Pin1 Plays Essential Roles in NASH Development by Modulating Multiple Target Proteins

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Prolyl Isomerase Pin1
3. NASH/NAFLD
3.1. Genetic Factors Contributing to NASH/NAFLD
3.2. “Two-Hit Theory” Versus “Multi-Hit Theory”
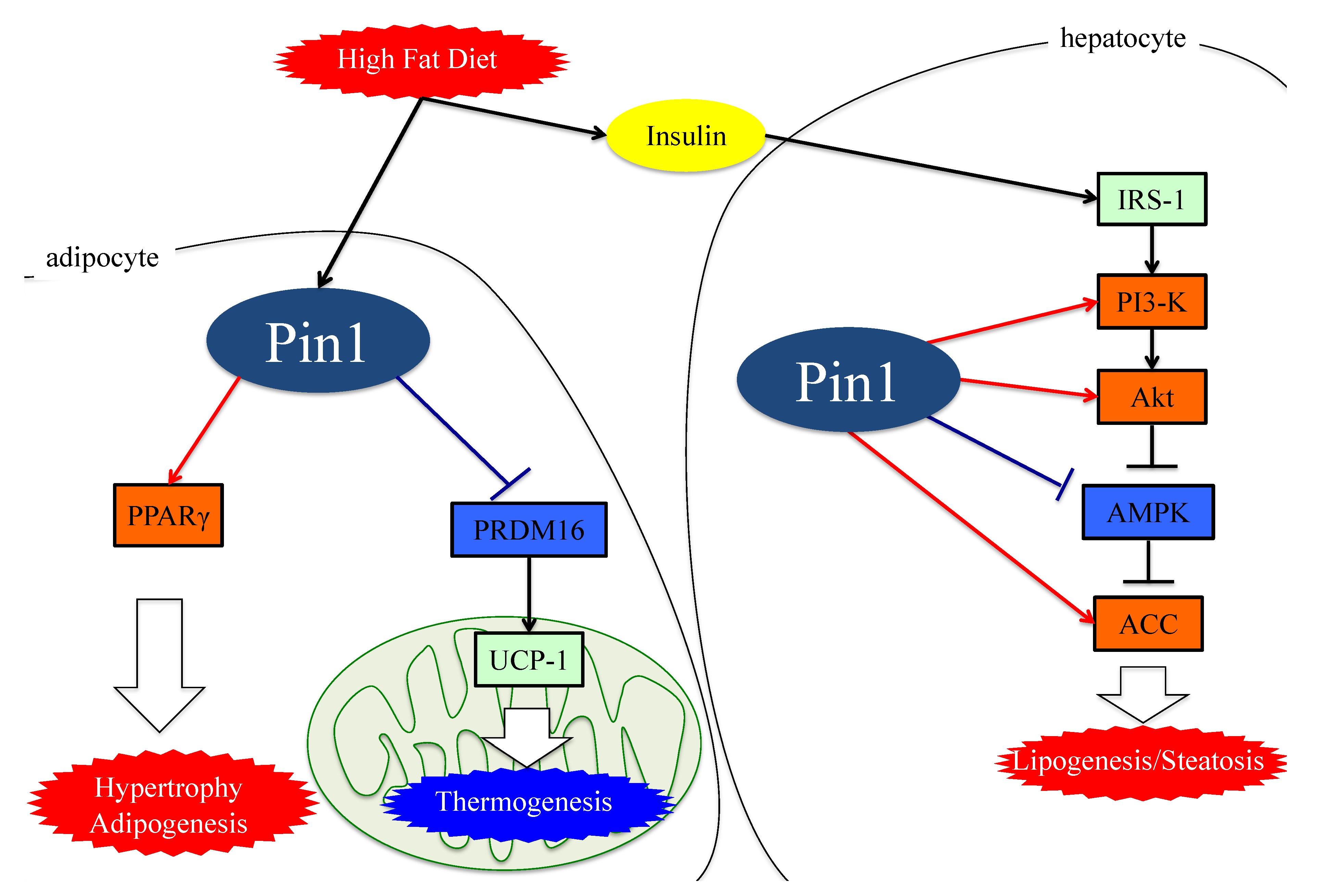
4. Role of Pin1 in the Pathogenesis of Hepatic Steatosis
5. Essential Role of Adipose Pin1 in Obesity and NASH Development
6. Pin1 Promotes the Generation of ROS by Associating with NADPH Oxidase
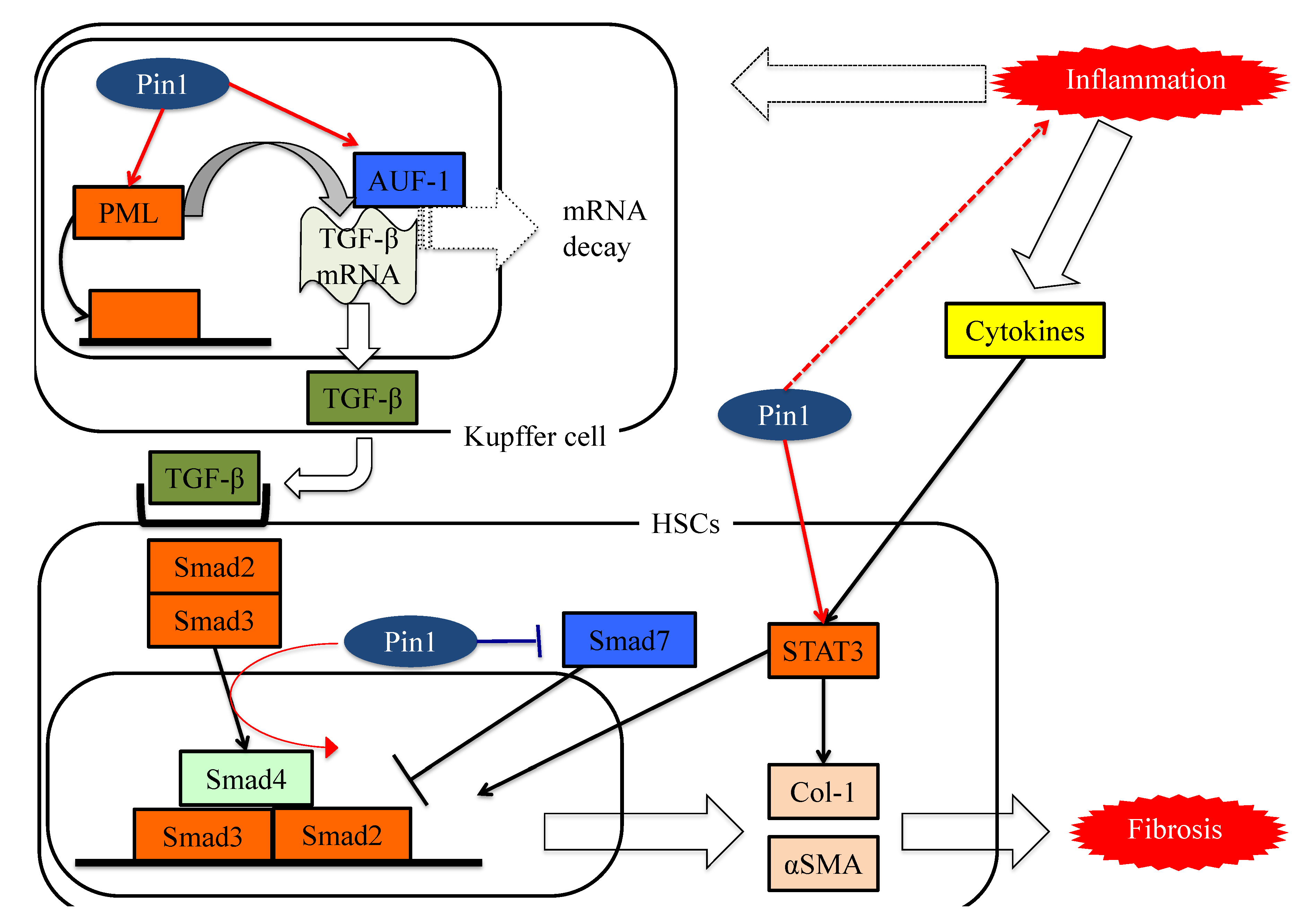
7. Pin1 Enhances Inflammation
8. Pin1 Activates the Pathways Leading to Fibrosis
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hashimoto, E.; Tokushige, K. Prevalence, gender, ethnic variations, and prognosis of NASH. J. Gastroenterol. 2011, 46, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Sherif, Z.A.; Saeed, A.; Ghavimi, S.; Nouraie, S.M.; Laiyemo, A.O.; Brim, H.; Ashktorab, H. Global Epidemiology of Nonalcoholic Fatty Liver Disease and Perspectives on US Minority Populations. Dig. Dis. Sci. 2016, 61, 1214–1225. [Google Scholar] [CrossRef] [PubMed]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Brunt, E.M.; Kleiner, D.E.; Wilson, L.A.; Belt, P.; Neuschwander-Tetri, B.A.; NASH Clinical Research Network (CRN). Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: Distinct clinicopathologic meanings. Hepatology 2011, 53, 810–820. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Zelber-Sagi, S.; Nitzan-Kaluski, D.; Halpern, Z.; Oren, R. Prevalence of primary non-alcoholic fatty liver disease in a population-based study and its association with biochemical and anthropometric measures. Liver Int. 2006, 26, 856–863. [Google Scholar] [CrossRef]
- Wang, R.T.; Koretz, R.L.; Yee Jr, H.F. Is weight reduction an effective therapy for nonalcoholic fatty liver? A systematic review. Am. J. Med. 2003, 115, 554–559. [Google Scholar] [CrossRef]
- Promrat, K.; Kleiner, D.E.; Niemeier, H.M.; Jackvony, E.; Kearns, M.; Wands, J.R.; Fava, J.L.; Wing, R.R. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology 2010, 51, 121–129. [Google Scholar] [CrossRef]
- Hamaguchi, M.; Kojima, T.; Takeda, N.; Nakagawa, T.; Taniguchi, H.; Fujii, K.; Omatsu, T.; Nakajima, T.; Sarui, H.; Shimazaki, M.; et al. The metabolic syndrome as a predictor of nonalcoholic fatty liver disease. Ann. Intern. Med. 2005, 143, 722–728. [Google Scholar] [CrossRef]
- Marchesini, G.; Brizi, M.; Morselli-Labate, A.M.; Bianchi, G.; Bugianesi, E.; McCullough, A.J.; Forlani, G.; Melchionda, N. Association of nonalcoholic fatty liver disease with insulin resistance. Am. J. Med. 1999, 107, 450–455. [Google Scholar] [CrossRef]
- Chen, C.H.; Huang, M.H.; Yang, J.C.; Nien, C.; Yang, C.C.; Yeh, Y.H.; Yueh, S.K. Prevalence and risk factors of nonalcoholic fatty liver disease in an adult population of taiwan: Metabolic significance of nonalcoholic fatty liver disease in nonobese adults. J. Clin. Gastroenterol. 2006, 40, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xia, B.; Ma, C.; Hu, Z.; Chen, X.; Cao, P. Prevalence and risk factors of fatty liver disease in the Shuiguohu district of Wuhan city, central China. Postgrad. Med. J. 2007, 83, 192–195. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fan, J.G.; Li, F.; Cai, X.B.; Peng, Y.D.; Ao, Q.H.; Gao, Y. The importance of metabolic factors for the increasing prevalence of fatty liver in Shanghai factory workers. J. Gastroenterol. Hepatol. 2007, 22, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.M.; deVera, M.E.; Fontes, P.; Shaikh, O.; Ahmad, J. Outcome after liver transplantation for NASH cirrhosis. Am. J. Transplant. 2009, 9, 782–793. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, Y.R.; Yang, H.Y.; Li, X.Z.; Jie, M.M.; Hu, C.J.; Wu, Y.Y.; Yang, S.M.; Yang, Y.B. Prolyl isomerase Pin1: A promoter of cancer and a target for therapy. Cell Death Dis. 2018, 9, 883. [Google Scholar] [CrossRef] [PubMed]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar]
- Morgan, D.O. Cyclin-dependent kinases: Engines, clocks, and microprocessors. Annu. Rev. Cell Dev. Biol. 1997, 13, 261–291. [Google Scholar] [CrossRef]
- Lu, K.P.; Liou, Y.C.; Zhou, X.Z. Pinning down proline-directed phosphorylation signaling. Trends Cell Biol. 2002, 12, 164–172. [Google Scholar] [CrossRef]
- Schmidpeter, P.A.; Koch, J.R.; Schmid, F.X. Control of protein function by prolyl isomerization. Biochim. Biophys. Acta 2015, 1850, 1973–1982. [Google Scholar] [CrossRef]
- Schmid, F.X. Prolyl isomerase: Enzymatic catalysis of slow protein-folding reactions. Annu. Rev. Biophys. Biomol. Struct. 1993, 22, 123–142. [Google Scholar] [CrossRef]
- Day, C.P.; Saksena, S. Non-alcoholic steatohepatitis: Definitions and pathogenesis. J. Gastroenterol. Hepatol. 2002, 17, S377–S384. [Google Scholar] [CrossRef] [PubMed]
- Yeh, E.S.; Means, A.R. PIN1, the cell cycle and cancer. Nat. Rev. Cancer 2007, 7, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Verdecia, M.A.; Bowman, M.E.; Lu, K.P.; Hunter, T.; Noel, J.P. Structural basis for phosphoserine-proline recognition by group IV WW domains. Nat. Struct. Mol. Biol. 2000, 7, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Matena, A.; Rehic, E.; Hönig, D.; Kamba, B.; Bayer, P. Structure and function of the human parvulins Pin1 and Par14/17. Biol. Chem. 2018, 399, 101–125. [Google Scholar] [CrossRef] [PubMed]
- Kuboki, S.; Sakai, N.; Clarke, C.; Schuster, R.; Blanchard, J.; Edwards, M.J.; Lentsch, A.B. The peptidyl-prolyl isomerase, Pin1, facilitates NF-kappaB binding in hepatocytes and protects against hepatic ischemia/reperfusion injury. J. Hepatol. 2009, 51, 296–306. [Google Scholar] [CrossRef][Green Version]
- Yang, J.W.; Hien, T.T.; Lim, S.C.; Jun, D.W.; Choi, H.S.; Yoon, J.H.; Cho, I.J.; Kang, K.W. Pin1 induction in the fibrotic liver and its roles in TGF-β1 expression and Smad2/3 phosphorylation. J. Hepatol. 2014, 60, 1235–1241. [Google Scholar] [CrossRef]
- Wulf, G.M.; Ryo, A.; Wulf, G.G.; Lee, S.W.; Niu, T.; Petkova, V.; Lu, K.P. Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c-Jun towards cyclin D1. EMBO J. 2001, 20, 3459–3472. [Google Scholar] [CrossRef]
- Rustighi, A.; Tiberi, L.; Soldano, A.; Napoli, M.; Nuciforo, P.; Rosato, A.; Kaplan, F.; Capobianco, A.; Pece, S.; Di Fiore, P.P.; et al. The prolyl-isomerase Pin1 is a Notch1 target that enhances Notch1 activation in cancer. Nat. Cell Biol. 2009, 11, 133–142. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Anstee, Q.M.; Valenti, L. Genetic Predisposition in NAFLD and NASH: Impact on Severity of Liver Disease and Response to Treatment. Curr. Pharm. Des. 2013, 19, 5219–5238. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Tiniakos, D.G.; Vos, M.B.; Brunt, E.M. Nonalcoholic fatty liver disease: Pathology and pathogenesis. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 145–171. [Google Scholar] [CrossRef]
- Feldstein, A.E.; Werneburg, N.W.; Canbay, A.; Guicciardi, M.E.; Bronk, S.F.; Rydzewski, R.; Burgart, L.J.; Gores, G.J. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology 2004, 40, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Marí, M.; Caballero, F.; Colell, A.; Morales, A.; Caballeria, J.; Fernandez, A.; Enrich, C.; Fernandez-Checa, J.C.; García-Ruiz, C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006, 4, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, Y.; Otani, Y.; Sakoda, H.; Zhang, J.; Guo, Y.; Okubo, H.; Kushiyama, A.; Fujishiro, M.; Kikuch, T.; Fukushima, T.; et al. Role of Pin1 protein in the pathogenesis of nonalcoholic steatohepatitis in a rodent model. J. Biol. Chem. 2012, 287, 44526–44535. [Google Scholar] [CrossRef]
- Nakatsu, Y.; Matsunaga, Y.; Yamamotoya, T.; Ueda, K.; Inoue, M.K.; Mizuno, Y.; Nakanishi, M.; Sano, T.; Yamawaki, Y.; Kushiyama, A.; et al. Prolyl Isomerase Pin1 Suppresses Thermogenic Programs in Adipocytes by Promoting Degradation of Transcriptional Co-activator PRDM16. Cell Rep. 2019, 26, 3221.e3–3230.e3. [Google Scholar] [CrossRef]
- Nakatsu, Y.; Matsunaga, Y.; Yamamotoya, T.; Ueda, K.; Inoue, Y.; Mori, K.; Sakoda, H.; Fujishiro, M.; Ono, H.; Kushiyama, A.; et al. Physiological and Pathogenic Roles of Prolyl Isomerase Pin1 in Metabolic Regulations via Multiple Signal Transduction Pathway Modulations. Int. J. Mol. Sci. 2016, 17, 1495. [Google Scholar] [CrossRef]
- Rosen, E.D.; MacDougald, O.A. Adipocyte differentiation from the inside out. Nat. Rev. Mol. Cell Biol. 2006, 7, 885–896. [Google Scholar] [CrossRef]
- Liao, Y.; Wei, Y.; Zhou, X.; Yang, J.Y.; Dai, C.; Chen, Y.J.; Agarwal, N.K.; Sarbassov, D.; Shi, D.; Yu, D.; et al. Peptidyl-prolyl cis/trans isomerase Pin1 is critical for the regulation of PKB/Akt stability and activation phosphorylation. Oncogene 2009, 28, 2436–2445. [Google Scholar] [CrossRef]
- Ueda, K.; Nakatsu, Y.; Yamamotoya, T.; Ono, H.; Inoue, Y.; Inoue, M.K.; Mizuno, Y.; Matsunaga, Y.; Kushiyama, A.; Sakoda, H.; et al. Prolyl isomerase Pin1 binds to and stabilizes acetyl CoA carboxylase 1 protein, thereby supporting cancer cell proliferation. Oncotarget 2019, 10, 1637–1648. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.J.; Kim, J.Y.; Kim, G.; Choi, H.S. Prolyl-isomerase Pin1 impairs trastuzumab sensitivity by up-regulating fatty acid synthase expression. Anticancer Res. 2014, 34, 1409–1416. [Google Scholar] [PubMed]
- Nakatsu, Y.; Iwashita, M.; Sakoda, H.; Ono, H.; Nagata, K.; Matsunaga, Y.; Fukushima, T.; Fujishiro, M.; Kushiyama, A.; Kamata, H.; et al. Prolyl isomerase Pin1 negatively regulates AMP-activated protein kinase (AMPK) by associating with the CBS domain in the γ subunit. J. Biol. Chem. 2015, 290, 24255–24266. [Google Scholar] [CrossRef] [PubMed]
- Ronnett, G.V.; Kleman, A.M.; Kim, E.K.; Landree, L.E.; Tu, Y. Fatty acid metabolism, the central nervous system, and feeding. Obesity 2006, 14, 201S–207S. [Google Scholar] [CrossRef]
- Hardie, D.G.; Pan, D.A. Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem. Soc. Trans. 2002, 30, 1064–1070. [Google Scholar] [CrossRef]
- Hardie, D.G.; Carling, D. The AMP-activated protein kinase—fuel gauge of the mammalian cell? Eur. J. Biochem. 1997, 246, 259–273. [Google Scholar] [CrossRef]
- Ruderman, N.B.; Saha, A.K.; Kraegen, E.W. Minireview: Malonyl CoA, AMP-activated protein kinase, and adiposity. Endocrinology 2003, 144, 5166–5171. [Google Scholar] [CrossRef]
- Reggio, S.; Pellegrinelli, V.; Clément, K.; Tordjman, J. Fibrosis as a Cause or a Consequence of White Adipose Tissue Inflammation in Obesity. Curr. Obes. Rep. 2013, 2, 1–9. [Google Scholar] [CrossRef]
- Nakatsu, Y.; Sakoda, H.; Kushiyama, A.; Zhang, J.; Ono, H.; Fujishiro, M.; Kikuchi, T.; Fukushima, T.; Yoneda, M.; Ohno, H.; et al. Peptidyl-prolyl cis/trans isomerase NIMA-interacting 1 associates with insulin receptor substrate-1 and enhances insulin actions and adipogenesis. J. Biol. Chem. 2011, 286, 20812–20822. [Google Scholar] [CrossRef]
- Nakatsu, Y.; Mori, K.; Matsunaga, Y.; Yamamotoya, T.; Ueda, K.; Inoue, Y.; Mitsuzaki-Miyoshi, K.; Sakoda, H.; Fujishiro, M.; Yamaguchi, S.; et al. The prolyl isomerase Pin1 increases β-cell proliferation and enhances insulin secretion. J. Biol. Chem. 2017, 292, 11886–11895. [Google Scholar] [CrossRef]
- Han, Y.; Lee, S.H.; Bahn, M.; Yeo, C.Y.; Lee, K.Y. Pin1 enhances adipocyte differentiation by positively regulating the transcriptional activity of PPARγ. Mol. Cell Endocrinol. 2016, 436, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.C.; Porter, K.E. Cellular and molecular mechanisms of endothelial dysfunction in diabetes. Diabetes Vasc. Dis. Res. 2013, 10, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Belambri, S.A.; Rolas, L.; Raad, H.; Hurtado-Nedelec, M.; Dang, P.M.; El-Benna, J. NADPH oxidase activation in neutrophils: Role of the phosphorylation of its subunits. Eur. J. Clin. Investig. 2018, 48, e12951. [Google Scholar] [CrossRef]
- Groemping, Y.; Rittinger, K. Activation and assembly of the NADPH oxidase: A structural perspective. Biochem. J. 2005, 386, 401–416. [Google Scholar] [CrossRef]
- Makni-Maalej, K.; Boussetta, T.; Hurtado-Nedelec, M.; Belambri, S.A.; Gougerot-Pocidalo, M.A.; El-Benna, J. The TLR7/8 agonist CL097 primes N-formyl-methionyl-leucyl-phenylalanine-stimulated NADPH oxidase activation in human neutrophils: Critical role of p47phox phosphorylation and the proline isomerase Pin1. J. Immunol. 2012, 189, 4657–4665. [Google Scholar] [CrossRef]
- Boussetta, T.; Gougerot-Pocidalo, M.A.; Hayem, G.; Ciappelloni, S.; Raad, H.; Arabi Derkawi, R.; Bournier, O.; Kroviarski, Y.; Zhou, X.Z.; Malter, J.S.; et al. The prolyl isomerase Pin1 acts as a novel molecular switch for TNF-alpha-induced priming of the NADPH oxidase in human neutrophils. Blood 2010, 116, 5795–5802. [Google Scholar] [CrossRef]
- Keune, W.J.; Jones, D.R.; Divecha, N. PtdIns5P and Pin1 in oxidative stress signaling. Adv. Biol. Regul. 2013, 53, 179–189. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef]
- Tothova, Z.; Kollipara, R.; Huntly, B.J.; Lee, B.H.; Castrillon, D.H.; Cullen, D.E.; McDowell, E.P.; Lazo-Kallanian, S.; Williams, I.R.; Sears, C.; et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 2007, 128, 325–339. [Google Scholar] [CrossRef]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; McMahon, M.; Chowdhry, S.; Dinkova-Kostova, A.T. Cancer chemoprevention mechanisms mediated through the Keap1-Nrf2 pathway. Antioxid. Redox Signal. 2010, 13, 1713–1748. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.E.; Lee, M.Y.; Lim, S.C.; Hien, T.T.; Kim, J.W.; Ahn, S.G.; Yoon, J.H.; Kim, S.K.; Choi, H.S.; Kang, K.W. Role of Pin1 in neointima formation: Down-regulation of Nrf2-dependent heme oxygenase-1 expression by Pin1. Free Radic. Biol. Med. 2010, 48, 1644–1653. [Google Scholar] [CrossRef]
- Keune, W.J.; Jones, D.R.; Bultsma, Y.; Sommer, L.; Zhou, X.Z.; Lu, K.P.; Divecha, N. Regulation of phosphatidylinositol-5-phosphate signaling by Pin1 determines sensitivity to oxidative stress. Sci. Signal. 2012, 5, ra86. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Sun, S.C.; Chang, J.H.; Jin, J. Regulation of nuclear factor-κB in autoimmunity. Trends Immunol. 2013, 34, 282–289. [Google Scholar] [CrossRef]
- Sun, S.C. Non-canonical NF-κB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef]
- Karin, M.; Delhase, M. The I kappa B kinase (IKK) and NF-kappa B: Key elements of proinflammatory signalling. Semin. Immunol. 2000, 12, 85–98. [Google Scholar] [CrossRef]
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: The control of NF-[kappa]B activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Verma, I.M. NF-kappaB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Karin, M. Missing pieces in the NF-kappaB puzzle. Cell 2002, 109, S81–S96. [Google Scholar] [CrossRef]
- Ryo, A.; Suizu, F.; Yoshida, Y.; Perrem, K.; Liou, Y.C.; Wulf, G.; Rottapel, R.; Yamaoka, S.; Lu, K.P. Regulation of NF-kappaB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol. Cell 2003, 12, 1413–1426. [Google Scholar] [CrossRef]
- Takino, J.; Nagamine, K.; Hori, T.; Sakasai-Sakai, A.; Takeuchi, M. Contribution of the toxic advanced glycation end-products-receptor axis in nonalcoholic steatohepatitis-related hepatocellular carcinoma. World J. Hepatol. 2015, 7, 2459–2469. [Google Scholar] [CrossRef]
- Yang, M.C.; Wang, C.J.; Liao, P.C.; Yen, C.J.; Shan, Y.S. Hepatic stellate cells secretes type I collagen to trigger epithelial mesenchymal transition of hepatoma cells. Am. J. Cancer Res. 2014, 4, 751–763. [Google Scholar]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef]
- Park, J.E.; Lee, J.A.; Park, S.G.; Lee, D.H.; Kim, S.J.; Kim, H.J.; Uchida, C.; Uchida, T.; Park, B.C.; Cho, S. A critical step for JNK activation: Isomerization by the prolyl isomerase Pin1. Cell Death Differ. 2012, 19, 153–161. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2001, 81, 807–869. [Google Scholar] [CrossRef]
- Fan, G.; Fan, Y.; Gupta, N.; Matsuura, I.; Liu, F.; Zhou, X.Z.; Lu, K.P.; Gélinas, C. Peptidyl-prolyl isomerase Pin1 markedly enhances the oncogenic activity of the rel proteins in the nuclear factor-kappaB family. Cancer Res. 2009, 69, 4589–4597. [Google Scholar] [CrossRef]
- Cengiz, M.; Ozenirler, S.; Yücel, A.A.; Yılmaz, G. Can serum pin1 level be regarded as an indicative marker of nonalcoholic steatohepatitis and fibrotic stages? Digestion 2014, 90, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Arjaans, M.; Munnink, T.H.O.; Timmer-Bosscha, H.; Reiss, M.; Walenkamp, A.M.; Lub-de Hooge, M.N.; de Vries, E.G.; Schröder, C.P. Transforming growth factor (TGF)-β expression and activation mechanisms as potential targets for anti-tumor therapy and tumor imaging. Pharmacol. Ther. 2012, 135, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Tucker, G.C. Integrins: Molecular targets in cancer therapy. Curr. Oncol. Rep. 2006, 8, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.; Hill, C.S. How the Smads regulate transcription. Int. J. Biochem. Cell Biol. 2008, 40, 383–408. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.M.; Bansal, A.; Melton, D.A. Xenopus Mad proteins transduce distinct subsets of signals for the TGF beta superfamily. Cell 1996, 85, 479–487. [Google Scholar] [CrossRef]
- Zhang, Y.; Feng, X.; We, R.; Derynck, R. Receptor-associated Mad homologues synergize as effectors of the TGF-beta response. Nature 1996, 383, 168–172. [Google Scholar] [CrossRef]
- Lagna, G.; Hata, A.; Hemmati-Brivanlou, A.; Massagué, J. Partnership between DPC4 and SMAD proteins in TGF-beta signalling pathways. Nature 1996, 383, 832–836. [Google Scholar] [CrossRef]
- Matsuura, I.; Chiang, K.N.; Lai, C.Y.; He, D.; Wang, G.; Ramkumar, R.; Uchida, T.; Ryo, A.; Lu, K.; Liu, F. Pin1 promotes transforming growth factor-beta-induced migration and invasion. J. Biol. Chem. 2010, 285, 1754–1764. [Google Scholar] [CrossRef]
- Aragón, E.; Goerner, N.; Zaromytidou, A.I.; Xi, Q.; Escobedo, A.; Massagué, J.; Macias, M.J. A Smad action turnover switch operated by WW domain readers of a phosphoserine code. Genes Dev. 2011, 25, 1275–1288. [Google Scholar] [CrossRef]
- Atkinson, G.P.; Nozell, S.E.; Harrison, D.K.; Stonecypher, M.S.; Chen, D.; Benveniste, E.N. The prolyl isomerase Pin1 regulates the NF-kappaB signaling pathway and interleukin-8 expression in glioblastoma. Oncogene 2009, 28, 3735–3745. [Google Scholar] [CrossRef]
- Shen, Z.J.; Esnault, S.; Malter, J.S. The peptidyl-prolyl isomerase Pin1 regulates the stability of granulocyte-macrophage colony-stimulating factor mRNA in activated eosinophils. Nat. Immunol. 2005, 6, 1280–1287. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Huang, D.; Li, L.L.; Ni, P.; Li, X.X.; Wang, B.; Han, Y.N.; Shao, X.Q.; Zhao, D.; Chu, W.F.; et al. TGF-β1-PML SUMOylation-peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 (Pin1) form a positive feedback loop to regulate cardiac fibrosis. J. Cell Physiol. 2019, 234, 6263–6273. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.J.; Braun, R.K.; Hu, J.; Xie, Q.; Chu, H.; Love, R.B.; Stodola, L.A.; Rosenthal, L.A.; Szakaly, R.J.; Sorkness, R.L.; et al. Pin1 protein regulates Smad protein signaling and pulmonary fibrosis. J. Biol. Chem. 2012, 287, 23294–23305. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Y.; Hu, J.J.; Shen, J.; Wang, M.L.; Zhang, Q.Q.; Qu, Y.; Lu, L.G. Stat3 signaling activation crosslinking of TGF-β1 in hepatic stellate cell exacerbates liver injury and fibrosis. Biochim. Biophys. Acta 2014, 1842, 2237–2245. [Google Scholar] [CrossRef]
- Saxena, N.K.; Ikeda, K.; Rockey, D.C.; Friedman, S.L.; Anania, F.A. Leptin in hepatic fibrosis: Evidence for increased collagen production in stellate cells and lean littermates of ob/ob mice. Hepatology 2002, 35, 762–771. [Google Scholar] [CrossRef]
- Papaioannou, I.; Xu, S.; Denton, C.P.; Abraham, D.J.; Ponticos, M. STAT3 controls COL1A2 enhancer activation cooperatively with JunB, regulates type I collagen synthesis posttranscriptionally, and is essential for lung myofibroblast differentiation. Mol. Biol. Cell 2018, 29, 84–95. [Google Scholar] [CrossRef]
- Kasembeli, M.M.; Bharadwaj, U.; Robinson, P.; Tweardy, D.J. Contribution of STAT3 to Inflammatory and Fibrotic Diseases and Prospects for its Targeting for Treatment. Int. J. Mol. Sci. 2018, 19, 2299. [Google Scholar] [CrossRef]
- O’Reilly, S.; Ciechomska, M.; Cant, R.; van Laar, J.M. Interleukin-6 (IL-6) trans signaling drives a STAT3-dependent pathway that leads to hyperactive transforming growth factor-β (TGF-β) signaling promoting SMAD3 activation and fibrosis via Gremlin protein. J. Biol. Chem. 2014, 289, 9952–9960. [Google Scholar] [CrossRef]
- Lufei, C.; Koh, T.H.; Uchida, T.; Cao, X. Pin1 is required for the Ser727 phosphorylation-dependent Stat3 activity. Oncogene 2007, 26, 7656–7664. [Google Scholar] [CrossRef]
- Nakatsu, Y.; Matsunaga, Y.; Ueda, K.; Yamamotoya, T.; Inoue, Y.; Inoue, M.K.; Mizuno, Y.; Kushiyama, A.; Ono, H.; Fujishiro, M.; et al. Development of Pin1 inhibitors and their potential as therapeutic agents. Curr. Med. Chem. 2018, 5. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inoue, M.-K.; Nakatsu, Y.; Yamamotoya, T.; Hasei, S.; Kanamoto, M.; Naitou, M.; Matsunaga, Y.; Sakoda, H.; Fujishiro, M.; Ono, H.; et al. Pin1 Plays Essential Roles in NASH Development by Modulating Multiple Target Proteins. Cells 2019, 8, 1545. https://doi.org/10.3390/cells8121545
Inoue M-K, Nakatsu Y, Yamamotoya T, Hasei S, Kanamoto M, Naitou M, Matsunaga Y, Sakoda H, Fujishiro M, Ono H, et al. Pin1 Plays Essential Roles in NASH Development by Modulating Multiple Target Proteins. Cells. 2019; 8(12):1545. https://doi.org/10.3390/cells8121545
Chicago/Turabian StyleInoue, Masa-Ki, Yusuke Nakatsu, Takeshi Yamamotoya, Shun Hasei, Mayu Kanamoto, Miki Naitou, Yasuka Matsunaga, Hideyuki Sakoda, Midori Fujishiro, Hiraku Ono, and et al. 2019. "Pin1 Plays Essential Roles in NASH Development by Modulating Multiple Target Proteins" Cells 8, no. 12: 1545. https://doi.org/10.3390/cells8121545
APA StyleInoue, M.-K., Nakatsu, Y., Yamamotoya, T., Hasei, S., Kanamoto, M., Naitou, M., Matsunaga, Y., Sakoda, H., Fujishiro, M., Ono, H., Kushiyama, A., & Asano, T. (2019). Pin1 Plays Essential Roles in NASH Development by Modulating Multiple Target Proteins. Cells, 8(12), 1545. https://doi.org/10.3390/cells8121545