A New Sensitive Method for the Detection of Mycoplasmas Using Fluorescence Microscopy

 ,
,
Abstract
:1. Introduction
- -
- histological staining
- -
- electron microscopy detection
- -
- biochemical methods (e.g., assays based on enzyme activity measurement)
- -
- immunological methods (e.g., ELISA or fluorescence detection in situ)
- -
- staining by DNA dyes (DAPI, Hoechst 33258)
- -
- microbiological approaches (agar/broth media assay)
- -
- hybridization (e.g., filter or liquid hybridization)
- -
- polymerase chain reaction (PCR) and real-time PCR
2. Materials and Methods
2.1. Cell Cultures
2.2. Mycoplasma Species, Inoculation of Cell Line with the Mycoplasma Strains
2.3. Bacterial Strains
2.4. Mycoplasma Detection by the Developed Approach
2.5. Nuclease Treatment
2.6. Alternative Approaches of Mycoplasma Detection
2.6.1. DAPI and Hoechst Staining
2.6.2. PCR Detection
2.6.3. MycoAlertTM PLUS Mycoplasma Detection Kit
2.7. Fluorescence Microscopy
2.8. Data Evaluation
3. Results and Discussion
3.1. Method Description
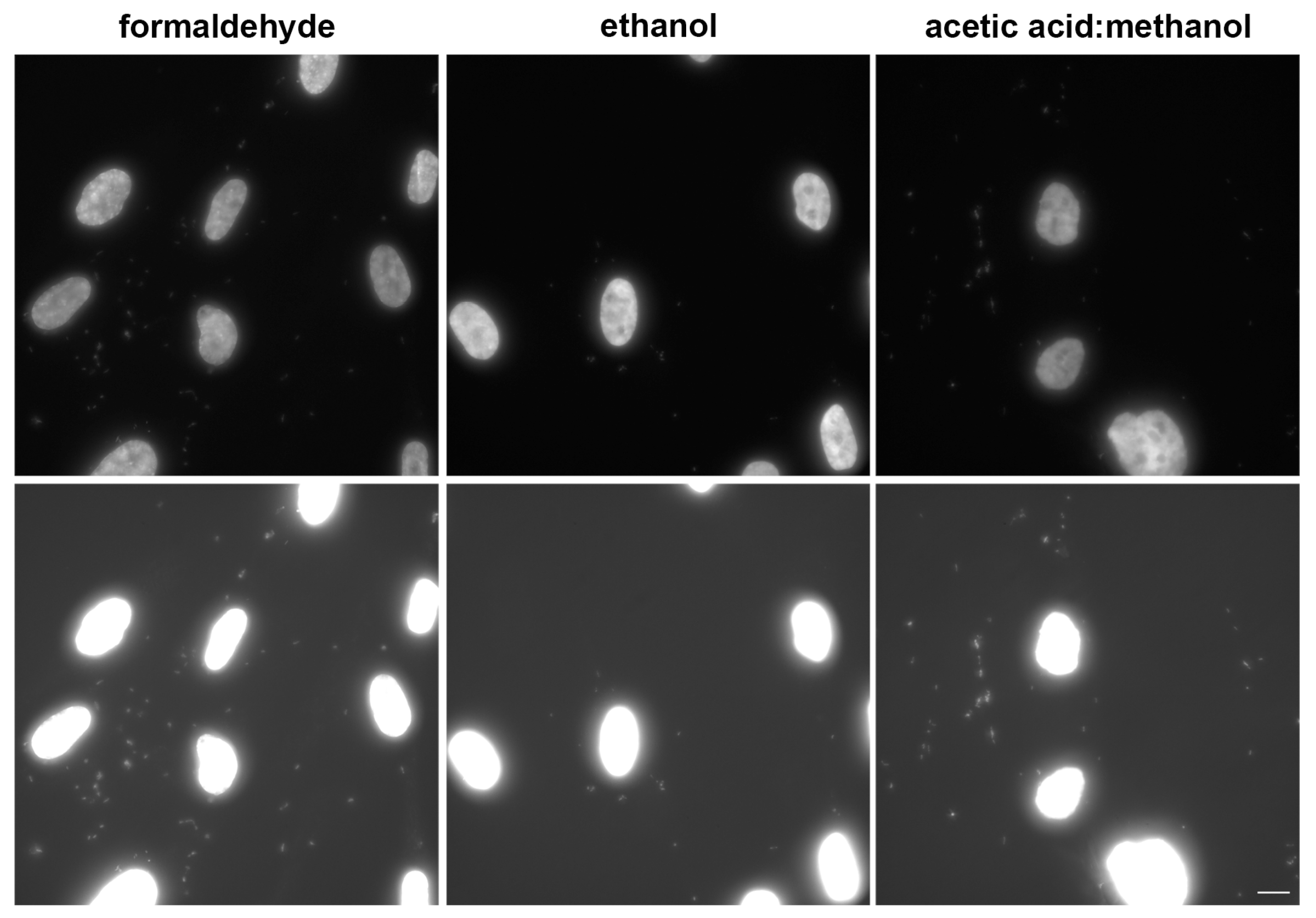
3.2. Procedure Optimisation
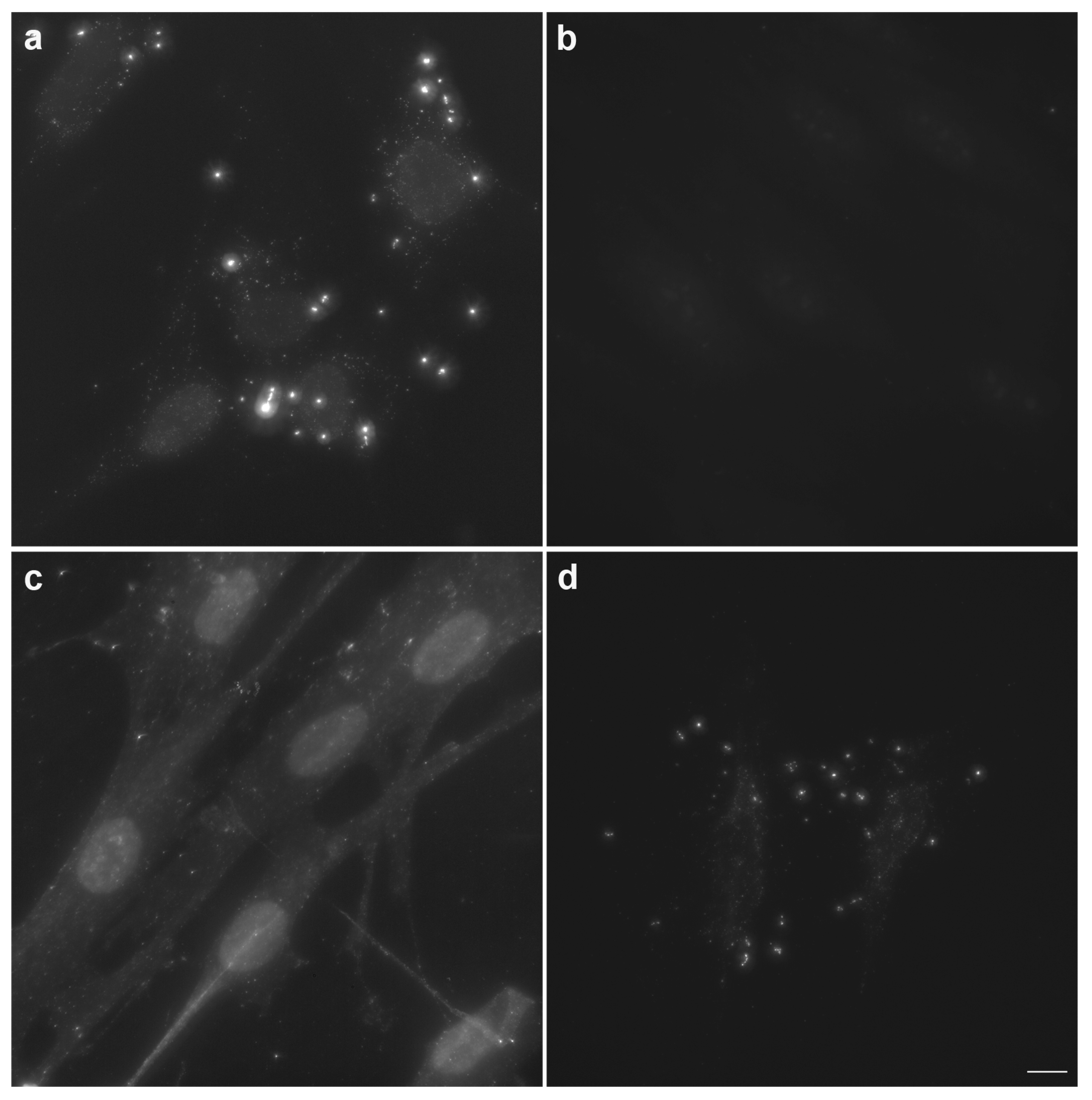
3.3. Labeling of Bacterial Cells Using the Developed Approach
3.4. Comparison of the Developed Approach with the Direct DAPI or Hoechst Staining
3.5. Sensitivity of the Developed Approach
4. Conclusions
- Fixation with 2% formaldehyde in 1× PBS (10 min, RT);
- Washing with 1× PBS (3 times);
- Permeabilization with 0.2% Triton X-100 in 1× PBS (10 min, RT);
- Washing with 1× PBS (3 times);
- Enzymatic labeling of samples (30 min, RT); the solution is composed of 1× buffer for DNA polymerase I, DNA polymerase I (the final concentration is 0.2 U/µL), mixture of dATP, dCTP, dGTP (the final concentration of all nucleotides is 0.05 mM), biotin-dUTP (the final concentration is 12.5 µM);
- Washing in 25 mM Tris-HCl, pH 7.5 + 150 mM NaCl (3 times);
- Primary antibody labeling with anti-biotin antibody (dilution - 1:100 in 25 mM Tris-HCl, pH 7.5 + 150 mM NaCl, 30 min, RT). Optionally, an antibody against the mitochondrial marker such as MTCO2 can be added;
- Washing in 25 mM Tris-HCl, pH 7.5 + 150 mM NaCl (3 times);
- Secondary antibody labeling with the fluorescently labeled antibody raised against the primary antibody (dilution - 1:100 in 25 mM Tris-HCl, pH 7.5 + 150 mM NaCl, 30 min, RT). If the mitochondrial marker is used, then a secondary antibody against this primary antibody should also be added to this solution. Optionally, DAPI can also be added;
- Washing in 25 mM Tris-HCl, pH 7.5 + 150 mM NaCl (3 times);
- Mounting in the mounting medium and observation.
5. Patents
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Thompson, C.C.; Vieira, N.M.; Vicente, A.C.P.; Thompson, F.L. Towards a genome based taxonomy of Mycoplasmas. Infect. Genet. Evol. 2011, 11, 1798–1804. [Google Scholar] [CrossRef] [PubMed]
- Volokhov, D.V.; Graham, L.J.; Brorson, K.A.; Chizhikov, V.E. Mycoplasma testing of cell substrates and biologics: Review of alternative non-microbiological techniques. Mol. Cell Probes 2011, 25, 69–77. [Google Scholar] [CrossRef]
- Halbedel, S.; Stulke, J. Tools for the genetic analysis of Mycoplasma. Int. J. Med. Microbiol. 2007, 297, 37–44. [Google Scholar] [CrossRef]
- Drexler, H.G.; Uphoff, C.C. Mycoplasma contamination of cell cultures: Incidence, sources, effects, detection, elimination, prevention. Cytotechnology 2002, 39, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Uphoff, C.C.; Brauer, S.; Grunicke, D.; Gignac, S.M.; MacLeod, R.A.; Quentmeier, H.; Steube, K.; Tummler, M.; Voges, M.; Wagner, B.; et al. Sensitivity and specificity of five different mycoplasma detection assays. Leukemia 1992, 6, 335–341. [Google Scholar] [PubMed]
- Young, L.; Sung, J.; Stacey, G.; Masters, J.R. Detection of Mycoplasma in cell cultures. Nat. Protoc. 2010, 5, 929–934. [Google Scholar] [CrossRef] [PubMed]
- Baum, S.G. Mycoplasma Infections. In Goldman’s Cecil Medicine, 24TH EDITION; Goldman, L., Schafer, A.I., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2012; pp. 1912–1916. [Google Scholar]
- Garner, C.M.; Hubbold, L.M.; Chakraborti, P.R. Mycoplasma detection in cell cultures: a comparison of four methods. Br. J. Biomed. Sci. 2000, 57, 295–301. [Google Scholar]
- Nikfarjam, L.; Farzaneh, P. Prevention and detection of Mycoplasma contamination in cell culture. Cell J. 2012, 13, 203–212. [Google Scholar]
- Rottem, S.; Barile, M.F. Beware of mycoplasmas. Trends Biotechnol. 1993, 11, 143–151. [Google Scholar] [CrossRef]
- Uphoff, C.C.; Drexler, H.G. Eradication of Mycoplasma contaminations from cell cultures. Curr. Protoc. Mol. Biol. 2014, 106, 28.5.1–28.5.12. [Google Scholar] [CrossRef]
- Vankuppeveld, F.J.M.; Johansson, K.E.; Galama, J.M.D.; Kissing, J.; Bolske, G.; Vanderlogt, J.T.M.; Melchers, W.J.G. Detection of Mycoplasma Contamination in Cell-Cultures by a Mycoplasma Group-Specific Pcr. Appl. Environ. Microb. 1994, 60, 149–152. [Google Scholar]
- Benisheva, T.; Loewer, J. Comparison of Three Methods for the Detection of Mycoplasms in Cell Cultures. Biotechnol. Biotechnol. Equip. 1994, 8, 42–45. [Google Scholar] [CrossRef]
- Gigahertz-Optik, Inc. II.6. Spectral Sensitivity of the Human Eye. Available online: https://light-measurement.com/spectral-sensitivity-of-eye/ (accessed on 7 October 2019).
- Spring, K.R.; Davidson, M.W. Concepts in Digital Imaging Technology: Quantum Efficiency. Available online: http://hamamatsu.magnet.fsu.edu/articles/quantumefficiency.html (accessed on 4 October 2019).
- Andor_Zyla_5.5_and_4.2_PLUS_Specifications. Available online: http://www.andor.com/pdfs/specifications/Andor_Zyla_5.5_and_4.2_PLUS_Specifications.pdf (accessed on 4 October 2019).
- Carpenter, A.E.; Jones, T.R.; Lamprecht, M.R.; Clarke, C.; Kang, I.H.; Friman, O.; Guertin, D.A.; Chang, J.H.; Lindquist, R.A.; Moffat, J.; et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006, 7, R100. [Google Scholar] [CrossRef] [PubMed]
- Kamentsky, L.; Jones, T.R.; Fraser, A.; Bray, M.A.; Logan, D.J.; Madden, K.L.; Ljosa, V.; Rueden, C.; Eliceiri, K.W.; Carpenter, A.E. Improved structure, function and compatibility for CellProfiler: modular high-throughput image analysis software. Bioinformatics 2011, 27, 1179–1180. [Google Scholar] [CrossRef]
- Baczynska, A.; Svenstrup, H.F.; Fedder, J.; Birkelund, S.; Christiansen, G. Development of real-time PCR for detection of Mycoplasma hominis. BMC Microbiol. 2004, 4, 35. [Google Scholar] [CrossRef] [PubMed]
- Ligasová, A.; Strunin, D.; Koberna, K. A New Method of the Visualization of the Double-Stranded Mitochondrial and Nuclear DNA. PLoS ONE 2013, 8, e66864. [Google Scholar] [CrossRef] [PubMed]
- Ligasová, A.; Strunin, D.; Liboska, R.; Rosenberg, I.; Koberna, K. Atomic scissors: a new method of tracking the 5-bromo-2’-deoxyuridine-labeled DNA in situ. PLoS ONE 2012, 7, e52584. [Google Scholar] [CrossRef] [PubMed]
- Minion, F.C.; Jarvill-Taylor, K.J.; Billings, D.E.; Tigges, E. Membrane-associated nuclease activities in mycoplasmas. J. Bacteriol. 1993, 175, 7842–7847. [Google Scholar] [CrossRef]
- Citti, C.; Blanchard, A. Mycoplasmas and their host: emerging and re-emerging minimal pathogens. Trends Microbiol. 2013, 21, 196–203. [Google Scholar] [CrossRef]
- Kapuscinski, J.; Szer, W. Interactions of 4’, 6-diamidine-2-phenylindole with synthetic polynucleotides. Nucleic Acids Res. 1979, 6, 3519–3534. [Google Scholar] [CrossRef]
- Uphoff, C.C.; Gignac, S.M.; Drexler, H.G. Mycoplasma contamination in human leukemia cell lines. I. Comparison of various detection methods. J. Immunol. Methods 1992, 149, 43–53. [Google Scholar] [CrossRef]
- Jean, A.; Tardy, F.; Allatif, O.; Grosjean, I.; Blanquier, B.; Gerlier, D. Assessing mycoplasma contamination of cell cultures by qPCR using a set of universal primer pairs targeting a 1.5 kb fragment of 16S rRNA genes. PLoS ONE 2017, 12, e0172358. [Google Scholar] [CrossRef] [PubMed]
- Geraghty, R.J.; Capes-Davis, A.; Davis, J.M.; Downward, J.; Freshney, R.I.; Knezevic, I.; Lovell-Badge, R.; Masters, J.R.W.; Meredith, J.; Stacey, G.N.; et al. Guidelines for the use of cell lines in biomedical research. Brit. J. Cancer 2014, 111, 1021–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Egan, W.; Chang, A.; Webber, K. Mycoplasma In-Process and Lot Release Testing: To PCR or Not to PCR. BioProcess. Int. 2005, 3, 30–39. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Buffer | Enzyme | Nucleotides | Marker Nucleotide |
|---|---|---|---|
| 1× buffer for DNA polymerase I 1 (ThermoFisher Scientific) | 0.2 U/µL DNA polymerase I (ThermoFisher Scientific) | 0.05 mM dATP, dCTP, dGTP (Promega) | 12.5 µM biotin-dUTP or digoxigenin-dUTP |
| Name | Sequence (5′→3′ Direction) | Length |
|---|---|---|
| Myco 1 forward primer | GCTGTGTGCCTAATACATGCAT | 22 bp |
| Myco 2 reverse primer | CACCATCTGTCATTCTGTTAACCT | 24 bp |
| Myco P probe | FAM-ATCCGCACAAGCGGTGGAGC-BHQ1 | 20 bp |
| Step | Temperature | Time | |
|---|---|---|---|
| Initial denaturation | 95 °C | 15 min | |
| 40 Cycles | Denaturation | 95 °C | 45 s |
| Annealing | 55 °C | 1 min 40 s | |
| Method | Advantages | Disadvantages | References |
|---|---|---|---|
| agar/broth media assay |
|
| [2,4,6,27,28] |
| indicator cell culture assay |
|
| [2,4,6,27,28] |
| direct DNA staining |
|
| [2,4,6,27,28] |
| PCR |
|
| [27,28] |
| biochemical detection using MycoAlert detection kit |
|
| [27,28] |
| the developed assay |
|
|
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ligasová, A.; Vydržalová, M.; Buriánová, R.; Brůčková, L.; Večeřová, R.; Janošťáková, A.; Koberna, K. A New Sensitive Method for the Detection of Mycoplasmas Using Fluorescence Microscopy. Cells 2019, 8, 1510. https://doi.org/10.3390/cells8121510
Ligasová A, Vydržalová M, Buriánová R, Brůčková L, Večeřová R, Janošťáková A, Koberna K. A New Sensitive Method for the Detection of Mycoplasmas Using Fluorescence Microscopy. Cells. 2019; 8(12):1510. https://doi.org/10.3390/cells8121510
Chicago/Turabian StyleLigasová, Anna, Markéta Vydržalová, Renata Buriánová, Lenka Brůčková, Renata Večeřová, Anna Janošťáková, and Karel Koberna. 2019. "A New Sensitive Method for the Detection of Mycoplasmas Using Fluorescence Microscopy" Cells 8, no. 12: 1510. https://doi.org/10.3390/cells8121510