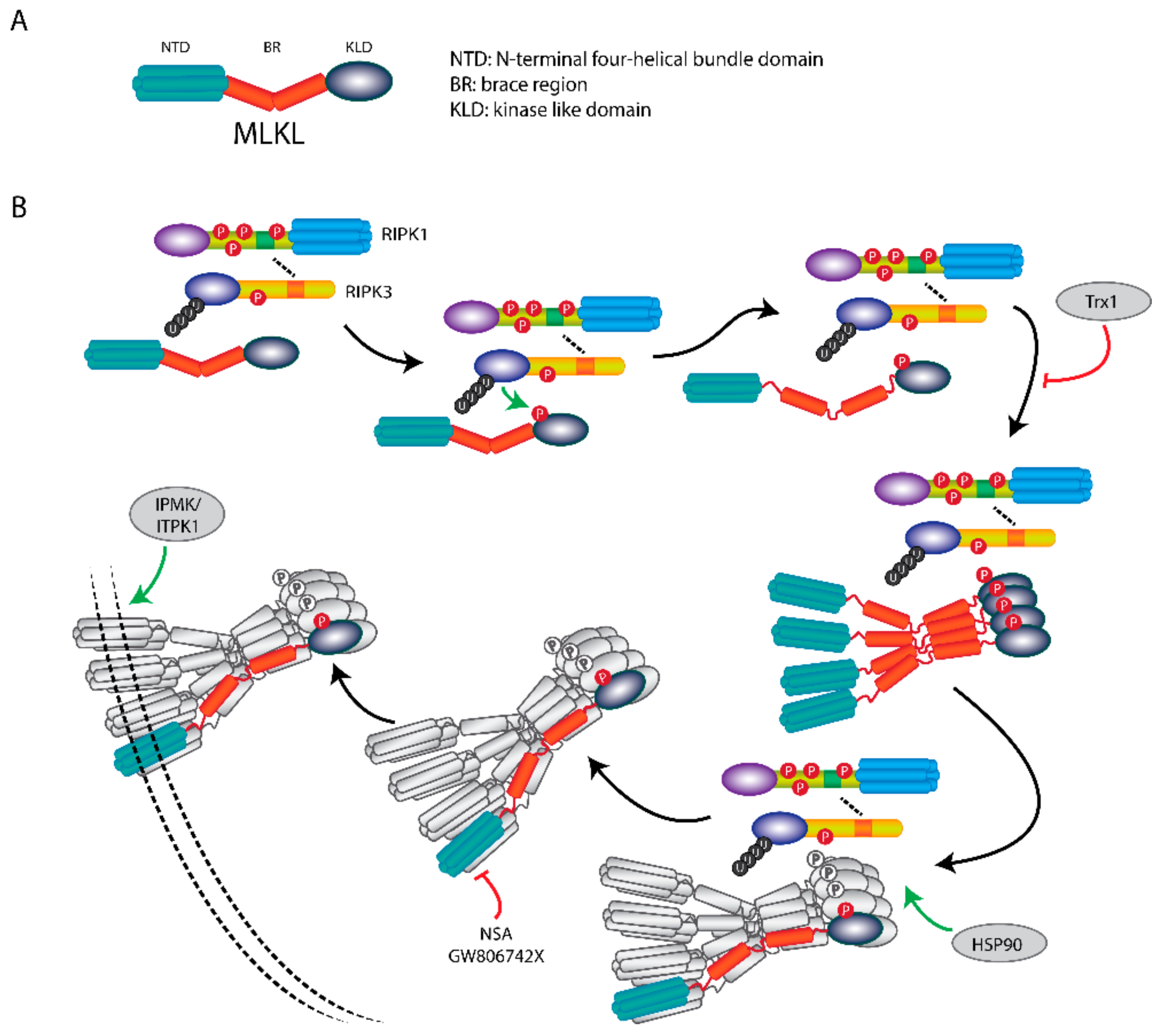
5. MLKL Is the Effector Protein in Necroptosis
6. The Necroptotic Pathway as Therapeutic Target
6.1. Necroptosis in Neurodegenerative Diseases
6.2. Necroptosis in Rheumatoid Arthritis
6.3. Necroptosis in Kidney Injury
6.4. Necroptosis in Circulatory System Diseases
6.5. Necroptosis in Diabetes
6.6. Necroptosis in Cancers
7. Conclusions
Funding
Conflicts of Interest
References
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe 2010, 7, 302–313. [Google Scholar] [CrossRef]
- de Almagro, M.C.; Vucic, D. Necroptosis: Pathway diversity and characteristics. Semin. Cell Dev. Biol. 2015, 39, 56–62. [Google Scholar] [CrossRef]
- Chan, F.K.; Luz, N.F.; Moriwaki, K. Programmed necrosis in the cross talk of cell death and inflammation. Annu. Rev. Immunol. 2015, 33, 79–106. [Google Scholar] [CrossRef]
- McQuade, T.; Cho, Y.; Chan, F.K. Positive and negative phosphorylation regulates RIP1- and RIP3-induced programmed necrosis. Biochem. J. 2013, 456, 409–415. [Google Scholar] [CrossRef]
- Grootjans, S.; Vanden Berghe, T.; Vandenabeele, P. Initiation and execution mechanisms of necroptosis: An overview. Cell Death Differ. 2017, 24, 1184–1195. [Google Scholar] [CrossRef]
- Silke, J.; Rickard, J.A.; Gerlic, M. The diverse role of RIP kinases in necroptosis and inflammation. Nat. Immunol. 2015, 16, 689–697. [Google Scholar] [CrossRef]
- Riegler, A.N.; Brissac, T.; Gonzalez-Juarbe, N.; Orihuela, C.J. Necroptotic Cell Death Promotes Adaptive Immunity Against Colonizing Pneumococci. Front. Immunol. 2019, 10, 615. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Chan, F.K.; Kroemer, G. Necroptosis: Mechanisms and Relevance to Disease. Annu. Rev. Pathol. 2017, 12, 103–130. [Google Scholar] [CrossRef]
- Koo, G.B.; Morgan, M.J.; Lee, D.G.; Kim, W.J.; Yoon, J.H.; Koo, J.S.; Kim, S.I.; Kim, S.J.; Son, M.K.; Hong, S.S.; et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 2015, 25, 707–725. [Google Scholar] [CrossRef]
- Hockendorf, U.; Yabal, M.; Herold, T.; Munkhbaatar, E.; Rott, S.; Jilg, S.; Kauschinger, J.; Magnani, G.; Reisinger, F.; Heuser, M.; et al. RIPK3 Restricts Myeloid Leukemogenesis by Promoting Cell Death and Differentiation of Leukemia Initiating Cells. Cancer Cell 2016, 30, 75–91. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Hsu, H.; Huang, J.; Shu, H.B.; Baichwal, V.; Goeddel, D.V. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity 1996, 4, 387–396. [Google Scholar] [CrossRef]
- Csomos, R.A.; Brady, G.F.; Duckett, C.S. Enhanced cytoprotective effects of the inhibitor of apoptosis protein cellular IAP1 through stabilization with TRAF2. J. Biol. Chem. 2009, 284, 20531–20539. [Google Scholar] [CrossRef]
- Ea, C.K.; Deng, L.; Xia, Z.P.; Pineda, G.; Chen, Z.J. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol. Cell 2006, 22, 245–257. [Google Scholar] [CrossRef]
- Li, H.; Kobayashi, M.; Blonska, M.; You, Y.; Lin, X. Ubiquitination of RIP is required for tumor necrosis factor alpha-induced NF-kappaB activation. J. Biol. Chem. 2006, 281, 13636–13643. [Google Scholar] [CrossRef]
- Dondelinger, Y.; Jouan-Lanhouet, S.; Divert, T.; Theatre, E.; Bertin, J.; Gough, P.J.; Giansanti, P.; Heck, A.J.; Dejardin, E.; Vandenabeele, P.; et al. NF-kappaB-Independent Role of IKKalpha/IKKbeta in Preventing RIPK1 Kinase-Dependent Apoptotic and Necroptotic Cell Death during TNF Signaling. Mol. Cell 2015, 60, 63–76. [Google Scholar] [CrossRef]
- Dondelinger, Y.; Delanghe, T.; Priem, D.; Wynosky-Dolfi, M.A.; Sorobetea, D.; Rojas-Rivera, D.; Giansanti, P.; Roelandt, R.; Gropengiesser, J.; Ruckdeschel, K.; et al. Serine 25 phosphorylation inhibits RIPK1 kinase-dependent cell death in models of infection and inflammation. Nat. Commun. 2019, 10, 1729. [Google Scholar] [CrossRef]
- Amin, P.; Florez, M.; Najafov, A.; Pan, H.; Geng, J.; Ofengeim, D.; Dziedzic, S.A.; Wang, H.; Barrett, V.J.; Ito, Y.; et al. Regulation of a distinct activated RIPK1 intermediate bridging complex I and complex II in TNFalpha-mediated apoptosis. Proc. Natl. Acad. Sci. USA 2018, 115, E5944–E5953. [Google Scholar] [CrossRef]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef]
- Wang, L.; Du, F.; Wang, X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008, 133, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Feoktistova, M.; Geserick, P.; Kellert, B.; Dimitrova, D.P.; Langlais, C.; Hupe, M.; Cain, K.; MacFarlane, M.; Hacker, G.; Leverkus, M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 2011, 43, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.M.; Li, Y.; Lu, A.; Li, Z.; Vajjhala, P.R.; Cruz, A.C.; Srivastava, D.B.; DiMaio, F.; Penczek, P.A.; Siegel, R.M.; et al. Cryo-EM Structure of Caspase-8 Tandem DED Filament Reveals Assembly and Regulation Mechanisms of the Death-Inducing Signaling Complex. Mol. Cell 2016, 64, 236–250. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.A.; Powley, I.R.; Jukes-Jones, R.; Horn, S.; Feoktistova, M.; Fairall, L.; Schwabe, J.W.; Leverkus, M.; Cain, K.; MacFarlane, M. Co-operative and Hierarchical Binding of c-FLIP and Caspase-8: A Unified Model Defines How c-FLIP Isoforms Differentially Control Cell Fate. Mol. Cell 2016, 61, 834–849. [Google Scholar] [CrossRef] [PubMed]
- Irmler, M.; Thome, M.; Hahne, M.; Schneider, P.; Hofmann, K.; Steiner, V.; Bodmer, J.L.; Schroter, M.; Burns, K.; Mattmann, C.; et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997, 388, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Dillon, C.P.; Oberst, A.; Weinlich, R.; Janke, L.J.; Kang, T.B.; Ben-Moshe, T.; Mak, T.W.; Wallach, D.; Green, D.R. Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Rep. 2012, 1, 401–407. [Google Scholar] [CrossRef]
- Zhang, D.; Lin, J.; Han, J. Receptor-interacting protein (RIP) kinase family. Cell. Mol. Immunol. 2010, 7, 243–249. [Google Scholar] [CrossRef]
- Duprez, L.; Bertrand, M.J.; Vanden Berghe, T.; Dondelinger, Y.; Festjens, N.; Vandenabeele, P. Intermediate domain of receptor-interacting protein kinase 1 (RIPK1) determines switch between necroptosis and RIPK1 kinase-dependent apoptosis. J. Biol. Chem. 2012, 287, 14863–14872. [Google Scholar] [CrossRef]
- Geng, J.; Ito, Y.; Shi, L.; Amin, P.; Chu, J.; Ouchida, A.T.; Mookhtiar, A.K.; Zhao, H.; Xu, D.; Shan, B.; et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat. Commun. 2017, 8, 359. [Google Scholar] [CrossRef]
- Shutinoski, B.; Alturki, N.A.; Rijal, D.; Bertin, J.; Gough, P.J.; Schlossmacher, M.G.; Sad, S. K45A mutation of RIPK1 results in poor necroptosis and cytokine signaling in macrophages, which impacts inflammatory responses in vivo. Cell Death Differ. 2016, 23, 1628–1637. [Google Scholar] [CrossRef]
- Polykratis, A.; Hermance, N.; Zelic, M.; Roderick, J.; Kim, C.; Van, T.M.; Lee, T.H.; Chan, F.K.M.; Pasparakis, M.; Kelliher, M.A. Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J. Immunol. 2014, 193, 1539–1543. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Dugger, D.L.; Wickliffe, K.E.; Kapoor, N.; de Almagro, M.C.; Vucic, D.; Komuves, L.; Ferrando, R.E.; French, D.M.; Webster, J.; et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 2014, 343, 1357–1360. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Liu, Z.; Li, X.; Wang, H.; Jin, T.; Wu, G.; Shan, B.; Christofferson, D.E.; Qi, C.; Yu, Q.; et al. Death-domain dimerization-mediated activation of RIPK1 controls necroptosis and RIPK1-dependent apoptosis. Proc. Natl. Acad. Sci. USA 2018, 115, E2001–E2009. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Meng, H.; Li, X.; Zhu, K.; Dong, K.; Mookhtiar, A.K.; Wei, H.; Li, Y.; Sun, S.C.; Yuan, J. PELI1 functions as a dual modulator of necroptosis and apoptosis by regulating ubiquitination of RIPK1 and mRNA levels of c-FLIP. Proc. Natl. Acad. Sci. USA 2017, 114, 11944–11949. [Google Scholar] [CrossRef]
- Wegner, K.W.; Saleh, D.; Degterev, A. Complex Pathologic Roles of RIPK1 and RIPK3: Moving Beyond Necroptosis. Trends Pharmacol. Sci. 2017, 38, 202–225. [Google Scholar] [CrossRef]
- Kearney, C.J.; Cullen, S.P.; Clancy, D.; Martin, S.J. RIPK1 can function as an inhibitor rather than an initiator of RIPK3-dependent necroptosis. FEBS J. 2014, 281, 4921–4934. [Google Scholar] [CrossRef]
- Lin, J.; Kumari, S.; Kim, C.; Van, T.M.; Wachsmuth, L.; Polykratis, A.; Pasparakis, M. RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature 2016, 540, 124–128. [Google Scholar] [CrossRef]
- Newton, K.; Wickliffe, K.E.; Maltzman, A.; Dugger, D.L.; Strasser, A.; Pham, V.C.; Lill, J.R.; Roose-Girma, M.; Warming, S.; Solon, M.; et al. RIPK1 inhibits ZBP1-driven necroptosis during development. Nature 2016, 540, 129–133. [Google Scholar] [CrossRef]
- Suda, J.; Dara, L.; Yang, L.; Aghajan, M.; Song, Y.; Kaplowitz, N.; Liu, Z.X. Knockdown of RIPK1 Markedly Exacerbates Murine Immune-Mediated Liver Injury through Massive Apoptosis of Hepatocytes, Independent of Necroptosis and Inhibition of NF-kappaB. J. Immunol. 2016, 197, 3120–3129. [Google Scholar] [CrossRef]
- Ren, Y.; Su, Y.; Sun, L.; He, S.; Meng, L.; Liao, D.; Liu, X.; Ma, Y.; Liu, C.; Li, S.; et al. Discovery of a Highly Potent, Selective, and Metabolically Stable Inhibitor of Receptor-Interacting Protein 1 (RIP1) for the Treatment of Systemic Inflammatory Response Syndrome. J. Med. Chem. 2017, 60, 972–986. [Google Scholar] [CrossRef]
- Caccamo, A.; Branca, C.; Piras, I.S.; Ferreira, E.; Huentelman, M.J.; Liang, W.S.; Readhead, B.; Dudley, J.T.; Spangenberg, E.E.; Green, K.N.; et al. Necroptosis activation in Alzheimer’s disease. Nat. Neurosci. 2017, 20, 1236–1246. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.A.; Berger, S.B.; Jeong, J.U.; Nagilla, R.; Bandyopadhyay, D.; Campobasso, N.; Capriotti, C.A.; Cox, J.A.; Dare, L.; Dong, X.; et al. Discovery of a First-in-Class Receptor Interacting Protein 1 (RIP1) Kinase Specific Clinical Candidate (GSK2982772) for the Treatment of Inflammatory Diseases. J. Med. Chem. 2017, 60, 1247–1261. [Google Scholar] [CrossRef] [PubMed]
- Fauster, A.; Rebsamen, M.; Huber, K.V.; Bigenzahn, J.W.; Stukalov, A.; Lardeau, C.H.; Scorzoni, S.; Bruckner, M.; Gridling, M.; Parapatics, K.; et al. A cellular screen identifies ponatinib and pazopanib as inhibitors of necroptosis. Cell Death Dis. 2015, 6, e1767. [Google Scholar] [CrossRef] [PubMed]
- Nogusa, S.; Thapa, R.J.; Dillon, C.P.; Liedmann, S.; Oguin III, T.H.; Ingram, J.P.; Rodriguez, D.A.; Kosoff, R.; Sharma, S.; Sturm, O.; et al. RIPK3 Activates Parallel Pathways of MLKL-Driven Necroptosis and FADD-Mediated Apoptosis to Protect against Influenza A Virus. Cell Host Microbe 2016, 20, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, W.J.; Sridharan, H.; Huang, C.; Mandal, P.; Upton, J.W.; Gough, P.J.; Sehon, C.A.; Marquis, R.W.; Bertin, J.; Mocarski, E.S. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 2013, 288, 31268–31279. [Google Scholar] [CrossRef] [PubMed]
- Li, J.X.; Feng, J.M.; Wang, Y.; Li, X.H.; Chen, X.X.; Su, Y.; Shen, Y.Y.; Chen, Y.; Xiong, B.; Yang, C.H.; et al. The B-Raf(V600E) inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury. Cell Death Dis. 2014, 5, e1278. [Google Scholar] [CrossRef]
- Park, H.H.; Park, S.Y.; Mah, S.; Park, J.H.; Hong, S.S.; Hong, S.; Kim, Y.S. HS-1371, a novel kinase inhibitor of RIP3-mediated necroptosis. Exp. Mol. Med. 2018, 50, 125. [Google Scholar] [CrossRef]
- Yan, B.; Liu, L.; Huang, S.; Ren, Y.; Wang, H.; Yao, Z.; Li, L.; Chen, S.; Wang, X.; Zhang, Z. Discovery of a new class of highly potent necroptosis inhibitors targeting the mixed lineage kinase domain-like protein. Chem. Commun. 2017, 53, 3637–3640. [Google Scholar] [CrossRef]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef]
- Jacobsen, A.V.; Lowes, K.N.; Tanzer, M.C.; Lucet, I.S.; Hildebrand, J.M.; Petrie, E.J.; van Delft, M.F.; Liu, Z.; Conos, S.A.; Zhang, J.G.; et al. HSP90 activity is required for MLKL oligomerisation and membrane translocation and the induction of necroptotic cell death. Cell Death Dis. 2016, 7, e2051. [Google Scholar] [CrossRef]
- Fuchslocher Chico, J.; Falk-Paulsen, M.; Luzius, A.; Saggau, C.; Ruder, B.; Bolik, J.; Schmidt-Arras, D.; Linkermann, A.; Becker, C.; Rosenstiel, P.; et al. The enhanced susceptibility of ADAM-17 hypomorphic mice to DSS-induced colitis is not ameliorated by loss of RIPK3, revealing an unexpected function of ADAM-17 in necroptosis. Oncotarget 2018, 9, 12941–12958. [Google Scholar] [CrossRef] [PubMed]
- Onizawa, M.; Oshima, S.; Schulze-Topphoff, U.; Oses-Prieto, J.A.; Lu, T.; Tavares, R.; Prodhomme, T.; Duong, B.; Whang, M.I.; Advincula, R.; et al. The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat. Immunol. 2015, 16, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Lee, E.W.; Sung, H.; Seong, D.; Dondelinger, Y.; Shin, J.; Jeong, M.; Lee, H.K.; Kim, J.H.; Han, S.Y.; et al. CHIP controls necroptosis through ubiquitylation- and lysosome-dependent degradation of RIPK3. Nat. Cell Biol. 2016, 18, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, K.; Chan, F.K. Regulation of RIPK3- and RHIM-dependent Necroptosis by the Proteasome. J. Biol. Chem. 2016, 291, 5948–5959. [Google Scholar] [CrossRef]
- Cook, W.D.; Moujalled, D.M.; Ralph, T.J.; Lock, P.; Young, S.N.; Murphy, J.M.; Vaux, D.L. RIPK1- and RIPK3-induced cell death mode is determined by target availability. Cell Death Differ. 2014, 21, 1600–1612. [Google Scholar] [CrossRef]
- Rodriguez, D.A.; Weinlich, R.; Brown, S.; Guy, C.; Fitzgerald, P.; Dillon, C.P.; Oberst, A.; Quarato, G.; Low, J.; Cripps, J.G.; et al. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 2016, 23, 76–88. [Google Scholar] [CrossRef]
- Murphy, J.M.; Czabotar, P.E.; Hildebrand, J.M.; Lucet, I.S.; Zhang, J.G.; Alvarez-Diaz, S.; Lewis, R.; Lalaoui, N.; Metcalf, D.; Webb, A.I.; et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 2013, 39, 443–453. [Google Scholar] [CrossRef]
- Dondelinger, Y.; Declercq, W.; Montessuit, S.; Roelandt, R.; Goncalves, A.; Bruggeman, I.; Hulpiau, P.; Weber, K.; Sehon, C.A.; Marquis, R.W.; et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014, 7, 971–981. [Google Scholar] [CrossRef]
- Hildebrand, J.M.; Tanzer, M.C.; Lucet, I.S.; Young, S.N.; Spall, S.K.; Sharma, P.; Pierotti, C.; Garnier, J.M.; Dobson, R.C.; Webb, A.I.; et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc. Natl. Acad. Sci. USA 2014, 111, 15072–15077. [Google Scholar] [CrossRef]
- Tanzer, M.C.; Tripaydonis, A.; Webb, A.I.; Young, S.N.; Varghese, L.N.; Hall, C.; Alexander, W.S.; Hildebrand, J.M.; Silke, J.; Murphy, J.M. Necroptosis signalling is tuned by phosphorylation of MLKL residues outside the pseudokinase domain activation loop. Biochem. J. 2015, 471, 255–265. [Google Scholar] [CrossRef]
- Quarato, G.; Guy, C.S.; Grace, C.R.; Llambi, F.; Nourse, A.; Rodriguez, D.A.; Wakefield, R.; Frase, S.; Moldoveanu, T.; Green, D.R. Sequential Engagement of Distinct MLKL Phosphatidylinositol-Binding Sites Executes Necroptosis. Mol. Cell 2016, 61, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, H.; Johnston, A.; Hanna-Addams, S.; Reynoso, E.; Xiang, Y.; Wang, Z. MLKL forms disulfide bond-dependent amyloid-like polymers to induce necroptosis. Proc. Natl. Acad. Sci. USA 2017, 114, E7450–E7459. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Zheng, X.; Wang, Z.A.; Chen, X.; He, W.T.; Zhang, Y.; Xu, J.G.; Zhao, H.; Shi, W.; Wang, X.; et al. The MLKL Channel in Necroptosis Is an Octamer Formed by Tetramers in a Dyadic Process. Mol. Cell. Biol. 2017, 37, e00497-16. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, W.; Ren, J.; Huang, D.; He, W.T.; Song, Y.; Yang, C.; Li, W.; Zheng, X.; Chen, P.; et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014, 24, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.G.; Liu, Z.G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Bogdanov, K.; Kovalenko, A.; Wallach, D. Necroptosis is preceded by nuclear translocation of the signaling proteins that induce it. Cell Death Differ. 2016, 23, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.; Roelandt, R.; Bruggeman, I.; Estornes, Y.; Vandenabeele, P. Nuclear RIPK3 and MLKL contribute to cytosolic necrosome formation and necroptosis. Commun. Biol. 2018, 1, 6. [Google Scholar] [CrossRef]
- Petrie, E.J.; Sandow, J.J.; Jacobsen, A.V.; Smith, B.J.; Griffin, M.D.W.; Lucet, I.S.; Dai, W.; Young, S.N.; Tanzer, M.C.; Wardak, A.; et al. Conformational switching of the pseudokinase domain promotes human MLKL tetramerization and cell death by necroptosis. Nat. Commun. 2018, 9, 2422. [Google Scholar] [CrossRef]
- Ma, B.; Marcotte, D.; Paramasivam, M.; Michelsen, K.; Wang, T.; Bertolotti-Ciarlet, A.; Jones, J.H.; Moree, B.; Butko, M.; Salafsky, J.; et al. ATP-Competitive MLKL Binders Have No Functional Impact on Necroptosis. PLoS ONE 2016, 11, e0165983. [Google Scholar] [CrossRef]
- Dovey, C.M.; Diep, J.; Clarke, B.P.; Hale, A.T.; McNamara, D.E.; Guo, H.; Brown, N.W., Jr.; Cao, J.Y.; Grace, C.R.; Gough, P.J.; et al. MLKL Requires the Inositol Phosphate Code to Execute Necroptosis. Mol. Cell 2018, 70, 936–948.e7. [Google Scholar] [CrossRef]
- Yoon, S.; Kovalenko, A.; Bogdanov, K.; Wallach, D. MLKL, the Protein that Mediates Necroptosis, Also Regulates Endosomal Trafficking and Extracellular Vesicle Generation. Immunity 2017, 47, 51–65.e57. [Google Scholar] [CrossRef] [PubMed]
- Reynoso, E.; Liu, H.; Li, L.; Yuan, A.L.; Chen, S.; Wang, Z. Thioredoxin-1 actively maintains the pseudokinase MLKL in a reduced state to suppress disulfide bond-dependent MLKL polymer formation and necroptosis. J. Biol. Chem. 2017, 292, 17514–17524. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.M.; Chen, Z.; Zhao, J.B.; Zhang, P.P.; Pu, Y.F.; Jiang, S.H.; Hou, J.J.; Cui, Y.M.; Jia, X.L.; Zhang, S.Q. Hsp90 modulates the stability of MLKL and is required for TNF-induced necroptosis. Cell Death Dis. 2016, 7, e2089. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.; Chang, H.Y.; Sang, T.K. Neuronal Cell Death Mechanisms in Major Neurodegenerative Diseases. Int. J. Mol. Sci. 2018, 19, 3082. [Google Scholar] [CrossRef]
- Ofengeim, D.; Mazzitelli, S.; Ito, Y.; DeWitt, J.P.; Mifflin, L.; Zou, C.; Das, S.; Adiconis, X.; Chen, H.; Zhu, H.; et al. RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E8788–E8797. [Google Scholar] [CrossRef]
- Ito, Y.; Ofengeim, D.; Najafov, A.; Das, S.; Saberi, S.; Li, Y.; Hitomi, J.; Zhu, H.; Chen, H.; Mayo, L.; et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 2016, 353, 603–608. [Google Scholar] [CrossRef]
- Iannielli, A.; Bido, S.; Folladori, L.; Segnali, A.; Cancellieri, C.; Maresca, A.; Massimino, L.; Rubio, A.; Morabito, G.; Caporali, L.; et al. Pharmacological Inhibition of Necroptosis Protects from Dopaminergic Neuronal Cell Death in Parkinson’s Disease Models. Cell Rep. 2018, 22, 2066–2079. [Google Scholar] [CrossRef]
- Lin, Q.S.; Chen, P.; Wang, W.X.; Lin, C.C.; Zhou, Y.; Yu, L.H.; Lin, Y.X.; Xu, Y.F.; Kang, D.Z. RIP1/RIP3/MLKL mediates dopaminergic neuron necroptosis in a mouse model of Parkinson disease. Lab. Invest. 2019. [Google Scholar] [CrossRef]
- Onate, M.; Catenaccio, A.; Salvadores, N.; Saquel, C.; Martinez, A.; Moreno-Gonzalez, I.; Gamez, N.; Soto, P.; Soto, C.; Hetz, C.; et al. The necroptosis machinery mediates axonal degeneration in a model of Parkinson disease. Cell Death Differ. 2019. [Google Scholar] [CrossRef]
- Ofengeim, D.; Ito, Y.; Najafov, A.; Zhang, Y.; Shan, B.; DeWitt, J.P.; Ye, J.; Zhang, X.; Chang, A.; Vakifahmetoglu-Norberg, H.; et al. Activation of necroptosis in multiple sclerosis. Cell Rep. 2015, 10, 1836–1849. [Google Scholar] [CrossRef]
- Zhang, S.; Su, Y.; Ying, Z.; Guo, D.; Pan, C.; Guo, J.; Zou, Z.; Wang, L.; Zhang, Z.; Jiang, Z.; et al. RIP1 kinase inhibitor halts the progression of an immune-induced demyelination disease at the stage of monocyte elevation. Proc. Natl. Acad. Sci. USA 2019, 116, 5675–5680. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Lv, X.; Hu, B.; Shao, Z.; Wang, B.; Ma, K.; Lin, H.; Cui, M. RIPK1/RIPK3/MLKL-mediated necroptosis contributes to compression-induced rat nucleus pulposus cells death. Apoptosis Int. J. Program. Cell Death 2017, 22, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kwon, J.Y.; Kim, S.Y.; Jung, K.; Cho, M.L. Interferon-gamma regulates inflammatory cell death by targeting necroptosis in experimental autoimmune arthritis. Sci. Rep. 2017, 7, 10133. [Google Scholar] [CrossRef] [PubMed]
- Jhun, J.; Lee, S.H.; Kim, S.Y.; Ryu, J.; Kwon, J.Y.; Na, H.S.; Jung, K.; Moon, S.J.; Cho, M.L.; Min, J.K. RIPK1 inhibition attenuates experimental autoimmune arthritis via suppression of osteoclastogenesis. J. Transl. Med. 2019, 17, 84. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Chen, B.; Wang, Y.; Meng, C.; Huang, H.; Huang, X.R.; Qin, J.; Mulay, S.R.; Anders, H.J.; Qiu, A.; et al. RGMb protects against acute kidney injury by inhibiting tubular cell necroptosis via an MLKL-dependent mechanism. Proc. Natl. Acad. Sci. USA 2018, 115, E1475–E1484. [Google Scholar] [CrossRef]
- Shen, B.; Mei, M.; Pu, Y.; Zhang, H.; Liu, H.; Tang, M.; Pan, Q.; He, Y.; Wu, X.; Zhao, H. Necrostatin-1 Attenuates Renal Ischemia and Reperfusion Injury via Meditation of HIF-1alpha/mir-26a/TRPC6/PARP1 Signaling. Mol. Ther. Nucleic Acids 2019, 17, 701–713. [Google Scholar] [CrossRef]
- Wang, J.N.; Liu, M.M.; Wang, F.; Wei, B.; Yang, Q.; Cai, Y.T.; Chen, X.; Liu, X.Q.; Jiang, L.; Li, C.; et al. RIPK1 inhibitor Cpd-71 attenuates renal dysfunction in cisplatin-treated mice via attenuating necroptosis, inflammation and oxidative stress. Clin. Sci. 2019, 133, 1609–1627. [Google Scholar] [CrossRef]
- Zhu, Y.; Cui, H.; Xia, Y.; Gan, H. RIPK3-Mediated Necroptosis and Apoptosis Contributes to Renal Tubular Cell Progressive Loss and Chronic Kidney Disease Progression in Rats. PLoS ONE 2016, 11, e0156729. [Google Scholar] [CrossRef]
- Mulay, S.R.; Desai, J.; Kumar, S.V.; Eberhard, J.N.; Thomasova, D.; Romoli, S.; Grigorescu, M.; Kulkarni, O.P.; Popper, B.; Vielhauer, V.; et al. Cytotoxicity of crystals involves RIPK3-MLKL-mediated necroptosis. Nat. Commun. 2016, 7, 10274. [Google Scholar] [CrossRef]
- Oerlemans, M.I.; Liu, J.; Arslan, F.; den Ouden, K.; van Middelaar, B.J.; Doevendans, P.A.; Sluijter, J.P. Inhibition of RIP1-dependent necrosis prevents adverse cardiac remodeling after myocardial ischemia-reperfusion in vivo. Basic Res. Cardiol. 2012, 107, 270. [Google Scholar] [CrossRef]
- Koshinuma, S.; Miyamae, M.; Kaneda, K.; Kotani, J.; Figueredo, V.M. Combination of necroptosis and apoptosis inhibition enhances cardioprotection against myocardial ischemia-reperfusion injury. J. Anesth. 2014, 28, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Hu, S.; Jin, Q.; Li, D.; Tian, F.; Toan, S.; Li, Y.; Zhou, H.; Chen, Y. Ripk3 promotes ER stress-induced necroptosis in cardiac IR injury: A mechanism involving calcium overload/XO/ROS/mPTP pathway. Redox Biol. 2018, 16, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, D.; Geoffrion, M.; Wei, L.; Gan, W.; Richards, L.; Shangari, P.; DeKemp, E.M.; Beanlands, R.A.; Perisic, L.; Maegdefessel, L.; et al. Targeting macrophage necroptosis for therapeutic and diagnostic interventions in atherosclerosis. Sci. Adv. 2016, 2, e1600224. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Li, H.; Yang, M.; Ren, J.; Huang, Z.; Han, F.; Huang, J.; Ma, J.; Zhang, D.; Zhang, Z.; et al. A role of RIP3-mediated macrophage necrosis in atherosclerosis development. Cell Rep. 2013, 3, 200–210. [Google Scholar] [CrossRef]
- Xu, H.; Du, X.; Liu, G.; Huang, S.; Du, W.; Zou, S.; Tang, D.; Fan, C.; Xie, Y.; Wei, Y.; et al. The pseudokinase MLKL regulates hepatic insulin sensitivity independently of inflammation. Mol. Metab. 2019, 23, 14–23. [Google Scholar] [CrossRef]
- Gautheron, J.; Vucur, M.; Schneider, A.T.; Severi, I.; Roderburg, C.; Roy, S.; Bartneck, M.; Schrammen, P.; Diaz, M.B.; Ehling, J.; et al. The necroptosis-inducing kinase RIPK3 dampens adipose tissue inflammation and glucose intolerance. Nat. Commun. 2016, 7, 11869. [Google Scholar] [CrossRef]
- Najafov, A.; Chen, H.; Yuan, J. Necroptosis and Cancer. Trends Cancer 2017, 3, 294–301. [Google Scholar] [CrossRef]
- Moriwaki, K.; Bertin, J.; Gough, P.J.; Orlowski, G.M.; Chan, F.K. Differential roles of RIPK1 and RIPK3 in TNF-induced necroptosis and chemotherapeutic agent-induced cell death. Cell Death Dis. 2015, 6, e1636. [Google Scholar] [CrossRef]
- Melo-Lima, S.; Celeste Lopes, M.; Mollinedo, F. Necroptosis is associated with low procaspase-8 and active RIPK1 and -3 in human glioma cells. Oncoscience 2014, 1, 649–664. [Google Scholar] [CrossRef]
- Zhou, J.; Du, X.; Chen, X.; Wang, J.; Zhou, N.; Wu, D.; Xu, B. Enzymatic Self-Assembly Confers Exceptionally Strong Synergism with NF-kappaB Targeting for Selective Necroptosis of Cancer Cells. J. Am. Chem. Soc. 2018, 140, 2301–2308. [Google Scholar] [CrossRef]
- Yao, C.; Li, G.; Cai, M.; Qian, Y.; Wang, L.; Xiao, L.; Thaiss, F.; Shi, B. Expression and genetic polymorphism of necroptosis related protein RIPK1 is correlated with severe hepatic ischemia-reperfusion injury and prognosis after hepatectomy in hepatocellular carcinoma patients. Cancer Biomark. Sect. A Dis. Markers 2017, 20, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Geserick, P.; Wang, J.; Schilling, R.; Horn, S.; Harris, P.A.; Bertin, J.; Gough, P.J.; Feoktistova, M.; Leverkus, M. Absence of RIPK3 predicts necroptosis resistance in malignant melanoma. Cell Death Dis. 2015, 6, e1884. [Google Scholar] [CrossRef]
- Bozec, D.; Iuga, A.C.; Roda, G.; Dahan, S.; Yeretssian, G. Critical function of the necroptosis adaptor RIPK3 in protecting from intestinal tumorigenesis. Oncotarget 2016, 7, 46384–46400. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.V.; Seibert, S.; Walch-Ruckheim, B.; Vicinus, B.; Kamionka, E.M.; Pahne-Zeppenfeld, J.; Solomayer, E.F.; Kim, Y.J.; Bohle, R.M.; Smola, S. RIPK3 expression in cervical cancer cells is required for PolyIC-induced necroptosis, IL-1alpha release, and efficient paracrine dendritic cell activation. Oncotarget 2015, 6, 8635–8647. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Apoptosis | Necroptosis | Necrosis |
|---|---|---|
| Regulated | Regulated | Unregulated |
| Triggered by specific cytokines | Triggered by specific cytokines | Induced by damaging environmental stress (such as extreme physiological stress and viral or toxin-mediated infections) |
| Shrinkage of cell | Rapid swelling of cell | Cell swelling |
| - | Swelling of organelles | Swelling of organelles |
| Blebbing of plasma membrane | Disruption of plasma membrane | Loss of membrane integrity |
| Formation of apoptotic bodies | Organelle breakdown | |
| - | Release of cellular contents | Release of cellular contents |
| - | Pro-inflammatory response | Pro-inflammatory response |
| Compound Name | Target | References |
|---|---|---|
| Necrostatins (necrostatin-1, 7-Cl-O-necrostatin) | RIPK1 | [1,41] |
| GSK2982772 | RIPK1 | [42] |
| GSK3145095 | RIPK1 | (Clinicaltrials.gov) |
| RIPA-56 | RIPK1 | [40] |
| Ponatinib | RIPK1/RIPK3 | [43] |
| Pazopanib | RIPK1 | [43] |
| DNL747 | RIPK1 | (Clinicaltrials.gov) |
| GSK’840, GSK’843 and GSK’872 | RIPK3 | [44,45] |
| Dabrafenib | RIPK3 | [46] |
| HS-1371 | RIPK3 | [47] |
| GW806742X | MLKL | [48] |
| Necrosulfonamide (NSA) | MLKL | [49] |
| 17AAG | HSP90 | [50] |
| Compound Name | Target | Phase | Disease Condition | Identifier | Study Type | Status (Oct 2019) |
|---|---|---|---|---|---|---|
| GSK2982772 | RIPK1 | II | Ulcerative colitis | NCT02903966 | With placebo | Completed |
| I | Rheumatoid arthritis | NCT02858492 | With placebo | Completed | ||
| II | Psoriasis | NCT02776033 | With placebo | Completed | ||
| GSK3145095 | I | Solid tumors | NCT03681951 | With Pembrolizumab | Terminated | |
| Dabrafenib | RIPK3 | IV | Melanoma Non-small cell lung cancer Solid tumor Rare cancers High grade glioma | NCT03340506 | With trametinib | Recruiting |
| II | Metastatic colorectal cancer | NCT03668431 | With trametinib and PDR001 | Recruiting | ||
| II | Melanoma | NCT01682213 | Single agent | Completed | ||
| II | BRAF Mutation-Positive Malignant Melanoma and Brain Metastases | NCT02974803 | With trametinib and stereotactic radiation | Active, not recruiting | ||
| II | Metastatic Melanoma (Carrying BRAF V600 Mutation) | NCT02052193 | With vemurafenib | Terminated | ||
| III | Malignant Melanoma | NCT03551626 | With trametinib | Recruiting | ||
| Ponatinib | RIPK/RIPK3 | Chronic Myeloid Leukemia (CML) Philadelphia Chromosome Positive Acute Lymphoblastic Leukemia (Ph+ ALL) | NCT01592136 | Expanded Access | Approved for marketing | |
| II | Non-small cell lung cancer | NCT01813734 | Single agent | Completed | ||
| II | Leukemia | NCT01570868 | Single agent | Terminated | ||
| II/III | Non-small cell lung cancer head and neck Cancer | NCT01761747 | Single agent | Terminated | ||
| I/II | Acute myeloid leukemia | NCT02428543 | With cytarabine | Recruiting | ||
| II | Medullary thyroid cancer | NCT03838692 | Single agent | Not yet recruiting | ||
| Pazopanib | RIPK1 | I | Renal cell carcinoma Soft tissue sarcoma Metastatic disease | NCT02795819 | With AR-42 | Terminated |
| II | Renal Cell carcinoma | NCT01545817 | Pazopanib followed by everolimus | Terminated | ||
| DNL747 | RIPK1 | I | AD | NCT03757325 | With Placebo | Recruiting |
| I | ALS | NCT03757351 | With Placebo | Recruiting | ||
| 17AAG | HSP90 | II | Anaplastic Large Cell Lymphoma Recurrent Adult Hodgkin Lymphoma Recurrent Mantle Cell Lymphoma | NCT00117988 | Single agent | Completed |
| I | Unspecified Adult Solid Tumor, Protocol Specific | NCT00121264 | With sorafenib tosylate | Completed | ||
| I | Relapsed or Refractory Hematologic Cancer | NCT00103272 | With bortezomib | Terminated | ||
| IPI-504 | HSP90 | I | Solid Tumors | NCT00606814 | With Docetaxel | Completed |
| I | Multiple Myeloma | NCT00113204 | Single agent | Completed | ||
| II | Prostate Cancer | NCT00564928 | Single agent | Completed |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Kos, R.; Garssen, J.; Redegeld, F. Molecular Insights into the Mechanism of Necroptosis: The Necrosome as a Potential Therapeutic Target. Cells 2019, 8, 1486. https://doi.org/10.3390/cells8121486
Chen J, Kos R, Garssen J, Redegeld F. Molecular Insights into the Mechanism of Necroptosis: The Necrosome as a Potential Therapeutic Target. Cells. 2019; 8(12):1486. https://doi.org/10.3390/cells8121486
Chicago/Turabian StyleChen, Jing, Renate Kos, Johan Garssen, and Frank Redegeld. 2019. "Molecular Insights into the Mechanism of Necroptosis: The Necrosome as a Potential Therapeutic Target" Cells 8, no. 12: 1486. https://doi.org/10.3390/cells8121486
APA StyleChen, J., Kos, R., Garssen, J., & Redegeld, F. (2019). Molecular Insights into the Mechanism of Necroptosis: The Necrosome as a Potential Therapeutic Target. Cells, 8(12), 1486. https://doi.org/10.3390/cells8121486