Targeting mTOR and Metabolism in Cancer: Lessons and Innovations



Abstract
:1. Introduction
2. mTOR Signaling and its Role in Metabolism
2.1. Signaling to mTOR
2.2. Protein Synthesis
2.3. Glucose Metabolism
2.4. Lipid Metabolism
2.5. Glutamine Metabolism
2.6. Pentose Phosphate Pathway and Nucleotide Synthesis
2.7. Other Metabolic Pathways
3. Targeting the mTOR Pathway in Cancer
3.1. Rapalogs: Targeting mTORC1 Activity
3.2. Co-targeting mTORC1 and Growth Factor Signaling
3.3. Targeting mTORC1 and mTORC2 with ATP-Competitive mTOR Kinase Inhibitors (TORKIs)
3.4. Dual PI3K/mTOR Inhibitors: Targeting PI3K and mTOR Signaling
3.5. Targeting mTORC2 Signaling
3.6. RapaLink1
3.7. Combining mTOR Inhibition with Other Protein Kinase Inhibitors
3.8. Combining mTOR Inhibition with Conventional Chemotherapies and Other Targeted Therapies
3.9. Co-targeting mTOR and Metabolism
3.10. Other Inhibitors of the mTOR Pathway
3.11. Resistance Mechanisms and Other Therapeutic Opportunities
3.12. Immunotherapy
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Heitman, J.; Movva, N.R.; Hall, M.N. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991, 253, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef] [PubMed]
- Easton, J.B.; Houghton, P.J. mTOR and cancer therapy. Oncogene 2006, 25, 6436–6446. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, D.; Colombi, M.; Moroni, C.; Hall, M.N. Rapamycin passes the torch: A new generation of mTOR inhibitors. Nat. Rev. Drug Discov. 2011, 10, 868–880. [Google Scholar] [CrossRef]
- Barbet, N.C.; Schneider, U.; Helliwell, S.B.; Stansfield, I.; Tuite, M.F.; Hall, M.N. TOR controls translation initiation and early G1 progression in yeast. Mol. Biol. Cell 1996, 7, 25–42. [Google Scholar] [CrossRef] [Green Version]
- Beck, T.; Hall, M.N. The TOR signalling pathway controls nuclear localization of nutrient- regulated transcription factors. Nature 1999, 402, 689–692. [Google Scholar] [CrossRef]
- Zhang, H.; Stallock, J.P.; Ng, J.C.; Reinhard, C.; Neufeld, T.P. Regulation of cellular growth by the Drosophila target of rapamycin dTOR. Genes Dev. 2000, 14, 2712–2724. [Google Scholar] [CrossRef] [Green Version]
- Dennis, P.B.; Fumagalli, S.; Thomas, G. Target of rapamycin (TOR): Balancing the opposing forces of protein synthesis and degradation. Curr. Opin. Genet. Dev. 1999, 9, 49–54. [Google Scholar] [CrossRef]
- Noda, T.; Ohsumi, Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 1998, 273, 3963–3966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardwick, J.S.; Kuruvilla, F.G.; Tong, J.K.; Shamji, A.F.; Schreiber, S.L. Rapamycin-modulated transcription defines the subset of nutrient- sensitive signaling pathways directly controlled by the Tor proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 14866–14870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, T.; Golub, T.R.; Sabatini, D.M. The immunosuppressant rapamycin mimics a starvation-like signal distinct from amino acid and glucose deprivation. Mol. Cell Biol. 2002, 22, 5575–5584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [Green Version]
- Cardenas, M.E.; Cutler, N.S.; Lorenz, M.C.; Di Como, C.J.; Heitman, J. The TOR signaling cascade regulates gene expression in response to nutrients. Genes Dev. 1999, 13, 3271–3279. [Google Scholar] [CrossRef] [Green Version]
- Lynch, T.; Moloughney, J.; Jacinto, E. The mTOR complexes in cancer cell metabolism. In Pi3k-mTOR Cancer and Cancer Therapy; Dey, N., De, P., Leyland-Jones, B., Eds.; Springer: New York, NY, USA, 2016; pp. 29–63. [Google Scholar]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Shimobayashi, M.; Hall, M.N. Making new contacts: The mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014, 15, 155–162. [Google Scholar] [CrossRef]
- Kim, J.; Guan, K.L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef]
- Fan, S.J.; Snell, C.; Turley, H.; Li, J.L.; McCormick, R.; Perera, S.M.; Heublein, S.; Kazi, S.; Azad, A.; Wilson, C.; et al. PAT4 levels control amino-acid sensitivity of rapamycin-resistant mTORC1 from the Golgi and affect clinical outcome in colorectal cancer. Oncogene 2016, 35, 3004–3015. [Google Scholar] [CrossRef] [Green Version]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Guan, K.L. Amino acid signaling in TOR activation. Annu. Rev. Biochem. 2011, 80, 1001–1032. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, B.C.; Bode, B.P. Amino acid transporters ASCT2 and LAT1 in cancer: Partners in crime? Semin. Cancer Biol. 2005, 15, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 2008, 10, 935–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfson, R.L.; Sabatini, D.M. The Dawn of the Age of Amino Acid Sensors for the mTORC1 Pathway. Cell Metab. 2017, 26, 301–309. [Google Scholar] [CrossRef] [Green Version]
- Rebsamen, M.; Pochini, L.; Stasyk, T.; de Araujo, M.E.; Galluccio, M.; Kandasamy, R.K.; Snijder, B.; Fauster, A.; Rudashevskaya, E.L.; Bruckner, M.; et al. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature 2015, 519, 477–481. [Google Scholar] [CrossRef]
- Wyant, G.A.; Abu-Remaileh, M.; Wolfson, R.L.; Chen, W.W.; Freinkman, E.; Danai, L.V.; Vander Heiden, M.G.; Sabatini, D.M. mTORC1 Activator SLC38A9 Is Required to Efflux Essential Amino Acids from Lysosomes and Use Protein as a Nutrient. Cell 2017, 171, 642–654. [Google Scholar] [CrossRef]
- Duran, R.V.; Oppliger, W.; Robitaille, A.M.; Heiserich, L.; Skendaj, R.; Gottlieb, E.; Hall, M.N. Glutaminolysis activates Rag-mTORC1 signaling. Mol. Cell 2012, 47, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Stracka, D.; Jozefczuk, S.; Rudroff, F.; Sauer, U.; Hall, M.N. Nitrogen source activates TOR (target of rapamycin) complex 1 via glutamine and independently of Gtr/Rag proteins. J. Biol. Chem. 2014, 289, 25010–25020. [Google Scholar] [CrossRef] [Green Version]
- Jewell, J.L.; Kim, Y.C.; Russell, R.C.; Yu, F.X.; Park, H.W.; Plouffe, S.W.; Tagliabracci, V.S.; Guan, K.L. Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science 2015, 347, 194–198. [Google Scholar] [CrossRef] [Green Version]
- Gan, L.; Seki, A.; Shen, K.; Iyer, H.; Han, K.; Hayer, A.; Wollman, R.; Ge, X.; Lin, J.R.; Dey, G.; et al. The lysosomal GPCR-like protein GPR137B regulates Rag and mTORC1 localization and activity. Nat. Cell Biol. 2019, 21, 614–626. [Google Scholar] [CrossRef] [PubMed]
- Son, S.M.; Park, S.J.; Lee, H.; Siddiqi, F.; Lee, J.E.; Menzies, F.M.; Rubinsztein, D.C. Leucine Signals to mTORC1 via Its Metabolite Acetyl-Coenzyme A. Cell Metab. 2019, 29, 192–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Tee, A.R.; Logsdon, M.N.; Blenis, J.; Cantley, L.C. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol. Cell 2002, 10, 151–162. [Google Scholar] [CrossRef]
- Tee, A.R.; Manning, B.D.; Roux, P.P.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. 2003, 13, 1259–1268. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Jiang, X.; Li, B.; Yang, H.J.; Miller, M.; Yang, A.; Dhar, A.; Pavletich, N.P. Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature 2017, 552, 368–373. [Google Scholar] [CrossRef]
- Garami, A.; Zwartkruis, F.J.; Nobukuni, T.; Joaquin, M.; Roccio, M.; Stocker, H.; Kozma, S.C.; Hafen, E.; Bos, J.L.; Thomas, G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 2003, 11, 1457–1466. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [Green Version]
- Jewell, J.L.; Fu, V.; Hong, A.W.; Yu, F.X.; Meng, D.; Melick, C.H.; Wang, H.; Lam, W.M.; Yuan, H.X.; Taylor, S.S.; et al. GPCR signaling inhibits mTORC1 via PKA phosphorylation of Raptor. eLife 2019, 8, e43038. [Google Scholar] [CrossRef]
- Thomas, J.D.; Zhang, Y.J.; Wei, Y.H.; Cho, J.H.; Morris, L.E.; Wang, H.Y.; Zheng, X.F. Rab1A is an mTORC1 activator and a colorectal oncogene. Cancer Cell 2014, 26, 754–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goberdhan, D.C.; Wilson, C.; Harris, A.L. Amino Acid Sensing by mTORC1: Intracellular Transporters Mark the Spot. Cell Metab. 2016, 23, 580–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, J.; Wang, X.; Proud, C.G. Who does TORC2 talk to? Biochem J. 2018, 475, 1721–1738. [Google Scholar] [CrossRef] [PubMed]
- Ebner, M.; Sinkovics, B.; Szczygiel, M.; Ribeiro, D.W.; Yudushkin, I. Localization of mTORC2 activity inside cells. J. Cell Biol. 2017, 216, 343–353. [Google Scholar] [CrossRef] [Green Version]
- Gan, X.; Wang, J.; Su, B.; Wu, D. Evidence for Direct Activation of mTORC2 Kinase Activity by Phosphatidylinositol 3,4,5-Trisphosphate. J. Biol. Chem. 2011, 286, 10998–11002. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Gan, W.; Chin, Y.R.; Ogura, K.; Guo, J.; Zhang, J.; Wang, B.; Blenis, J.; Cantley, L.C.; Toker, A.; et al. PtdIns(3,4,5)P3-Dependent Activation of the mTORC2 Kinase Complex. Cancer Discov. 2015, 5, 1194–1209. [Google Scholar] [CrossRef] [Green Version]
- Ikenoue, T.; Inoki, K.; Yang, Q.; Zhou, X.; Guan, K.L. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. Embo J. 2008, 27, 1919–1931. [Google Scholar] [CrossRef] [Green Version]
- Facchinetti, V.; Ouyang, W.; Wei, H.; Soto, N.; Lazorchak, A.; Gould, C.; Lowry, C.; Newton, A.C.; Mao, Y.; Miao, R.Q.; et al. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. Embo J. 2008, 27, 1932–1943. [Google Scholar] [CrossRef]
- Garcia-Martinez, J.M.; Alessi, D.R. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem J. 2008, 416, 375–385. [Google Scholar] [CrossRef] [Green Version]
- Cameron, A.J.; Linch, M.D.; Saurin, A.T.; Escribano, C.; Parker, P.J. mTORC2 targets AGC kinases through Sin1-dependent recruitment. Biochem J. 2011, 439, 287–297. [Google Scholar] [CrossRef] [Green Version]
- Oh, W.J.; Wu, C.C.; Kim, S.J.; Facchinetti, V.; Julien, L.A.; Finlan, M.; Roux, P.P.; Su, B.; Jacinto, E. mTORC2 can associate with ribosomes to promote cotranslational phosphorylation and stability of nascent Akt polypeptide. Embo J. 2010, 29, 3939–3951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinzalla, V.; Stracka, D.; Oppliger, W.; Hall, M.N. Activation of mTORC2 by Association with the Ribosome. Cell 2011, 144, 757–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moloughney, J.G.; Kim, P.K.; Vega-Cotto, N.M.; Wu, C.C.; Zhang, S.; Adlam, M.; Lynch, T.; Chou, P.C.; Rabinowitz, J.D.; Werlen, G.; et al. mTORC2 Responds to Glutamine Catabolite Levels to Modulate the Hexosamine Biosynthesis Enzyme GFAT1. Mol. Cell 2016, 63, 811–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazyken, D.; Magnuson, B.; Bodur, C.; Acosta-Jaquez, H.A.; Zhang, D.; Tong, X.; Barnes, T.M.; Steinl, G.K.; Patterson, N.E.; Altheim, C.H.; et al. AMPK directly activates mTORC2 to promote cell survival during acute energetic stress. Sci. Signal 2019, 12, eaav3249. [Google Scholar] [CrossRef] [PubMed]
- Albert, V.; Svensson, K.; Shimobayashi, M.; Colombi, M.; Munoz, S.; Jimenez, V.; Handschin, C.; Bosch, F.; Hall, M.N. mTORC2 sustains thermogenesis via Akt-induced glucose uptake and glycolysis in brown adipose tissue. Embo Mol. Med 2016, 8, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.M.; Hung, C.M.; Hildebrand, S.R.; Sanchez-Gurmaches, J.; Martinez-Pastor, B.; Gengatharan, J.M.; Wallace, M.; Mukhopadhyay, D.; Martinez Calejman, C.; Luciano, A.K.; et al. Non-canonical mTORC2 Signaling Regulates Brown Adipocyte Lipid Catabolism through SIRT6-FoxO1. Mol. Cell 2019, 75, 807–822. [Google Scholar] [CrossRef]
- Sato, M.; Evans, B.A.; Sandstrom, A.L.; Chia, L.Y.; Mukaida, S.; Thai, B.S.; Nguyen, A.; Lim, L.; Tan, C.Y.R.; Baltos, J.A.; et al. alpha1A-Adrenoceptors activate mTOR signalling and glucose uptake in cardiomyocytes. Biochem. Pharm. 2018, 148, 27–40. [Google Scholar] [CrossRef]
- Tavares, M.R.; Pavan, I.C.; Amaral, C.L.; Meneguello, L.; Luchessi, A.D.; Simabuco, F.M. The S6K protein family in health and disease. Life Sci. 2015, 131, 1–10. [Google Scholar] [CrossRef]
- Dennis, M.D.; Jefferson, L.S.; Kimball, S.R. Role of p70S6K1-mediated phosphorylation of eIF4B and PDCD4 proteins in the regulation of protein synthesis. J. Biol. Chem. 2012, 287, 42890–42899. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.M.; Yoon, S.O.; Richardson, C.J.; Julich, K.; Blenis, J. SKAR links pre-mRNA splicing to mTOR/S6K1-mediated enhanced translation efficiency of spliced mRNAs. Cell 2008, 133, 303–313. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.H.; Martinez, T.F.; Kim, S.; Donaldson, C.; Shokhirev, M.N.; Saghatelian, A.; Jones, K.A. CDK12 phosphorylates 4E-BP1 to enable mTORC1-dependent translation and mitotic genome stability. Genes Dev. 2019, 33, 418–435. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Jiang, B.; Zhang, Y. 4E-BP1, a multifactor regulated multifunctional protein. Cell Cycle 2016, 15, 781–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toda, K.; Kawada, K.; Iwamoto, M.; Inamoto, S.; Sasazuki, T.; Shirasawa, S.; Hasegawa, S.; Sakai, Y. Metabolic Alterations Caused by KRAS Mutations in Colorectal Cancer Contribute to Cell Adaptation to Glutamine Depletion by Upregulation of Asparagine Synthetase. Neoplasia 2016, 18, 654–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwinn, D.M.; Lee, A.G.; Briones-Martin-Del-Campo, M.; Conn, C.S.; Simpson, D.R.; Scott, A.I.; Le, A.; Cowan, T.M.; Ruggero, D.; Sweet-Cordero, E.A. Oncogenic KRAS Regulates Amino Acid Homeostasis and Asparagine Biosynthesis via ATF4 and Alters Sensitivity to L-Asparaginase. Cancer Cell 2018, 33, 91–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Y.; Albuquerque, C.P.; Braas, D.; Zhang, W.; Villa, G.R.; Bi, J.; Ikegami, S.; Masui, K.; Gini, B.; Yang, H.; et al. mTORC2 Regulates Amino Acid Metabolism in Cancer by Phosphorylation of the Cystine-Glutamate Antiporter xCT. Mol. Cell 2017, 67, 128–138. [Google Scholar] [CrossRef] [Green Version]
- Gentilella, A.; Kozma, S.C.; Thomas, G. A liaison between mTOR signaling, ribosome biogenesis and cancer. Biochim. Biophys Acta 2015, 1849, 812–820. [Google Scholar] [CrossRef] [Green Version]
- Prakash, V.; Carson, B.B.; Feenstra, J.M.; Dass, R.A.; Sekyrova, P.; Hoshino, A.; Petersen, J.; Guo, Y.; Parks, M.M.; Kurylo, C.M.; et al. Ribosome biogenesis during cell cycle arrest fuels EMT in development and disease. Nat. Commun. 2019, 10, 2110. [Google Scholar] [CrossRef]
- Bywater, M.J.; Poortinga, G.; Sanij, E.; Hein, N.; Peck, A.; Cullinane, C.; Wall, M.; Cluse, L.; Drygin, D.; Anderes, K.; et al. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell 2012, 22, 51–65. [Google Scholar] [CrossRef] [Green Version]
- Devlin, J.R.; Hannan, K.M.; Hein, N.; Cullinane, C.; Kusnadi, E.; Ng, P.Y.; George, A.J.; Shortt, J.; Bywater, M.J.; Poortinga, G.; et al. Combination Therapy Targeting Ribosome Biogenesis and mRNA Translation Synergistically Extends Survival in MYC-Driven Lymphoma. Cancer Discov. 2016, 6, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef] [Green Version]
- Toschi, A.; Lee, E.; Gadir, N.; Ohh, M.; Foster, D.A. Differential dependence of hypoxia-inducible factors 1 alpha and 2 alpha on mTORC1 and mTORC2. J. Biol. Chem. 2008, 283, 34495–34499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.J.; Wang, L.Y.; Chodosh, L.A.; Keith, B.; Simon, M.C. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol. Cell Biol. 2003, 23, 9361–9374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, H.; Chiles, K.; Feldser, D.; Laughner, E.; Hanrahan, C.; Georgescu, M.M.; Simons, J.W.; Semenza, G.L. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: Implications for tumor angiogenesis and therapeutics. Cancer Res. 2000, 60, 1541–1545. [Google Scholar] [PubMed]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol. Cell Biol. 2002, 22, 7004–7014. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Gomes, A.P.; Wang, X.; Yoon, S.O.; Lee, G.; Nagiec, M.J.; Cho, S.; Chavez, A.; Islam, T.; Yu, Y.; et al. mTORC1 Promotes Metabolic Reprogramming by the Suppression of GSK3-Dependent Foxk1 Phosphorylation. Mol. Cell 2018, 70, 949–960. [Google Scholar] [CrossRef] [Green Version]
- Brugarolas, J.B.; Vazquez, F.; Reddy, A.; Sellers, W.R.; Kaelin, W.G., Jr. TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell 2003, 4, 147–158. [Google Scholar] [CrossRef] [Green Version]
- Laughner, E.; Taghavi, P.; Chiles, K.; Mahon, P.C.; Semenza, G.L. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: Novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol. Cell Biol. 2001, 21, 3995–4004. [Google Scholar] [CrossRef] [Green Version]
- Thomas, G.V.; Tran, C.; Mellinghoff, I.K.; Welsbie, D.S.; Chan, E.; Fueger, B.; Czernin, J.; Sawyers, C.L. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat. Med. 2006, 12, 122–127. [Google Scholar] [CrossRef]
- Grolleau, A.; Bowman, J.; Pradet-Balade, B.; Puravs, E.; Hanash, S.; Garcia-Sanz, J.A.; Beretta, L. Global and specific translational control by rapamycin in T cells uncovered by microarrays and proteomics. J. Biol. Chem. 2002, 9, 9. [Google Scholar] [CrossRef] [Green Version]
- Majumder, P.K.; Febbo, P.G.; Bikoff, R.; Berger, R.; Xue, Q.; McMahon, L.M.; Manola, J.; Brugarolas, J.; McDonnell, T.J.; Golub, T.R.; et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat. Med. 2004, 10, 594–601. [Google Scholar] [CrossRef]
- Sun, Q.; Chen, X.; Ma, J.; Peng, H.; Wang, F.; Zha, X.; Wang, Y.; Jing, Y.; Yang, H.; Chen, R.; et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc. Natl. Acad. Sci. USA 2011, 108, 4129–4134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broecker-Preuss, M.; Becher-Boveleth, N.; Bockisch, A.; Duhrsen, U.; Muller, S. Regulation of glucose uptake in lymphoma cell lines by c-MYC- and PI3K-dependent signaling pathways and impact of glycolytic pathways on cell viability. J. Transl. Med. 2017, 15, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Kenerson, H.; Aicher, L.; Miyaoka, R.; Eary, J.; Bissler, J.; Yeung, R.S. The tuberous sclerosis complex regulates trafficking of glucose transporters and glucose uptake. Am. J. Pathol. 2008, 172, 1748–1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrington, L.S.; Findlay, G.M.; Lamb, R.F. Restraining PI3K: mTOR signalling goes back to the membrane. Trends Biochem. Sci. 2005, 30, 35–42. [Google Scholar] [CrossRef] [PubMed]
- West, M.J.; Stoneley, M.; Willis, A.E. Translational induction of the c-myc oncogene via activation of the FRAP/TOR signalling pathway. Oncogene 1998, 17, 769–780. [Google Scholar] [CrossRef] [Green Version]
- Gordan, J.D.; Thompson, C.B.; Simon, M.C. HIF and c-Myc: Sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell 2007, 12, 108–113. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V. A Time for MYC: Metabolism and Therapy. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 79–83. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Dang, C.V.; O’Donnell, K.A.; Zeller, K.I.; Nguyen, T.; Osthus, R.C.; Li, F. The c-Myc target gene network. Semin. Cancer Biol. 2006, 16, 253–264. [Google Scholar] [CrossRef]
- van der Poel, H.G.; Hanrahan, C.; Zhong, H.; Simons, J.W. Rapamycin induces Smad activity in prostate cancer cell lines. Urol. Res. 2003, 30, 380–386. [Google Scholar]
- Zha, X.; Wang, F.; Wang, Y.; He, S.; Jing, Y.; Wu, X.; Zhang, H. Lactate dehydrogenase B is critical for hyperactive mTOR-mediated tumorigenesis. Cancer Res. 2011, 71, 13–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tandon, P.; Gallo, C.A.; Khatri, S.; Barger, J.F.; Yepiskoposyan, H.; Plas, D.R. Requirement for ribosomal protein S6 kinase 1 to mediate glycolysis and apoptosis resistance induced by Pten deficiency. Proc. Natl. Acad. Sci. USA 2011, 108, 2361–2365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pourdehnad, M.; Truitt, M.L.; Siddiqi, I.N.; Ducker, G.S.; Shokat, K.M.; Ruggero, D. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proc. Natl. Acad. Sci. USA 2013, 110, 11988–11993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutchak, P.A.; Estill-Terpack, S.J.; Plec, A.A.; Zhao, X.; Yang, C.; Chen, J.; Ko, B.; Deberardinis, R.J.; Yu, Y.; Tu, B.P. Loss of a Negative Regulator of mTORC1 Induces Aerobic Glycolysis and Altered Fiber Composition in Skeletal Muscle. Cell Rep. 2018, 23, 1907–1914. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.W.; Esposito, C.; Stephen, J.M.; Costa, A.S.H.; Frezza, C.; Blacker, T.S.; Szabadkai, G.; Ashcroft, M. CHCHD4 regulates tumour proliferation and EMT-related phenotypes, through respiratory chain-mediated metabolism. Cancer Metab. 2019, 7, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, M.; Gravel, S.P.; Chenard, V.; Sikstrom, K.; Zheng, L.; Alain, T.; Gandin, V.; Avizonis, D.; Arguello, M.; Zakaria, C.; et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013, 18, 698–711. [Google Scholar] [CrossRef] [Green Version]
- Gottlob, K.; Majewski, N.; Kennedy, S.; Kandel, E.; Robey, R.B.; Hay, N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001, 15, 1406–1418. [Google Scholar] [CrossRef] [Green Version]
- Deprez, J.; Vertommen, D.; Alessi, D.R.; Hue, L.; Rider, M.H. Phosphorylation and activation of heart 6-phosphofructo-2-kinase by protein kinase B and other protein kinases of the insulin signaling cascades. J. Biol. Chem. 1997, 272, 17269–17275. [Google Scholar] [CrossRef] [Green Version]
- Barthel, A.; Okino, S.T.; Liao, J.; Nakatani, K.; Li, J.; Whitlock, J.P., Jr.; Roth, R.A. Regulation of GLUT1 gene transcription by the serine/threonine kinase Akt1. J. Biol. Chem. 1999, 274, 20281–20286. [Google Scholar] [CrossRef] [Green Version]
- Robey, R.B.; Hay, N. Is Akt the “Warburg kinase”?-Akt-energy metabolism interactions and oncogenesis. Semin. Cancer Biol. 2009, 19, 25–31. [Google Scholar] [CrossRef] [Green Version]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.L.; Xu, P.Z.; Peng, X.D.; Chen, W.S.; Guzman, G.; Yang, X.; Di Cristofano, A.; Pandolfi, P.P.; Hay, N. The deficiency of Akt1 is sufficient to suppress tumor development in Pten+/- mice. Genes Dev. 2006, 20, 1569–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagiwara, A.; Cornu, M.; Cybulski, N.; Polak, P.; Betz, C.; Trapani, F.; Terracciano, L.; Heim, M.H.; Ruegg, M.A.; Hall, M.N. Hepatic mTORC2 Activates Glycolysis and Lipogenesis through Akt, Glucokinase, and SREBP1c. Cell Metab. 2012, 15, 725–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerniglia, G.J.; Dey, S.; Gallagher-Colombo, S.M.; Daurio, N.A.; Tuttle, S.; Busch, T.M.; Lin, A.; Sun, R.; Esipova, T.V.; Vinogradov, S.A.; et al. The PI3K/Akt Pathway Regulates Oxygen Metabolism via Pyruvate Dehydrogenase (PDH)-E1alpha Phosphorylation. Mol. Cancer 2015, 14, 1928–1938. [Google Scholar] [CrossRef] [Green Version]
- Pore, N.; Jiang, Z.; Shu, H.K.; Bernhard, E.; Kao, G.D.; Maity, A. Akt1 activation can augment hypoxia-inducible factor-1alpha expression by increasing protein translation through a mammalian target of rapamycin-independent pathway. Mol. Cancer Res. 2006, 4, 471–479. [Google Scholar] [CrossRef] [Green Version]
- Masui, K.; Tanaka, K.; Akhavan, D.; Babic, I.; Gini, B.; Matsutani, T.; Iwanami, A.; Liu, F.; Villa, G.R.; Gu, Y.; et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab. 2013, 18, 726–739. [Google Scholar] [CrossRef] [Green Version]
- Vadla, R.; Haldar, D. Mammalian target of rapamycin complex 2 (mTORC2) controls glycolytic gene expression by regulating Histone H3 Lysine 56 acetylation. Cell Cycle 2018, 17, 110–123. [Google Scholar] [CrossRef]
- Li, Y.; He, Z.C.; Liu, Q.; Zhou, K.; Shi, Y.; Yao, X.H.; Zhang, X.; Kung, H.F.; Ping, Y.F.; Bian, X.W. Large Intergenic Non-coding RNA-RoR Inhibits Aerobic Glycolysis of Glioblastoma Cells via Akt Pathway. J. Cancer 2018, 9, 880–889. [Google Scholar] [CrossRef] [Green Version]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.L.; Schulze, A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef] [Green Version]
- Owen, J.L.; Zhang, Y.; Bae, S.H.; Farooqi, M.S.; Liang, G.; Hammer, R.E.; Goldstein, J.L.; Brown, M.S. Insulin stimulation of SREBP-1c processing in transgenic rat hepatocytes requires p70 S6-kinase. Proc. Natl. Acad. Sci. USA 2012, 109, 16184–16189. [Google Scholar] [CrossRef] [Green Version]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yecies, J.L.; Zhang, H.H.; Menon, S.; Liu, S.; Yecies, D.; Lipovsky, A.I.; Gorgun, C.; Kwiatkowski, D.J.; Hotamisligil, G.S.; Lee, C.H.; et al. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab. 2011, 14, 21–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatt, A.P.; Jacobs, S.R.; Freemerman, A.J.; Makowski, L.; Rathmell, J.C.; Dittmer, D.P.; Damania, B. Dysregulation of fatty acid synthesis and glycolysis in non-Hodgkin lymphoma. Proc. Natl. Acad. Sci. USA 2012, 109, 11818–11823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Che, L.; Li, L.; Pilo, M.G.; Cigliano, A.; Ribback, S.; Li, X.; Latte, G.; Mela, M.; Evert, M.; et al. Co-activation of AKT and c-Met triggers rapid hepatocellular carcinoma development via the mTORC1/FASN pathway in mice. Sci. Rep. 2016, 6, 20484. [Google Scholar] [CrossRef] [PubMed]
- Stepanova, D.S.; Semenova, G.; Kuo, Y.M.; Andrews, A.J.; Ammoun, S.; Hanemann, C.O.; Chernoff, J. An Essential Role for the Tumor-Suppressor Merlin in Regulating Fatty Acid Synthesis. Cancer Res. 2017, 77, 5026–5038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luyimbazi, D.; Akcakanat, A.; McAuliffe, P.F.; Zhang, L.; Singh, G.; Gonzalez-Angulo, A.M.; Chen, H.; Do, K.A.; Zheng, Y.; Hung, M.C.; et al. Rapamycin regulates stearoyl CoA desaturase 1 expression in breast cancer. Mol. Cancer 2010, 9, 2770–2784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barger, J.F.; Gallo, C.A.; Tandon, P.; Liu, H.; Sullivan, A.; Grimes, H.L.; Plas, D.R. S6K1 determines the metabolic requirements for BCR-ABL survival. Oncogene 2013, 32, 453–461. [Google Scholar] [CrossRef] [Green Version]
- Zaugg, K.; Yao, Y.; Reilly, P.T.; Kannan, K.; Kiarash, R.; Mason, J.; Huang, P.; Sawyer, S.K.; Fuerth, B.; Faubert, B.; et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 2011, 25, 1041–1051. [Google Scholar] [CrossRef] [Green Version]
- Guaita-Esteruelas, S.; Bosquet, A.; Saavedra, P.; Guma, J.; Girona, J.; Lam, E.W.; Amillano, K.; Borras, J.; Masana, L. Exogenous FABP4 increases breast cancer cell proliferation and activates the expression of fatty acid transport proteins. Mol. Carcinog. 2017, 56, 208–217. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Fan, Z.; Wang, Z.; Dai, Q.; Xiang, Z.; Yuan, F.; Yan, M.; Zhu, Z.; Liu, B.; Li, C. CD36 mediates palmitate acid-induced metastasis of gastric cancer via AKT/GSK-3beta/beta-catenin pathway. J. Exp. Clin. Cancer Res. 2019, 38, 52. [Google Scholar] [CrossRef] [Green Version]
- Guri, Y.; Colombi, M.; Dazert, E.; Hindupur, S.K.; Roszik, J.; Moes, S.; Jenoe, P.; Heim, M.H.; Riezman, I.; Riezman, H.; et al. mTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell 2017, 32, 807–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhutia, Y.D.; Ganapathy, V. Glutamine transporters in mammalian cells and their functions in physiology and cancer. Biochim. Biophys Acta 2016, 1863, 2531–2539. [Google Scholar] [CrossRef] [PubMed]
- van Geldermalsen, M.; Wang, Q.; Nagarajah, R.; Marshall, A.D.; Thoeng, A.; Gao, D.; Ritchie, W.; Feng, Y.; Bailey, C.G.; Deng, N.; et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene 2016, 35, 3201–3208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Hardie, R.A.; Hoy, A.J.; van Geldermalsen, M.; Gao, D.; Fazli, L.; Sadowski, M.C.; Balaban, S.; Schreuder, M.; Nagarajah, R.; et al. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J. Pathol. 2015, 236, 278–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willems, L.; Jacque, N.; Jacquel, A.; Neveux, N.; Maciel, T.T.; Lambert, M.; Schmitt, A.; Poulain, L.; Green, A.S.; Uzunov, M.; et al. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood 2013, 122, 3521–3532. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Chen, M.; Tao, Z.; Gao, S.; Li, Y.; Cao, Y.; Lu, C.; Zou, X. Effects of targeting SLC1A5 on inhibiting gastric cancer growth and tumor development in vitro and in vivo. Oncotarget 2017, 8, 76458–76467. [Google Scholar] [CrossRef] [Green Version]
- Ni, F.; Yu, W.M.; Li, Z.; Graham, D.K.; Jin, L.; Kang, S.; Rossi, M.R.; Li, S.; Broxmeyer, H.E.; Qu, C.K. Critical role of ASCT2-mediated amino acid metabolism in promoting leukaemia development and progression. Nat. Metab. 2019, 1, 390–403. [Google Scholar] [CrossRef]
- Digomann, D.; Kurth, I.; Tyutyunnykova, A.; Chen, O.; Lock, S.; Gorodetska, I.; Peitzsch, C.; Skvortsova, I.I.; Negro, G.; Aschenbrenner, B.; et al. The CD98 Heavy Chain Is a Marker and Regulator of Head and Neck Squamous Cell Carcinoma Radiosensitivity. Clin. Cancer Res. 2019, 25, 3152–3163. [Google Scholar] [CrossRef]
- Daye, D.; Wellen, K.E. Metabolic reprogramming in cancer: Unraveling the role of glutamine in tumorigenesis. Semin. Cell Dev. Biol. 2012, 23, 362–369. [Google Scholar] [CrossRef]
- Pusapati, R.V.; Daemen, A.; Wilson, C.; Sandoval, W.; Gao, M.; Haley, B.; Baudy, A.R.; Hatzivassiliou, G.; Evangelista, M.; Settleman, J. mTORC1-Dependent Metabolic Reprogramming Underlies Escape from Glycolysis Addiction in Cancer Cells. Cancer Cell 2016, 29, 548–562. [Google Scholar] [CrossRef] [Green Version]
- Qie, S.; Yoshida, A.; Parnham, S.; Oleinik, N.; Beeson, G.C.; Beeson, C.C.; Ogretmen, B.; Bass, A.J.; Wong, K.K.; Rustgi, A.K.; et al. Targeting glutamine-addiction and overcoming CDK4/6 inhibitor resistance in human esophageal squamous cell carcinoma. Nat. Commun. 2019, 10, 1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csibi, A.; Lee, G.; Yoon, S.O.; Tong, H.; Ilter, D.; Elia, I.; Fendt, S.M.; Roberts, T.M.; Blenis, J. The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr. Biol. 2014, 24, 2274–2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Momcilovic, M.; Bailey, S.T.; Lee, J.T.; Fishbein, M.C.; Braas, D.; Go, J.; Graeber, T.G.; Parlati, F.; Demo, S.; Li, R.; et al. The GSK3 Signaling Axis Regulates Adaptive Glutamine Metabolism in Lung Squamous Cell Carcinoma. Cancer Cell 2018, 33, 905–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demas, D.M.; Demo, S.; Fallah, Y.; Clarke, R.; Nephew, K.P.; Althouse, S.; Sandusky, G.; He, W.; Shajahan-Haq, A.N. Glutamine Metabolism Drives Growth in Advanced Hormone Receptor Positive Breast Cancer. Front. Oncol. 2019, 9, 686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csibi, A.; Fendt, S.M.; Li, C.; Poulogiannis, G.; Choo, A.Y.; Chapski, D.J.; Jeong, S.M.; Dempsey, J.M.; Parkhitko, A.; Morrison, T.; et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 2013, 153, 840–854. [Google Scholar] [CrossRef] [Green Version]
- Adebayo Michael, A.O.; Ko, S.; Tao, J.; Moghe, A.; Yang, H.; Xu, M.; Russell, J.O.; Pradhan-Sundd, T.; Liu, S.; Singh, S.; et al. Inhibiting Glutamine-Dependent mTORC1 Activation Ameliorates Liver Cancers Driven by beta-Catenin Mutations. Cell Metab. 2019, 29, 1135–1150. [Google Scholar] [CrossRef]
- Lie, S.; Wang, T.; Forbes, B.; Proud, C.G.; Petersen, J. The ability to utilise ammonia as nitrogen source is cell type specific and intricately linked to GDH, AMPK and mTORC1. Sci. Rep. 2019, 9, 1461. [Google Scholar] [CrossRef] [Green Version]
- Byun, J.K.; Choi, Y.K.; Kim, J.H.; Jeong, J.Y.; Jeon, H.J.; Kim, M.K.; Hwang, I.; Lee, S.Y.; Lee, Y.M.; Lee, I.K.; et al. A Positive Feedback Loop between Sestrin2 and mTORC2 Is Required for the Survival of Glutamine-Depleted Lung Cancer Cells. Cell Rep. 2017, 20, 586–599. [Google Scholar] [CrossRef] [Green Version]
- Denzel, M.S.; Antebi, A. Hexosamine pathway and (ER) protein quality control. Curr. Opin. Cell Biol. 2015, 33, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Moloughney, J.G.; Vega-Cotto, N.M.; Liu, S.; Patel, C.; Kim, P.K.; Wu, C.C.; Albaciete, D.; Magaway, C.; Chang, A.; Rajput, S.; et al. mTORC2 modulates the amplitude and duration of GFAT1 Ser-243 phosphorylation to maintain flux through the hexosamine pathway during starvation. J. Biol. Chem. 2018, 293, 16464–16478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.R.; Keune, W.J.; Anderson, K.E.; Stephens, L.R.; Hawkins, P.T.; Divecha, N. The hexosamine biosynthesis pathway and O-GlcNAcylation maintain insulin-stimulated PI3K-PKB phosphorylation and tumour cell growth after short-term glucose deprivation. Febs. J. 2014, 281, 3591–3608. [Google Scholar] [CrossRef] [PubMed]
- Stanton, R.C. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. Iubmb. Life 2012, 64, 362–369. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Yuan, W.; Chen, Z.; Wu, S.; Chen, J.; Ge, J.; Hou, F.; Chen, Z. Overexpression of G6PD is associated with poor clinical outcome in gastric cancer. Tumour Biol. 2012, 33, 95–101. [Google Scholar] [CrossRef]
- Yang, C.A.; Huang, H.Y.; Lin, C.L.; Chang, J.G. G6PD as a predictive marker for glioma risk, prognosis and chemosensitivity. J. Neurooncol. 2018, 139, 661–670. [Google Scholar] [CrossRef]
- Benito, A.; Polat, I.H.; Noe, V.; Ciudad, C.J.; Marin, S.; Cascante, M. Glucose-6-phosphate dehydrogenase and transketolase modulate breast cancer cell metabolic reprogramming and correlate with poor patient outcome. Oncotarget 2017, 8, 106693–106706. [Google Scholar] [CrossRef]
- Chen, X.; Xu, Z.; Zhu, Z.; Chen, A.; Fu, G.; Wang, Y.; Pan, H.; Jin, B. Modulation of G6PD affects bladder cancer via ROS accumulation and the AKT pathway in vitro. Int. J. Oncol. 2018, 53, 1703–1712. [Google Scholar] [CrossRef] [Green Version]
- Best, S.A.; Ding, S.; Kersbergen, A.; Dong, X.; Song, J.Y.; Xie, Y.; Reljic, B.; Li, K.; Vince, J.E.; Rathi, V.; et al. Distinct initiating events underpin the immune and metabolic heterogeneity of KRAS-mutant lung adenocarcinoma. Nat. Commun. 2019, 10, 4190. [Google Scholar] [CrossRef]
- Parkhitko, A.A.; Priolo, C.; Coloff, J.L.; Yun, J.; Wu, J.J.; Mizumura, K.; Xu, W.; Malinowska, I.A.; Yu, J.; Kwiatkowski, D.J.; et al. Autophagy-dependent metabolic reprogramming sensitizes TSC2-deficient cells to the antimetabolite 6-aminonicotinamide. Mol. Cancer Res. 2014, 12, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Ben-Sahra, I.; Howell, J.J.; Asara, J.M.; Manning, B.D. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 2013, 339, 1323–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robitaille, A.M.; Christen, S.; Shimobayashi, M.; Cornu, M.; Fava, L.L.; Moes, S.; Prescianotto-Baschong, C.; Sauer, U.; Jenoe, P.; Hall, M.N. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science 2013, 339, 1320–1323. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, J.T.; Moreno, M.V.; Lodi, A.; Ronen, S.M.; Ruggero, D. Protein and nucleotide biosynthesis are coupled by a single rate-limiting enzyme, PRPS2, to drive cancer. Cell 2014, 157, 1088–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.; Asara, J.M.; Manning, B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016, 351, 728–733. [Google Scholar] [CrossRef] [Green Version]
- Evert, M.; Calvisi, D.F.; Evert, K.; De Murtas, V.; Gasparetti, G.; Mattu, S.; Destefanis, G.; Ladu, S.; Zimmermann, A.; Delogu, S.; et al. V-AKT murine thymoma viral oncogene homolog/mammalian target of rapamycin activation induces a module of metabolic changes contributing to growth in insulin-induced hepatocarcinogenesis. Hepatology 2012, 55, 1473–1484. [Google Scholar] [CrossRef]
- Kliegman, J.I.; Fiedler, D.; Ryan, C.J.; Xu, Y.F.; Su, X.Y.; Thomas, D.; Caccese, M.C.; Cheng, A.; Shales, M.; Rabinowitz, J.D.; et al. Chemical genetics of rapamycin-insensitive TORC2 in S. cerevisiae. Cell Rep. 2013, 5, 1725–1736. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Fridman, A.; Blackledge, W.; Connelly, S.; Wilson, I.A.; Pilz, R.B.; Boss, G.R. The phosphatidylinositol 3-kinase/akt cassette regulates purine nucleotide synthesis. J. Biol. Chem. 2009, 284, 3521–3528. [Google Scholar] [CrossRef] [Green Version]
- Saha, A.; Connelly, S.; Jiang, J.; Zhuang, S.; Amador, D.T.; Phan, T.; Pilz, R.B.; Boss, G.R. Akt phosphorylation and regulation of transketolase is a nodal point for amino acid control of purine synthesis. Mol. Cell 2014, 55, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.; Orozco, J.M.; Saxton, R.A.; Condon, K.J.; Liu, G.Y.; Krawczyk, P.A.; Scaria, S.M.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science 2017, 358, 813–818. [Google Scholar] [CrossRef] [Green Version]
- Kottakis, F.; Nicolay, B.N.; Roumane, A.; Karnik, R.; Gu, H.; Nagle, J.M.; Boukhali, M.; Hayward, M.C.; Li, Y.Y.; Chen, T.; et al. LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature 2016, 539, 390–395. [Google Scholar] [CrossRef] [Green Version]
- Reina-Campos, M.; Linares, J.F.; Duran, A.; Cordes, T.; L’Hermitte, A.; Badur, M.G.; Bhangoo, M.S.; Thorson, P.K.; Richards, A.; Rooslid, T.; et al. Increased Serine and One-Carbon Pathway Metabolism by PKClambda/iota Deficiency Promotes Neuroendocrine Prostate Cancer. Cancer Cell 2019, 35, 385–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabiner, B.C.; Nardi, V.; Birsoy, K.; Possemato, R.; Shen, K.; Sinha, S.; Jordan, A.; Beck, A.H.; Sabatini, D.M. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov. 2014, 4, 554–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagle, N.; Grabiner, B.C.; Van Allen, E.M.; Hodis, E.; Jacobus, S.; Supko, J.G.; Stewart, M.; Choueiri, T.K.; Gandhi, L.; Cleary, J.M.; et al. Activating mTOR mutations in a patient with an extraordinary response on a phase I trial of everolimus and pazopanib. Cancer Discov. 2014, 4, 546–553. [Google Scholar] [CrossRef] [Green Version]
- Murugan, A.K. mTOR: Role in cancer, metastasis and drug resistance. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K.; Engelman, J.A.; Cantley, L.C. Targeting the PI3K signaling pathway in cancer. Curr. Opin. Genet. Dev. 2010, 20, 87–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef]
- Gustafson, W.C.; Weiss, W.A. Myc proteins as therapeutic targets. Oncogene 2010, 29, 1249–1259. [Google Scholar] [CrossRef] [Green Version]
- Meng, L.H.; Zheng, X.F. Toward rapamycin analog (rapalog)-based precision cancer therapy. Acta Pharm. Sin. 2015, 36, 1163–1169. [Google Scholar] [CrossRef] [Green Version]
- Le Tourneau, C.; Faivre, S.; Serova, M.; Raymond, E. mTORC1 inhibitors: Is temsirolimus in renal cancer telling us how they really work? Br. J. Cancer 2008, 99, 1197–1203. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; Oudard, S.; Hutson, T.E.; Porta, C.; Bracarda, S.; Grunwald, V.; Thompson, J.A.; Figlin, R.A.; Hollaender, N.; et al. Efficacy of everolimus in advanced renal cell carcinoma: A double-blind, randomised, placebo-controlled phase III trial. Lancet 2008, 372, 449–456. [Google Scholar] [CrossRef]
- Buti, S.; Leonetti, A.; Dallatomasina, A.; Bersanelli, M. Everolimus in the management of metastatic renal cell carcinoma: An evidence-based review of its place in therapy. Core Evid. 2016, 11, 23–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, D.A.; Care, M.M.; Holland, K.; Agricola, K.; Tudor, C.; Mangeshkar, P.; Wilson, K.A.; Byars, A.; Sahmoud, T.; Franz, D.N. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N. Engl. J. Med. 2010, 363, 1801–1811. [Google Scholar] [CrossRef] [PubMed]
- Bissler, J.J.; Kingswood, J.C.; Radzikowska, E.; Zonnenberg, B.A.; Frost, M.; Belousova, E.; Sauter, M.; Nonomura, N.; Brakemeier, S.; de Vries, P.J.; et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): A multicentre, randomised, double-blind, placebo-controlled trial. Lancet 2013, 381, 817–824. [Google Scholar] [CrossRef]
- Franz, D.N.; Belousova, E.; Sparagana, S.; Bebin, E.M.; Frost, M.D.; Kuperman, R.; Witt, O.; Kohrman, M.H.; Flamini, J.R.; Wu, J.Y.; et al. Long-Term Use of Everolimus in Patients with Tuberous Sclerosis Complex: Final Results from the EXIST-1 Study. PLoS ONE 2016, 11, e0158476. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.C.; Shah, M.H.; Ito, T.; Bohas, C.L.; Wolin, E.M.; Van Cutsem, E.; Hobday, T.J.; Okusaka, T.; Capdevila, J.; de Vries, E.G.; et al. Everolimus for advanced pancreatic neuroendocrine tumors. N. Engl. J. Med. 2011, 364, 514–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, J.C.; Fazio, N.; Singh, S.; Buzzoni, R.; Carnaghi, C.; Wolin, E.; Tomasek, J.; Raderer, M.; Lahner, H.; Voi, M.; et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): A randomised, placebo-controlled, phase 3 study. Lancet 2016, 387, 968–977. [Google Scholar] [CrossRef]
- Hudes, G.; Carducci, M.; Tomczak, P.; Dutcher, J.; Figlin, R.; Kapoor, A.; Staroslawska, E.; Sosman, J.; McDermott, D.; Bodrogi, I.; et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 2271–2281. [Google Scholar] [CrossRef] [Green Version]
- Kwitkowski, V.E.; Prowell, T.M.; Ibrahim, A.; Farrell, A.T.; Justice, R.; Mitchell, S.S.; Sridhara, R.; Pazdur, R. FDA approval summary: Temsirolimus as treatment for advanced renal cell carcinoma. Oncologist 2010, 15, 428–435. [Google Scholar] [CrossRef] [Green Version]
- Hess, G.; Herbrecht, R.; Romaguera, J.; Verhoef, G.; Crump, M.; Gisselbrecht, C.; Laurell, A.; Offner, F.; Strahs, A.; Berkenblit, A.; et al. Phase III study to evaluate temsirolimus compared with investigator’s choice therapy for the treatment of relapsed or refractory mantle cell lymphoma. J. Clin. Oncol. 2009, 27, 3822–3829. [Google Scholar] [CrossRef]
- Schneider, T.C.; de Wit, D.; Links, T.P.; van Erp, N.P.; van der Hoeven, J.J.; Gelderblom, H.; Roozen, I.C.; Bos, M.; Corver, W.E.; van Wezel, T.; et al. Everolimus in Patients With Advanced Follicular-Derived Thyroid Cancer: Results of a Phase II Clinical Trial. J. Clin. Endocrinol. Metab. 2017, 102, 698–707. [Google Scholar] [CrossRef]
- Chawla, S.P.; Staddon, A.P.; Baker, L.H.; Schuetze, S.M.; Tolcher, A.W.; D’Amato, G.Z.; Blay, J.Y.; Mita, M.M.; Sankhala, K.K.; Berk, L.; et al. Phase II study of the mammalian target of rapamycin inhibitor ridaforolimus in patients with advanced bone and soft tissue sarcomas. J. Clin. Oncol. 2012, 30, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; Chawla, S.P.; Ray-Coquard, I.; Le Cesne, A.; Staddon, A.P.; Milhem, M.M.; Penel, N.; Riedel, R.F.; Bui-Nguyen, B.; Cranmer, L.D.; et al. Results of an international randomized phase III trial of the mammalian target of rapamycin inhibitor ridaforolimus versus placebo to control metastatic sarcomas in patients after benefit from prior chemotherapy. J. Clin. Oncol. 2013, 31, 2485–2492. [Google Scholar] [CrossRef] [PubMed]
- Colombo, N.; McMeekin, D.S.; Schwartz, P.E.; Sessa, C.; Gehrig, P.A.; Holloway, R.; Braly, P.; Matei, D.; Morosky, A.; Dodion, P.F.; et al. Ridaforolimus as a single agent in advanced endometrial cancer: Results of a single-arm, phase 2 trial. Br. J. Cancer 2013, 108, 1021–1026. [Google Scholar] [CrossRef] [PubMed]
- Oza, A.M.; Pignata, S.; Poveda, A.; McCormack, M.; Clamp, A.; Schwartz, B.; Cheng, J.; Li, X.; Campbell, K.; Dodion, P.; et al. Randomized Phase II Trial of Ridaforolimus in Advanced Endometrial Carcinoma. J. Clin. Oncol. 2015, 33, 3576–3582. [Google Scholar] [CrossRef]
- Pearson, A.D.; Federico, S.M.; Aerts, I.; Hargrave, D.R.; DuBois, S.G.; Iannone, R.; Geschwindt, R.D.; Wang, R.; Haluska, F.G.; Trippett, T.M.; et al. A phase 1 study of oral ridaforolimus in pediatric patients with advanced solid tumors. Oncotarget 2016, 7, 84736–84747. [Google Scholar] [CrossRef] [Green Version]
- Tsoref, D.; Welch, S.; Lau, S.; Biagi, J.; Tonkin, K.; Martin, L.A.; Ellard, S.; Ghatage, P.; Elit, L.; Mackay, H.J.; et al. Phase II study of oral ridaforolimus in women with recurrent or metastatic endometrial cancer. Gynecol. Oncol. 2014, 135, 184–189. [Google Scholar] [CrossRef]
- Lucchesi, M.; Chiappa, E.; Giordano, F.; Mari, F.; Genitori, L.; Sardi, I. Sirolimus in Infants with Multiple Cardiac Rhabdomyomas Associated with Tuberous Sclerosis Complex. Case Rep. Oncol. 2018, 11, 425–430. [Google Scholar] [CrossRef]
- Park, H.; Garrido-Laguna, I.; Naing, A.; Fu, S.; Falchook, G.S.; Piha-Paul, S.A.; Wheler, J.J.; Hong, D.S.; Tsimberidou, A.M.; Subbiah, V.; et al. Phase I dose-escalation study of the mTOR inhibitor sirolimus and the HDAC inhibitor vorinostat in patients with advanced malignancy. Oncotarget 2016, 7, 67521–67531. [Google Scholar] [CrossRef] [Green Version]
- Ohtsu, A.; Ajani, J.A.; Bai, Y.X.; Bang, Y.J.; Chung, H.C.; Pan, H.M.; Sahmoud, T.; Shen, L.; Yeh, K.H.; Chin, K.; et al. Everolimus for previously treated advanced gastric cancer: Results of the randomized, double-blind, phase III GRANITE-1 study. J. Clin. Oncol. 2013, 31, 3935–3943. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kudo, M.; Assenat, E.; Cattan, S.; Kang, Y.K.; Lim, H.Y.; Poon, R.T.; Blanc, J.F.; Vogel, A.; Chen, C.L.; et al. Effect of everolimus on survival in advanced hepatocellular carcinoma after failure of sorafenib: The EVOLVE-1 randomized clinical trial. JAMA 2014, 312, 57–67. [Google Scholar] [CrossRef]
- Fenner, M.; Oing, C.; Dieing, A.; Gauler, T.; Oechsle, K.; Lorch, A.; Hentrich, M.; Kopp, H.G.; Bokemeyer, C.; Honecker, F. Everolimus in patients with multiply relapsed or cisplatin refractory germ cell tumors: Results of a phase II, single-arm, open-label multicenter trial (RADIT) of the German Testicular Cancer Study Group. J. Cancer Res. Clin. Oncol. 2019, 145, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Geiger, J.L.; Bauman, J.E.; Gibson, M.K.; Gooding, W.E.; Varadarajan, P.; Kotsakis, A.; Martin, D.; Gutkind, J.S.; Hedberg, M.L.; Grandis, J.R.; et al. Phase II trial of everolimus in patients with previously treated recurrent or metastatic head and neck squamous cell carcinoma. Head Neck 2016, 38, 1759–1764. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Grabiner, B.C.; Van Allen, E.M.; Amin-Mansour, A.; Taylor-Weiner, A.; Rosenberg, M.; Gray, N.; Barletta, J.A.; Guo, Y.; Swanson, S.J.; et al. Response and acquired resistance to everolimus in anaplastic thyroid cancer. N. Engl. J. Med. 2014, 371, 1426–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emons, G.; Kurzeder, C.; Schmalfeldt, B.; Neuser, P.; de Gregorio, N.; Pfisterer, J.; Park-Simon, T.W.; Mahner, S.; Schroder, W.; Luck, H.J.; et al. Temsirolimus in women with platinum-refractory/resistant ovarian cancer or advanced/recurrent endometrial carcinoma. A phase II study of the AGO-study group (AGO-GYN8). Gynecol. Oncol. 2016, 140, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Huang, H.; Zhao, R.; Li, P.; Li, M.; Miao, H.; Chen, N.; Chen, M. AZD8055 induces cell death associated with autophagy and activation of AMPK in hepatocellular carcinoma. Oncol. Rep. 2014, 31, 649–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Teng, B.; Wen, L.; Feng, Q.; Wang, H.; Li, N.; Wang, Y.; Liang, Z. mTOR inhibitor AZD8055 inhibits proliferation and induces apoptosis in laryngeal carcinoma. Int J. Clin. Exp. Med. 2014, 7, 337–347. [Google Scholar]
- Naing, A.; Aghajanian, C.; Raymond, E.; Olmos, D.; Schwartz, G.; Oelmann, E.; Grinsted, L.; Burke, W.; Taylor, R.; Kaye, S.; et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of AZD8055 in advanced solid tumours and lymphoma. Br. J. Cancer 2012, 107, 1093–1099. [Google Scholar] [CrossRef] [Green Version]
- Asahina, H.; Nokihara, H.; Yamamoto, N.; Yamada, Y.; Tamura, Y.; Honda, K.; Seki, Y.; Tanabe, Y.; Shimada, H.; Shi, X.; et al. Safety and tolerability of AZD8055 in Japanese patients with advanced solid tumors; a dose-finding phase I study. Investig. New Drugs 2013, 31, 677–684. [Google Scholar] [CrossRef]
- Basu, B.; Dean, E.; Puglisi, M.; Greystoke, A.; Ong, M.; Burke, W.; Cavallin, M.; Bigley, G.; Womack, C.; Harrington, E.A.; et al. First-in-Human Pharmacokinetic and Pharmacodynamic Study of the Dual m-TORC 1/2 Inhibitor AZD2014. Clin. Cancer Res. 2015, 21, 3412–3419. [Google Scholar] [CrossRef] [Green Version]
- Powles, T.; Wheater, M.; Din, O.; Geldart, T.; Boleti, E.; Stockdale, A.; Sundar, S.; Robinson, A.; Ahmed, I.; Wimalasingham, A.; et al. A Randomised Phase 2 Study of AZD2014 Versus Everolimus in Patients with VEGF-Refractory Metastatic Clear Cell Renal Cancer. Eur. Urol. 2016, 69, 450–456. [Google Scholar] [CrossRef]
- Kim, S.T.; Kim, S.Y.; Klempner, S.J.; Yoon, J.; Kim, N.; Ahn, S.; Bang, H.; Kim, K.M.; Park, W.; Park, S.H.; et al. Rapamycin-insensitive companion of mTOR (RICTOR) amplification defines a subset of advanced gastric cancer and is sensitive to AZD2014-mediated mTORC1/2 inhibition. Ann. Oncol. 2017, 28, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Sakre, N.; Wildey, G.; Behtaj, M.; Kresak, A.; Yang, M.; Fu, P.; Dowlati, A. RICTOR amplification identifies a subgroup in small cell lung cancer and predicts response to drugs targeting mTOR. Oncotarget 2017, 8, 5992–6002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burris, H.A., 3rd; Kurkjian, C.D.; Hart, L.; Pant, S.; Murphy, P.B.; Jones, S.F.; Neuwirth, R.; Patel, C.G.; Zohren, F.; Infante, J.R. TAK-228 (formerly MLN0128), an investigational dual TORC1/2 inhibitor plus paclitaxel, with/without trastuzumab, in patients with advanced solid malignancies. Cancer Chemother. Pharm. 2017, 80, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Ghobrial, I.M.; Siegel, D.S.; Vij, R.; Berdeja, J.G.; Richardson, P.G.; Neuwirth, R.; Patel, C.G.; Zohren, F.; Wolf, J.L. TAK-228 (formerly MLN0128), an investigational oral dual TORC1/2 inhibitor: A phase I dose escalation study in patients with relapsed or refractory multiple myeloma, non-Hodgkin lymphoma, or Waldenstrom’s macroglobulinemia. Am. J. Hematol. 2016, 91, 400–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateo, J.; Olmos, D.; Dumez, H.; Poondru, S.; Samberg, N.L.; Barr, S.; Van Tornout, J.M.; Jie, F.; Sandhu, S.; Tan, D.S.; et al. A first in man, dose-finding study of the mTORC1/mTORC2 inhibitor OSI-027 in patients with advanced solid malignancies. Br. J. Cancer 2016, 114, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Graham, L.; Banda, K.; Torres, A.; Carver, B.S.; Chen, Y.; Pisano, K.; Shelkey, G.; Curley, T.; Scher, H.I.; Lotan, T.L.; et al. A phase II study of the dual mTOR inhibitor MLN0128 in patients with metastatic castration resistant prostate cancer. Investig. New Drugs 2018, 36, 458–467. [Google Scholar] [CrossRef]
- Cheng, H.; Zou, Y.; Ross, J.S.; Wang, K.; Liu, X.; Halmos, B.; Ali, S.M.; Liu, H.; Verma, A.; Montagna, C.; et al. RICTOR Amplification Defines a Novel Subset of Patients with Lung Cancer Who May Benefit from Treatment with mTORC1/2 Inhibitors. Cancer Discov. 2015, 5, 1262–1270. [Google Scholar] [CrossRef] [Green Version]
- Engelman, J.A.; Chen, L.; Tan, X.; Crosby, K.; Guimaraes, A.R.; Upadhyay, R.; Maira, M.; McNamara, K.; Perera, S.A.; Song, Y.; et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat. Med. 2008, 14, 1351–1356. [Google Scholar] [CrossRef] [Green Version]
- Maira, S.M.; Stauffer, F.; Brueggen, J.; Furet, P.; Schnell, C.; Fritsch, C.; Brachmann, S.; Chene, P.; De Pover, A.; Schoemaker, K.; et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer 2008, 7, 1851–1863. [Google Scholar] [CrossRef] [Green Version]
- Serra, V.; Markman, B.; Scaltriti, M.; Eichhorn, P.J.; Valero, V.; Guzman, M.; Botero, M.L.; Llonch, E.; Atzori, F.; Di Cosimo, S.; et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008, 68, 8022–8030. [Google Scholar] [CrossRef] [Green Version]
- Brachmann, S.M.; Hofmann, I.; Schnell, C.; Fritsch, C.; Wee, S.; Lane, H.; Wang, S.; Garcia-Echeverria, C.; Maira, S.M. Specific apoptosis induction by the dual PI3K/mTor inhibitor NVP-BEZ235 in HER2 amplified and PIK3CA mutant breast cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 22299–22304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, P.; Mandl-Weber, S.; Oduncu, F.; Schmidmaier, R. The novel orally bioavailable inhibitor of phosphoinositol-3-kinase and mammalian target of rapamycin, NVP-BEZ235, inhibits growth and proliferation in multiple myeloma. Exp. Cell Res. 2009, 315, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Kurkjian, C.; Infante, J.R.; Bauer, T.M.; Burris, H.A., 3rd; Greco, F.A.; Shih, K.C.; Thompson, D.S.; Lane, C.M.; Finney, L.H.; et al. A phase 1 study of the sachet formulation of the oral dual PI3K/mTOR inhibitor BEZ235 given twice daily (BID) in patients with advanced solid tumors. Investig. New Drugs 2015, 33, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Carlo, M.I.; Molina, A.M.; Lakhman, Y.; Patil, S.; Woo, K.; DeLuca, J.; Lee, C.H.; Hsieh, J.J.; Feldman, D.R.; Motzer, R.J.; et al. A Phase Ib Study of BEZ235, a Dual Inhibitor of Phosphatidylinositol 3-Kinase (PI3K) and Mammalian Target of Rapamycin (mTOR), in Patients With Advanced Renal Cell Carcinoma. Oncologist 2016, 21, 787–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salazar, R.; Garcia-Carbonero, R.; Libutti, S.K.; Hendifar, A.E.; Custodio, A.; Guimbaud, R.; Lombard-Bohas, C.; Ricci, S.; Klumpen, H.J.; Capdevila, J.; et al. Phase II Study of BEZ235 versus Everolimus in Patients with Mammalian Target of Rapamycin Inhibitor-Naive Advanced Pancreatic Neuroendocrine Tumors. Oncologist 2018, 23, 766-e90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodon, J.; Perez-Fidalgo, A.; Krop, I.E.; Burris, H.; Guerrero-Zotano, A.; Britten, C.D.; Becerra, C.; Schellens, J.; Richards, D.A.; Schuler, M.; et al. Phase 1/1b dose escalation and expansion study of BEZ235, a dual PI3K/mTOR inhibitor, in patients with advanced solid tumors including patients with advanced breast cancer. Cancer Chemother. Pharm. 2018, 82, 285–298. [Google Scholar] [CrossRef]
- Seront, E.; Rottey, S.; Filleul, B.; Glorieux, P.; Goeminne, J.C.; Verschaeve, V.; Vandenbulcke, J.M.; Sautois, B.; Boegner, P.; Gillain, A.; et al. Phase II study of dual phosphoinositol-3-kinase (PI3K) and mammalian target of rapamycin (mTOR) inhibitor BEZ235 in patients with locally advanced or metastatic transitional cell carcinoma. Bju. Int. 2016, 118, 408–415. [Google Scholar] [CrossRef]
- Wise-Draper, T.M.; Moorthy, G.; Salkeni, M.A.; Karim, N.A.; Thomas, H.E.; Mercer, C.A.; Beg, M.S.; O’Gara, S.; Olowokure, O.; Fathallah, H.; et al. A Phase Ib Study of the Dual PI3K/mTOR Inhibitor Dactolisib (BEZ235) Combined with Everolimus in Patients with Advanced Solid Malignancies. Target. Oncol. 2017, 12, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Massard, C.; Chi, K.N.; Castellano, D.; de Bono, J.; Gravis, G.; Dirix, L.; Machiels, J.P.; Mita, A.; Mellado, B.; Turri, S.; et al. Phase Ib dose-finding study of abiraterone acetate plus buparlisib (BKM120) or dactolisib (BEZ235) in patients with castration-resistant prostate cancer. Eur. J. Cancer 2017, 76, 36–44. [Google Scholar] [CrossRef]
- Freitag, H.; Christen, F.; Lewens, F.; Grass, I.; Briest, F.; Iwaszkiewicz, S.; Siegmund, B.; Grabowski, P. Inhibition of mTOR’s Catalytic Site by PKI-587 Is a Promising Therapeutic Option for Gastroenteropancreatic Neuroendocrine Tumor Disease. Neuroendocrinology 2017, 105, 90–104. [Google Scholar] [CrossRef]
- Del Campo, J.M.; Birrer, M.; Davis, C.; Fujiwara, K.; Gollerkeri, A.; Gore, M.; Houk, B.; Lau, S.; Poveda, A.; Gonzalez-Martin, A.; et al. A randomized phase II non-comparative study of PF-04691502 and gedatolisib (PF-05212384) in patients with recurrent endometrial cancer. Gynecol. Oncol. 2016, 142, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Dolly, S.O.; Wagner, A.J.; Bendell, J.C.; Kindler, H.L.; Krug, L.M.; Seiwert, T.Y.; Zauderer, M.G.; Lolkema, M.P.; Apt, D.; Yeh, R.F.; et al. Phase I Study of Apitolisib (GDC-0980), Dual Phosphatidylinositol-3-Kinase and Mammalian Target of Rapamycin Kinase Inhibitor, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2016, 22, 2874–2884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powles, T.; Lackner, M.R.; Oudard, S.; Escudier, B.; Ralph, C.; Brown, J.E.; Hawkins, R.E.; Castellano, D.; Rini, B.I.; Staehler, M.D.; et al. Randomized Open-Label Phase II Trial of Apitolisib (GDC-0980), a Novel Inhibitor of the PI3K/Mammalian Target of Rapamycin Pathway, Versus Everolimus in Patients With Metastatic Renal Cell Carcinoma. J. Clin. Oncol 2016, 34, 1660–1668. [Google Scholar] [CrossRef] [PubMed]
- Makker, V.; Recio, F.O.; Ma, L.; Matulonis, U.A.; Lauchle, J.O.; Parmar, H.; Gilbert, H.N.; Ware, J.A.; Zhu, R.; Lu, S.; et al. A multicenter, single-arm, open-label, phase 2 study of apitolisib (GDC-0980) for the treatment of recurrent or persistent endometrial carcinoma (MAGGIE study). Cancer 2016, 122, 3519–3528. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; Hamadani, M.; Hayslip, J.; Janssens, A.; Wagner-Johnston, N.; Ottmann, O.; Arnason, J.; Tilly, H.; Millenson, M.; Offner, F.; et al. Voxtalisib (XL765) in patients with relapsed or refractory non-Hodgkin lymphoma or chronic lymphocytic leukaemia: An open-label, phase 2 trial. Lancet Haematol. 2018, 5, e170–e180. [Google Scholar] [CrossRef]
- Markman, B.; Tabernero, J.; Krop, I.; Shapiro, G.I.; Siu, L.; Chen, L.C.; Mita, M.; Melendez Cuero, M.; Stutvoet, S.; Birle, D.; et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of the oral phosphatidylinositol-3-kinase and mTOR inhibitor BGT226 in patients with advanced solid tumors. Ann. Oncol. 2012, 23, 2399–2408. [Google Scholar] [CrossRef]
- Kim, M.N.; Lee, S.M.; Kim, J.S.; Hwang, S.G. Preclinical efficacy of a novel dual PI3K/mTOR inhibitor, CMG002, alone and in combination with sorafenib in hepatocellular carcinoma. Cancer Chemother. Pharm. 2019. [Google Scholar] [CrossRef]
- Grilley-Olson, J.E.; Bedard, P.L.; Fasolo, A.; Cornfeld, M.; Cartee, L.; Razak, A.R.; Stayner, L.A.; Wu, Y.; Greenwood, R.; Singh, R.; et al. A phase Ib dose-escalation study of the MEK inhibitor trametinib in combination with the PI3K/mTOR inhibitor GSK2126458 in patients with advanced solid tumors. Investig. New Drugs 2016, 34, 740–749. [Google Scholar] [CrossRef]
- Benavides-Serrato, A.; Lee, J.; Holmes, B.; Landon, K.A.; Bashir, T.; Jung, M.E.; Lichtenstein, A.; Gera, J. Specific blockade of Rictor-mTOR association inhibits mTORC2 activity and is cytotoxic in glioblastoma. PLoS ONE 2017, 12, e0176599. [Google Scholar] [CrossRef]
- Rodrik-Outmezguine, V.S.; Okaniwa, M.; Yao, Z.; Novotny, C.J.; McWhirter, C.; Banaji, A.; Won, H.; Wong, W.; Berger, M.; de Stanchina, E.; et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature 2016, 534, 272–276. [Google Scholar] [CrossRef] [Green Version]
- Fan, Q.; Aksoy, O.; Wong, R.A.; Ilkhanizadeh, S.; Novotny, C.J.; Gustafson, W.C.; Truong, A.Y.; Cayanan, G.; Simonds, E.F.; Haas-Kogan, D.; et al. A Kinase Inhibitor Targeted to mTORC1 Drives Regression in Glioblastoma. Cancer Cell 2017, 31, 424–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oricchio, E.; Katanayeva, N.; Donaldson, M.C.; Sungalee, S.; Pasion, J.P.; Beguelin, W.; Battistello, E.; Sanghvi, V.R.; Jiang, M.; Jiang, Y.; et al. Genetic and epigenetic inactivation of SESTRIN1 controls mTORC1 and response to EZH2 inhibition in follicular lymphoma. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choo, A.Y.; Yoon, S.O.; Kim, S.G.; Roux, P.P.; Blenis, J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc. Natl. Acad. Sci. USA 2008, 105, 17414–17419. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, A.C.; Costa, M.; Zollo, O.; Davis, C.; Feldman, M.E.; Testa, J.R.; Meyuhas, O.; Shokat, K.M.; Ruggero, D. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell 2010, 17, 249–261. [Google Scholar] [CrossRef] [Green Version]
- Mallya, S.; Fitch, B.A.; Lee, J.S.; So, L.; Janes, M.R.; Fruman, D.A. Resistance to mTOR kinase inhibitors in lymphoma cells lacking 4EBP1. PLoS ONE 2014, 9, e88865. [Google Scholar] [CrossRef]
- Bi, C.; Zhang, X.; Lu, T.; Zhang, X.; Wang, X.; Meng, B.; Zhang, H.; Wang, P.; Vose, J.M.; Chan, W.C.; et al. Inhibition of 4EBP phosphorylation mediates the cytotoxic effect of mechanistic target of rapamycin kinase inhibitors in aggressive B-cell lymphomas. Haematologica 2017, 102, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Iyer, G.; Hanrahan, A.J.; Milowsky, M.I.; Al-Ahmadie, H.; Scott, S.N.; Janakiraman, M.; Pirun, M.; Sander, C.; Socci, N.D.; Ostrovnaya, I.; et al. Genome sequencing identifies a basis for everolimus sensitivity. Science 2012, 338, 221. [Google Scholar] [CrossRef] [Green Version]
- Voss, M.H.; Hakimi, A.A.; Pham, C.G.; Brannon, A.R.; Chen, Y.B.; Cunha, L.F.; Akin, O.; Liu, H.; Takeda, S.; Scott, S.N.; et al. Tumor genetic analyses of patients with metastatic renal cell carcinoma and extended benefit from mTOR inhibitor therapy. Clin. Cancer Res. 2014, 20, 1955–1964. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Jegede, O.; Gray, C.; Catalano, P.J.; Novak, J.; Kwiatkowski, D.J.; McKay, R.R.; George, D.J.; Choueiri, T.K.; McDermott, D.F.; et al. Comprehensive Genomic Profiling of Metastatic Tumors in a Phase 2 Biomarker Study of Everolimus in Advanced Renal Cell Carcinoma. Clin. Genitourin. Cancer 2018, 16, 341–348. [Google Scholar] [CrossRef]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem Sci. 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andre, F.; O’Regan, R.; Ozguroglu, M.; Toi, M.; Xu, B.; Jerusalem, G.; Masuda, N.; Wilks, S.; Arena, F.; Isaacs, C.; et al. Everolimus for women with trastuzumab-resistant, HER2-positive, advanced breast cancer (BOLERO-3): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet. Oncol. 2014, 15, 580–591. [Google Scholar] [CrossRef]
- Seiler, M.; Ray-Coquard, I.; Melichar, B.; Yardley, D.A.; Wang, R.X.; Dodion, P.F.; Lee, M.A. Oral ridaforolimus plus trastuzumab for patients with HER2+ trastuzumab-refractory metastatic breast cancer. Clin. Breast Cancer 2015, 15, 60–65. [Google Scholar] [CrossRef]
- Gandhi, L.; Bahleda, R.; Tolaney, S.M.; Kwak, E.L.; Cleary, J.M.; Pandya, S.S.; Hollebecque, A.; Abbas, R.; Ananthakrishnan, R.; Berkenblit, A.; et al. Phase I study of neratinib in combination with temsirolimus in patients with human epidermal growth factor receptor 2-dependent and other solid tumors. J. Clin. Oncol 2014, 32, 68–75. [Google Scholar] [CrossRef]
- Kyriakopoulos, C.E.; Braden, A.M.; Kolesar, J.M.; Eickhoff, J.C.; Bailey, H.H.; Heideman, J.; Liu, G.; Wisinski, K.B. A phase I study of tivantinib in combination with temsirolimus in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 290–297. [Google Scholar] [CrossRef] [Green Version]
- Hollebecque, A.; Bahleda, R.; Faivre, L.; Adam, J.; Poinsignon, V.; Paci, A.; Gomez-Roca, C.; Thery, J.C.; Le Deley, M.C.; Varga, A.; et al. Phase I study of temsirolimus in combination with cetuximab in patients with advanced solid tumours. Eur. J. Cancer 2017, 81, 81–89. [Google Scholar] [CrossRef]
- Liu, X.; Kambrick, S.; Fu, S.; Naing, A.; Subbiah, V.; Blumenschein, G.R.; Glisson, B.S.; Kies, M.S.; Tsimberidou, A.M.; Wheler, J.J.; et al. Advanced malignancies treated with a combination of the VEGF inhibitor bevacizumab, anti-EGFR antibody cetuximab, and the mTOR inhibitor temsirolimus. Oncotarget 2016, 7, 23227–23238. [Google Scholar] [CrossRef]
- Mahoney, K.M.; Jacobus, S.; Bhatt, R.S.; Song, J.; Carvo, I.; Cheng, S.C.; Simpson, M.; Fay, A.P.; Puzanov, I.; Michaelson, M.D.; et al. Phase 2 Study of Bevacizumab and Temsirolimus After VEGFR TKI in Metastatic Renal Cell Carcinoma. Clin. Genitourin Cancer 2016, 14, 304–313. [Google Scholar] [CrossRef]
- Baselga, J.; Morales, S.M.; Awada, A.; Blum, J.L.; Tan, A.R.; Ewertz, M.; Cortes, J.; Moy, B.; Ruddy, K.J.; Haddad, T.; et al. A phase II study of combined ridaforolimus and dalotuzumab compared with exemestane in patients with estrogen receptor-positive breast cancer. Breast Cancer Res. Treat. 2017, 163, 535–544. [Google Scholar] [CrossRef]
- Wagner, L.M.; Fouladi, M.; Ahmed, A.; Krailo, M.D.; Weigel, B.; DuBois, S.G.; Doyle, L.A.; Chen, H.; Blaney, S.M. Phase II study of cixutumumab in combination with temsirolimus in pediatric patients and young adults with recurrent or refractory sarcoma: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2015, 62, 440–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, S.F.; Chisholm, S.; Borger, D.; Foster, R.; Rueda, B.R.; Growdon, W.B. Ridaforolimus improves the anti-tumor activity of dual HER2 blockade in uterine serous carcinoma in vivo models with HER2 gene amplification and PIK3CA mutation. Gynecol. Oncol. 2016, 141, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Schiff, D.; Jaeckle, K.A.; Anderson, S.K.; Galanis, E.; Giannini, C.; Buckner, J.C.; Stella, P.; Flynn, P.J.; Erickson, B.J.; Schwerkoske, J.F.; et al. Phase 1/2 trial of temsirolimus and sorafenib in the treatment of patients with recurrent glioblastoma: North Central Cancer Treatment Group Study/Alliance N0572. Cancer 2018, 124, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Harzstark, A.L.; Small, E.J.; Weinberg, V.K.; Sun, J.; Ryan, C.J.; Lin, A.M.; Fong, L.; Brocks, D.R.; Rosenberg, J.E. A phase 1 study of everolimus and sorafenib for metastatic clear cell renal cell carcinoma. Cancer 2011, 117, 4194–4200. [Google Scholar] [CrossRef] [PubMed]
- Amato, R.J.; Flaherty, A.L.; Stepankiw, M. Phase I trial of everolimus plus sorafenib for patients with advanced renal cell cancer. Clin. Genitourin. Cancer 2012, 10, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Jin, N.; Jiang, T.; Rosen, D.M.; Nelkin, B.D.; Ball, D.W. Synergistic action of a RAF inhibitor and a dual PI3K/mTOR inhibitor in thyroid cancer. Clin. Cancer Res. 2011, 17, 6482–6489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caumanns, J.J.; van Wijngaarden, A.; Kol, A.; Meersma, G.J.; Jalving, M.; Bernards, R.; van der Zee, A.G.J.; Wisman, G.B.A.; de Jong, S. Low-dose triple drug combination targeting the PI3K/AKT/mTOR pathway and the MAPK pathway is an effective approach in ovarian clear cell carcinoma. Cancer Lett. 2019, 461, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, Z.; Tawbi, H.A.; Hu, J.; Guan, M.; Frankel, P.H.; Ruel, N.H.; Wilczynski, S.; Christensen, S.; Gandara, D.R.; Chow, W.A. A randomised phase II trial of selumetinib vs selumetinib plus temsirolimus for soft-tissue sarcomas. Br. J. Cancer 2015, 112, 1644–1651. [Google Scholar] [CrossRef] [Green Version]
- Teo, T.; Yu, M.; Yang, Y.; Gillam, T.; Lam, F.; Sykes, M.J.; Wang, S. Pharmacologic co-inhibition of Mnks and mTORC1 synergistically suppresses proliferation and perturbs cell cycle progression in blast crisis-chronic myeloid leukemia cells. Cancer Lett. 2015, 357, 612–623. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Bendell, J.C.; Papadopoulos, K.P.; Burris, H.A., 3rd; Patnaik, A.; Jones, S.F.; Rasco, D.; Cox, D.S.; Durante, M.; Bellew, K.M.; et al. A phase IB trial of the oral MEK inhibitor trametinib (GSK1120212) in combination with everolimus in patients with advanced solid tumors. Ann. Oncol. 2015, 26, 58–64. [Google Scholar] [CrossRef]
- Schram, A.M.; Gandhi, L.; Mita, M.M.; Damstrup, L.; Campana, F.; Hidalgo, M.; Grande, E.; Hyman, D.M.; Heist, R.S. A phase Ib dose-escalation and expansion study of the oral MEK inhibitor pimasertib and PI3K/MTOR inhibitor voxtalisib in patients with advanced solid tumours. Br. J. Cancer 2018, 119, 1471–1476. [Google Scholar] [CrossRef] [PubMed]
- Mita, M.; Fu, S.; Piha-Paul, S.A.; Janku, F.; Mita, A.; Natale, R.; Guo, W.; Zhao, C.; Kurzrock, R.; Naing, A. Phase I trial of MEK 1/2 inhibitor pimasertib combined with mTOR inhibitor temsirolimus in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 616–626. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Novik, Y.; Stein, S.; Volm, M.; Meyers, M.; Smith, J.; Omene, C.; Speyer, J.; Schneider, R.; Jhaveri, K.; et al. Phase 2 trial of everolimus and carboplatin combination in patients with triple negative metastatic breast cancer. Breast Cancer Res. 2014, 16, R32. [Google Scholar] [CrossRef] [Green Version]
- Vaishampayan, U.; Shevrin, D.; Stein, M.; Heilbrun, L.; Land, S.; Stark, K.; Li, J.; Dickow, B.; Heath, E.; Smith, D.; et al. Phase II Trial of Carboplatin, Everolimus, and Prednisone in Metastatic Castration-resistant Prostate Cancer Pretreated With Docetaxel Chemotherapy: A Prostate Cancer Clinical Trial Consortium Study. Urology 2015, 86, 1206–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chon, H.S.; Kang, S.; Lee, J.K.; Apte, S.M.; Shahzad, M.M.; Williams-Elson, I.; Wenham, R.M. Phase I study of oral ridaforolimus in combination with paclitaxel and carboplatin in patients with solid tumor cancers. BMC Cancer 2017, 17, 407. [Google Scholar] [CrossRef] [PubMed]
- Christopoulos, P.; Engel-Riedel, W.; Grohe, C.; Kropf-Sanchen, C.; von Pawel, J.; Gutz, S.; Kollmeier, J.; Eberhardt, W.; Ukena, D.; Baum, V.; et al. Everolimus with paclitaxel and carboplatin as first-line treatment for metastatic large-cell neuroendocrine lung carcinoma: A multicenter phase II trial. Ann. Oncol. 2017, 28, 1898–1902. [Google Scholar] [CrossRef]
- Jovanovic, B.; Mayer, I.A.; Mayer, E.L.; Abramson, V.G.; Bardia, A.; Sanders, M.E.; Kuba, M.G.; Estrada, M.V.; Beeler, J.S.; Shaver, T.M.; et al. A Randomized Phase II Neoadjuvant Study of Cisplatin, Paclitaxel With or Without Everolimus in Patients with Stage II/III Triple-Negative Breast Cancer (TNBC): Responses and Long-term Outcome Correlated with Increased Frequency of DNA Damage Response Gene Mutations, TNBC Subtype, AR Status, and Ki67. Clin. Cancer Res. 2017, 23, 4035–4045. [Google Scholar]
- Park, I.H.; Kong, S.Y.; Kwon, Y.; Kim, M.K.; Sim, S.H.; Joo, J.; Lee, K.S. Phase I/II clinical trial of everolimus combined with gemcitabine/cisplatin for metastatic triple-negative breast cancer. J. Cancer 2018, 9, 1145–1151. [Google Scholar] [CrossRef] [Green Version]
- Decker, T.; Marschner, N.; Muendlein, A.; Welt, A.; Hagen, V.; Rauh, J.; Schroder, H.; Jaehnig, P.; Potthoff, K.; Lerchenmuller, C. VicTORia: A randomised phase II study to compare vinorelbine in combination with the mTOR inhibitor everolimus versus vinorelbine monotherapy for second-line chemotherapy in advanced HER2-negative breast cancer. Breast Cancer Res. Treat. 2019, 176, 637–647. [Google Scholar] [CrossRef]
- Kim, S.J.; Shin, D.Y.; Kim, J.S.; Yoon, D.H.; Lee, W.S.; Lee, H.; Do, Y.R.; Kang, H.J.; Eom, H.S.; Ko, Y.H.; et al. A phase II study of everolimus (RAD001), an mTOR inhibitor plus CHOP for newly diagnosed peripheral T-cell lymphomas. Ann. Oncol. 2016, 27, 712–718. [Google Scholar] [CrossRef]
- Trucco, M.M.; Meyer, C.F.; Thornton, K.A.; Shah, P.; Chen, A.R.; Wilky, B.A.; Carrera-Haro, M.A.; Boyer, L.C.; Ferreira, M.F.; Shafique, U.; et al. A phase II study of temsirolimus and liposomal doxorubicin for patients with recurrent and refractory bone and soft tissue sarcomas. Clin. Sarcoma Res. 2018, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Place, A.E.; Pikman, Y.; Stevenson, K.E.; Harris, M.H.; Pauly, M.; Sulis, M.L.; Hijiya, N.; Gore, L.; Cooper, T.M.; Loh, M.L.; et al. Phase I trial of the mTOR inhibitor everolimus in combination with multi-agent chemotherapy in relapsed childhood acute lymphoblastic leukemia. Pediatr. Blood Cancer 2018, 65, e27062. [Google Scholar] [CrossRef] [PubMed]
- Emmenegger, U.; Booth, C.M.; Berry, S.; Sridhar, S.S.; Winquist, E.; Bandali, N.; Chow, A.; Lee, C.; Xu, P.; Man, S.; et al. Temsirolimus Maintenance Therapy After Docetaxel Induction in Castration-Resistant Prostate Cancer. Oncologist 2015, 20, 1351–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, P.Y.; Omuro, A.; Ahluwalia, M.S.; Fathallah-Shaykh, H.M.; Mohile, N.; Lager, J.J.; Laird, A.D.; Tang, J.; Jiang, J.; Egile, C.; et al. Phase I dose-escalation study of the PI3K/mTOR inhibitor voxtalisib (SAR245409, XL765) plus temozolomide with or without radiotherapy in patients with high-grade glioma. Neuro-Oncol. 2015, 17, 1275–1283. [Google Scholar] [CrossRef] [Green Version]
- Chung, V.; Frankel, P.; Lim, D.; Yeon, C.; Leong, L.; Chao, J.; Ruel, N.; Luevanos, E.; Koehler, S.; Chung, S.; et al. Phase Ib Trial of mFOLFOX6 and Everolimus (NSC-733504) in Patients with Metastatic Gastroesophageal Adenocarcinoma. Oncology 2016, 90, 307–312. [Google Scholar] [CrossRef]
- Baselga, J.; Campone, M.; Piccart, M.; Burris, H.A., 3rd; Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnant, M.; Pritchard, K.I.; Lebrun, F.; et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N. Engl. J. Med. 2012, 366, 520–529. [Google Scholar] [CrossRef] [Green Version]
- Blackwell, K.; Burris, H.; Gomez, P.; Lynn Henry, N.; Isakoff, S.; Campana, F.; Gao, L.; Jiang, J.; Mace, S.; Tolaney, S.M. Phase I/II dose-escalation study of PI3K inhibitors pilaralisib or voxtalisib in combination with letrozole in patients with hormone-receptor-positive and HER2-negative metastatic breast cancer refractory to a non-steroidal aromatase inhibitor. Breast Cancer Res. Treat. 2015, 154, 287–297. [Google Scholar] [CrossRef]
- Kornblum, N.; Zhao, F.; Manola, J.; Klein, P.; Ramaswamy, B.; Brufsky, A.; Stella, P.J.; Burnette, B.; Telli, M.; Makower, D.F.; et al. Randomized Phase II Trial of Fulvestrant Plus Everolimus or Placebo in Postmenopausal Women With Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Metastatic Breast Cancer Resistant to Aromatase Inhibitor Therapy: Results of PrE0102. J. Clin. Oncol. 2018, 36, 1556–1563. [Google Scholar]
- Rugo, H.S.; Tredan, O.; Ro, J.; Morales, S.M.; Campone, M.; Musolino, A.; Afonso, N.; Ferreira, M.; Park, K.H.; Cortes, J.; et al. A randomized phase II trial of ridaforolimus, dalotuzumab, and exemestane compared with ridaforolimus and exemestane in patients with advanced breast cancer. Breast Cancer Res. Treat. 2017, 165, 601–609. [Google Scholar] [CrossRef]
- Jacinto, E.; Lorberg, A. TOR regulation of AGC kinases in yeast and mammals. Biochem. J. 2008, 410, 19–37. [Google Scholar] [CrossRef]
- Feldman, M.E.; Apsel, B.; Uotila, A.; Loewith, R.; Knight, Z.A.; Ruggero, D.; Shokat, K.M. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009, 7, e38. [Google Scholar] [CrossRef] [PubMed]
- Willems, L.; Chapuis, N.; Puissant, A.; Maciel, T.T.; Green, A.S.; Jacque, N.; Vignon, C.; Park, S.; Guichard, S.; Herault, O.; et al. The dual mTORC1 and mTORC2 inhibitor AZD8055 has anti-tumor activity in acute myeloid leukemia. Leukemia 2012, 26, 1195–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evangelisti, C.; Ricci, F.; Tazzari, P.; Tabellini, G.; Battistelli, M.; Falcieri, E.; Chiarini, F.; Bortul, R.; Melchionda, F.; Pagliaro, P.; et al. Targeted inhibition of mTORC1 and mTORC2 by active-site mTOR inhibitors has cytotoxic effects in T-cell acute lymphoblastic leukemia. Leukemia 2011, leu201120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caumanns, J.J.; Berns, K.; Wisman, G.B.A.; Fehrmann, R.S.N.; Tomar, T.; Klip, H.; Meersma, G.J.; Hijmans, E.M.; Gennissen, A.M.C.; Duiker, E.W.; et al. Integrative Kinome Profiling Identifies mTORC1/2 Inhibition as Treatment Strategy in Ovarian Clear Cell Carcinoma. Clin. Cancer Res. 2018, 24, 3928–3940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borsari, C.; Rageot, D.; Dall’Asen, A.; Bohnacker, T.; Melone, A.; Sele, A.M.; Jackson, E.; Langlois, J.B.; Beaufils, F.; Hebeisen, P.; et al. A Conformational Restriction Strategy for the Identification of a Highly Selective Pyrimido-pyrrolo-oxazine mTOR Inhibitor. J. Med. Chem. 2019. [Google Scholar] [CrossRef]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef] [Green Version]
- Morrison Joly, M.; Hicks, D.J.; Jones, B.; Sanchez, V.; Estrada, M.V.; Young, C.; Williams, M.; Rexer, B.N.; Sarbassov dos, D.; Muller, W.J.; et al. Rictor/mTORC2 Drives Progression and Therapeutic Resistance of HER2-Amplified Breast Cancers. Cancer Res. 2016, 76, 4752–4764. [Google Scholar] [CrossRef] [Green Version]
- Verreault, M.; Weppler, S.A.; Stegeman, A.; Warburton, C.; Strutt, D.; Masin, D.; Bally, M.B. Combined RNAi-mediated suppression of Rictor and EGFR resulted in complete tumor regression in an orthotopic glioblastoma tumor model. PLoS ONE 2013, 8, e59597. [Google Scholar] [CrossRef]
- Wang, L.; Qi, J.; Yu, J.; Chen, H.; Zou, Z.; Lin, X.; Guo, L. Overexpression of Rictor protein in colorectal cancer is correlated with tumor progression and prognosis. Oncol. Lett. 2017, 14, 6198–6202. [Google Scholar] [CrossRef] [Green Version]
- Lang, S.A.; Hackl, C.; Moser, C.; Fichtner-Feigl, S.; Koehl, G.E.; Schlitt, H.J.; Geissler, E.K.; Stoeltzing, O. Implication of RICTOR in the mTOR inhibitor-mediated induction of insulin-like growth factor-I receptor (IGF-IR) and human epidermal growth factor receptor-2 (Her2) expression in gastrointestinal cancer cells. Biochim. Biophys. Acta 2010, 1803, 435–442. [Google Scholar] [CrossRef] [Green Version]
- Werfel, T.A.; Wang, S.; Jackson, M.A.; Kavanaugh, T.E.; Joly, M.M.; Lee, L.H.; Hicks, D.J.; Sanchez, V.; Ericsson, P.G.; Kilchrist, K.V.; et al. Selective mTORC2 Inhibitor Therapeutically Blocks Breast Cancer Cell Growth and Survival. Cancer Res. 2018, 78, 1845–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moraitis, D.; Karanikou, M.; Liakou, C.; Dimas, K.; Tzimas, G.; Tseleni-Balafouta, S.; Patsouris, E.; Rassidakis, G.Z.; Kouvaraki, M.A. SIN1, a critical component of the mTOR-Rictor complex, is overexpressed and associated with AKT activation in medullary and aggressive papillary thyroid carcinomas. Surgery 2014, 156, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hong, X.; Wang, J.; Yin, Y.; Zhang, Y.; Zhou, Y.; Piao, H.L.; Liang, Z.; Zhang, L.; Li, G.; et al. Inhibition of MAPK pathway is essential for suppressing Rheb-Y35N driven tumor growth. Oncogene 2017, 36, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Lasithiotakis, K.G.; Sinnberg, T.W.; Schittek, B.; Flaherty, K.T.; Kulms, D.; Maczey, E.; Garbe, C.; Meier, F.E. Combined inhibition of MAPK and mTOR signaling inhibits growth, induces cell death, and abrogates invasive growth of melanoma cells. J. Investig. Derm. 2008, 128, 2013–2023. [Google Scholar] [CrossRef] [Green Version]
- Molhoek, K.R.; Brautigan, D.L.; Slingluff, C.L., Jr. Synergistic inhibition of human melanoma proliferation by combination treatment with B-Raf inhibitor BAY43-9006 and mTOR inhibitor Rapamycin. J. Transl. Med. 2005, 3, 39. [Google Scholar] [CrossRef] [Green Version]
- Ruder, D.; Papadimitrakopoulou, V.; Shien, K.; Behrens, C.; Kalhor, N.; Chen, H.; Shen, L.; Lee, J.J.; Hong, W.K.; Tang, X.; et al. Concomitant targeting of the mTOR/MAPK pathways: Novel therapeutic strategy in subsets of RICTOR/KRAS-altered non-small cell lung cancer. Oncotarget 2018, 9, 33995–34008. [Google Scholar] [CrossRef] [Green Version]
- Setsu, N.; Yamamoto, H.; Kohashi, K.; Endo, M.; Matsuda, S.; Yokoyama, R.; Nishiyama, K.; Iwamoto, Y.; Dobashi, Y.; Oda, Y. The Akt/mammalian target of rapamycin pathway is activated and associated with adverse prognosis in soft tissue leiomyosarcomas. Cancer 2012, 118, 1637–1648. [Google Scholar] [CrossRef]
- Malhotra, V.; Perry, M.C. Classical chemotherapy: Mechanisms, toxicities and the therapeutic window. Cancer Biol. 2003, 2, S2–S4. [Google Scholar] [CrossRef] [Green Version]
- Daver, N.; Boumber, Y.; Kantarjian, H.; Ravandi, F.; Cortes, J.; Rytting, M.E.; Kawedia, J.D.; Basnett, J.; Culotta, K.S.; Zeng, Z.; et al. A Phase I/II Study of the mTOR Inhibitor Everolimus in Combination with HyperCVAD Chemotherapy in Patients with Relapsed/Refractory Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2015, 21, 2704–2714. [Google Scholar] [CrossRef] [Green Version]
- Rheingold, S.R.; Tasian, S.K.; Whitlock, J.A.; Teachey, D.T.; Borowitz, M.J.; Liu, X.; Minard, C.G.; Fox, E.; Weigel, B.J.; Blaney, S.M. A phase 1 trial of temsirolimus and intensive re-induction chemotherapy for 2nd or greater relapse of acute lymphoblastic leukaemia: A Children’s Oncology Group study (ADVL1114). Br. J. Haematol. 2017, 177, 467–474. [Google Scholar] [CrossRef]
- Vidal, G.A.; Chen, M.; Sheth, S.; Svahn, T.; Guardino, E. Phase I Trial of Everolimus and Capecitabine in Metastatic HER2(-) Breast Cancer. Clin. Breast Cancer 2017, 17, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Deenen, M.J.; Klumpen, H.J.; Richel, D.J.; Sparidans, R.W.; Weterman, M.J.; Beijnen, J.H.; Schellens, J.H.; Wilmink, J.W. Phase I and pharmacokinetic study of capecitabine and the oral mTOR inhibitor everolimus in patients with advanced solid malignancies. Investig. New Drugs 2012, 30, 1557–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Broto, J.; Redondo, A.; Valverde, C.; Vaz, M.A.; Mora, J.; Garcia Del Muro, X.; Gutierrez, A.; Tous, C.; Carnero, A.; Marcilla, D.; et al. Gemcitabine plus sirolimus for relapsed and progressing osteosarcoma patients after standard chemotherapy: A multicenter, single-arm phase II trial of Spanish Group for Research on Sarcoma (GEIS). Ann. Oncol. 2017, 28, 2994–2999. [Google Scholar] [CrossRef] [PubMed]
- Karavasilis, V.; Samantas, E.; Koliou, G.A.; Kalogera-Fountzila, A.; Pentheroudakis, G.; Varthalitis, I.; Linardou, H.; Rallis, G.; Skondra, M.; Papadopoulos, G.; et al. Gemcitabine Combined with the mTOR Inhibitor Temsirolimus in Patients with Locally Advanced or Metastatic Pancreatic Cancer. A Hellenic Cooperative Oncology Group Phase I/II Study. Target. Oncol. 2018, 13, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.; Tiong, I.S.; Fleming, S.; Pomilio, G.; Cummings, N.; Droogleever, M.; McManus, J.; Schwarer, A.; Catalano, J.; Patil, S.; et al. The mTOR inhibitor everolimus in combination with azacitidine in patients with relapsed/refractory acute myeloid leukemia: A phase Ib/II study. Oncotarget 2017, 8, 52269–52280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valvezan, A.J.; Turner, M.; Belaid, A.; Lam, H.C.; Miller, S.K.; McNamara, M.C.; Baglini, C.; Housden, B.E.; Perrimon, N.; Kwiatkowski, D.J.; et al. mTORC1 Couples Nucleotide Synthesis to Nucleotide Demand Resulting in a Targetable Metabolic Vulnerability. Cancer Cell 2017, 32, 624–638. [Google Scholar] [CrossRef] [Green Version]
- Yan, C.; Wei, H.; Minjuan, Z.; Yan, X.; Jingyue, Y.; Wenchao, L.; Sheng, H. The mTOR inhibitor rapamycin synergizes with a fatty acid synthase inhibitor to induce cytotoxicity in ER/HER2-positive breast cancer cells. PLoS ONE 2014, 9, e97697. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.S.; Matsuura, K.; Huang, N.J.; Robeson, A.C.; Huang, B.; Zhang, L.; Kornbluth, S. Fatty acid synthase inhibition engages a novel caspase-2 regulatory mechanism to induce ovarian cancer cell death. Oncogene 2015, 34, 3264–3272. [Google Scholar] [CrossRef] [Green Version]
- Khawaja, M.R.; Nick, A.M.; Madhusudanannair, V.; Fu, S.; Hong, D.; McQuinn, L.M.; Ng, C.S.; Piha-Paul, S.A.; Janku, F.; Subbiah, V.; et al. Phase I dose escalation study of temsirolimus in combination with metformin in patients with advanced/refractory cancers. Cancer Chemother. Pharm. 2016, 77, 973–977. [Google Scholar] [CrossRef]
- MacKenzie, M.J.; Ernst, S.; Johnson, C.; Winquist, E. A phase I study of temsirolimus and metformin in advanced solid tumours. Investig. New Drugs 2012, 30, 647–652. [Google Scholar] [CrossRef]
- Molenaar, R.J.; van de Venne, T.; Weterman, M.J.; Mathot, R.A.; Klumpen, H.J.; Richel, D.J.; Wilmink, J.W. A phase Ib study of everolimus combined with metformin for patients with advanced cancer. Investig. New Drugs 2018, 36, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Sehdev, A.; Karrison, T.; Zha, Y.; Janisch, L.; Turcich, M.; Cohen, E.E.W.; Maitland, M.; Polite, B.N.; Gajewski, T.F.; Salgia, R.; et al. A pharmacodynamic study of sirolimus and metformin in patients with advanced solid tumors. Cancer Chemother. Pharm. 2018, 82, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Chiche, J.; Reverso-Meinietti, J.; Mouchotte, A.; Rubio-Patino, C.; Mhaidly, R.; Villa, E.; Bossowski, J.P.; Proics, E.; Grima-Reyes, M.; Paquet, A.; et al. GAPDH Expression Predicts the Response to R-CHOP, the Tumor Metabolic Status, and the Response of DLBCL Patients to Metabolic Inhibitors. Cell Metab. 2019, 29, 1243–1257. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Niu, B.; Wang, L.; Chen, M.; Kang, X.; Wang, L.; Ji, Y.; Zhong, J. Autophagy inhibition enhances colorectal cancer apoptosis induced by dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235. Oncol. Lett. 2016, 12, 102–106. [Google Scholar] [CrossRef] [Green Version]
- Rangwala, R.; Chang, Y.C.; Hu, J.; Algazy, K.M.; Evans, T.L.; Fecher, L.A.; Schuchter, L.M.; Torigian, D.A.; Panosian, J.T.; Troxel, A.B.; et al. Combined MTOR and autophagy inhibition: Phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Haas, N.B.; Appleman, L.J.; Stein, M.; Redlinger, M.; Wilks, M.; Xu, X.; Onorati, A.; Kalavacharla, A.; Kim, T.; Zhen, C.J.; et al. Autophagy Inhibition to Augment mTOR Inhibition: A Phase I/II Trial of Everolimus and Hydroxychloroquine in Patients with Previously Treated Renal Cell Carcinoma. Clin. Cancer Res. 2019, 25, 2080–2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahoney, S.J.; Narayan, S.; Molz, L.; Berstler, L.A.; Kang, S.A.; Vlasuk, G.P.; Saiah, E. A small molecule inhibitor of Rheb selectively targets mTORC1 signaling. Nat. Commun. 2018, 9, 548. [Google Scholar] [CrossRef]
- Niessner, H.; Beck, D.; Sinnberg, T.; Lasithiotakis, K.; Maczey, E.; Gogel, J.; Venturelli, S.; Berger, A.; Mauthe, M.; Toulany, M.; et al. The farnesyl transferase inhibitor lonafarnib inhibits mTOR signaling and enforces sorafenib-induced apoptosis in melanoma cells. J. Investig. Derm. 2011, 131, 468–479. [Google Scholar] [CrossRef] [Green Version]
- Becher, O.J.; Gilheeney, S.W.; Khakoo, Y.; Lyden, D.C.; Haque, S.; De Braganca, K.C.; Kolesar, J.M.; Huse, J.T.; Modak, S.; Wexler, L.H.; et al. A phase I study of perifosine with temsirolimus for recurrent pediatric solid tumors. Pediatr. Blood Cancer 2017, 64. [Google Scholar] [CrossRef]
- Gupta, S.; Argiles, G.; Munster, P.N.; Hollebecque, A.; Dajani, O.; Cheng, J.D.; Wang, R.; Swift, A.; Tosolini, A.; Piha-Paul, S.A. A Phase I Trial of Combined Ridaforolimus and MK-2206 in Patients with Advanced Malignancies. Clin. Cancer Res. 2015, 21, 5235–5244. [Google Scholar] [CrossRef] [Green Version]
- Xing, Y.; Lin, N.U.; Maurer, M.A.; Chen, H.; Mahvash, A.; Sahin, A.; Akcakanat, A.; Li, Y.; Abramson, V.; Litton, J.; et al. Phase II trial of AKT inhibitor MK-2206 in patients with advanced breast cancer who have tumors with PIK3CA or AKT mutations, and/or PTEN loss/PTEN mutation. Breast Cancer Res. 2019, 21, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonasch, E.; Hasanov, E.; Corn, P.G.; Moss, T.; Shaw, K.R.; Stovall, S.; Marcott, V.; Gan, B.; Bird, S.; Wang, X.; et al. A randomized phase 2 study of MK-2206 versus everolimus in refractory renal cell carcinoma. Ann. Oncol. 2017, 28, 804–808. [Google Scholar] [CrossRef] [PubMed]
- Oki, Y.; Fanale, M.; Romaguera, J.; Fayad, L.; Fowler, N.; Copeland, A.; Samaniego, F.; Kwak, L.W.; Neelapu, S.; Wang, M.; et al. Phase II study of an AKT inhibitor MK2206 in patients with relapsed or refractory lymphoma. Br. J. Haematol. 2015, 171, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.T.; McEwen, R.; Crafter, C.; McDermott, U.; Garnett, M.J.; Barry, S.T.; Davies, B.R. Identification of differential PI3K pathway target dependencies in T-cell acute lymphoblastic leukemia through a large cancer cell panel screen. Oncotarget 2016, 7, 22128–22139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.; Lan, C.; Jiao, G.; Fu, W.; Long, X.; An, Y.; Wang, K.; Zhou, J.; Chen, T.; Li, Y.; et al. Therapeutic inhibition of SGK1 suppresses colorectal cancer. Exp. Mol. Med. 2017, 49, e399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matschke, J.; Wiebeck, E.; Hurst, S.; Rudner, J.; Jendrossek, V. Role of SGK1 for fatty acid uptake, cell survival and radioresistance of NCI-H460 lung cancer cells exposed to acute or chronic cycling severe hypoxia. Radiat. Oncol. 2016, 11, 75. [Google Scholar] [CrossRef] [Green Version]
- Talarico, C.; D’Antona, L.; Scumaci, D.; Barone, A.; Gigliotti, F.; Fiumara, C.V.; Dattilo, V.; Gallo, E.; Visca, P.; Ortuso, F.; et al. Preclinical model in HCC: The SGK1 kinase inhibitor SI113 blocks tumor progression in vitro and in vivo and synergizes with radiotherapy. Oncotarget 2015, 6, 37511–37525. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Huang, X.; Liu, H.; Xiao, F.; Wei, J.; You, L.; Qian, W. PDK1 inhibitor GSK2334470 exerts antitumor activity in multiple myeloma and forms a novel multitargeted combination with dual mTORC1/C2 inhibitor PP242. Oncotarget 2017, 8, 39185–39197. [Google Scholar] [CrossRef]
- Hopkins, B.D.; Pauli, C.; Du, X.; Wang, D.G.; Li, X.; Wu, D.; Amadiume, S.C.; Goncalves, M.D.; Hodakoski, C.; Lundquist, M.R.; et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 2018, 560, 499–503. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Lan, C.; Zhou, J.; Fu, W.; Long, X.; An, Y.; Jiao, G.; Wang, K.; Li, Y.; Xu, J.; et al. Development of a new analog of SGK1 inhibitor and its evaluation as a therapeutic molecule of colorectal cancer. J. Cancer 2017, 8, 2256–2262. [Google Scholar] [CrossRef] [Green Version]
- Earwaker, P.; Anderson, C.; Willenbrock, F.; Harris, A.L.; Protheroe, A.S.; Macaulay, V.M. RAPTOR up-regulation contributes to resistance of renal cancer cells to PI3K-mTOR inhibition. PLoS ONE 2018, 13, e0191890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamhamedi-Cherradi, S.E.; Menegaz, B.A.; Ramamoorthy, V.; Vishwamitra, D.; Wang, Y.; Maywald, R.L.; Buford, A.S.; Fokt, I.; Skora, S.; Wang, J.; et al. IGF-1R and mTOR Blockade: Novel Resistance Mechanisms and Synergistic Drug Combinations for Ewing Sarcoma. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.; Benavides-Serrato, A.; Saunders, J.T.; Landon, K.A.; Schreck, A.J.; Nishimura, R.N.; Gera, J. The protein arginine methyltransferase PRMT5 confers therapeutic resistance to mTOR inhibition in glioblastoma. J. Neurooncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Makinoshima, H.; Umemura, S.; Suzuki, A.; Nakanishi, H.; Maruyama, A.; Udagawa, H.; Mimaki, S.; Matsumoto, S.; Niho, S.; Ishii, G.; et al. Metabolic Determinants of Sensitivity to Phosphatidylinositol 3-Kinase Pathway Inhibitor in Small-Cell Lung Carcinoma. Cancer Res. 2018, 78, 2179–2190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, W.; Shin, Y.S.; Xue, M.; Matsutani, T.; Masui, K.; Yang, H.; Ikegami, S.; Gu, Y.; Herrmann, K.; Johnson, D.; et al. Single-Cell Phosphoproteomics Resolves Adaptive Signaling Dynamics and Informs Targeted Combination Therapy in Glioblastoma. Cancer Cell 2016, 29, 563–573. [Google Scholar] [CrossRef] [Green Version]
- Jacinto, E.; Werlen, G. mTOR: The mTOR complexes in T cell development and immunity. In Encyclopedia of Inflammatory Diseases; Springer: Berlin, Germany, 2015. [Google Scholar]
- Thomson, A.W.; Turnquist, H.R.; Raimondi, G. Immunoregulatory functions of mTOR inhibition. Nat. Rev. Immunol. 2009, 9, 324–337. [Google Scholar] [CrossRef] [Green Version]
- Patel, C.H.; Leone, R.D.; Horton, M.R.; Powell, J.D. Targeting metabolism to regulate immune responses in autoimmunity and cancer. Nat. Rev. Drug Discov. 2019, 18, 669–688. [Google Scholar] [CrossRef]
- Frauwirth, K.A.; Riley, J.L.; Harris, M.H.; Parry, R.V.; Rathmell, J.C.; Plas, D.R.; Elstrom, R.L.; June, C.H.; Thompson, C.B. The CD28 signaling pathway regulates glucose metabolism. Immunity 2002, 16, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [Green Version]
- Geltink, R.I.K.; Kyle, R.L.; Pearce, E.L. Unraveling the Complex Interplay Between T Cell Metabolism and Function. Annu. Rev. Immunol. 2018, 36, 461–488. [Google Scholar] [CrossRef]
- Huijts, C.M.; Lougheed, S.M.; Bodalal, Z.; van Herpen, C.M.; Hamberg, P.; Tascilar, M.; Haanen, J.B.; Verheul, H.M.; de Gruijl, T.D.; van der Vliet, H.J.; et al. The effect of everolimus and low-dose cyclophosphamide on immune cell subsets in patients with metastatic renal cell carcinoma: Results from a phase I clinical trial. Cancer Immunol. Immunother. 2019, 68, 503–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battaglia, M.; Stabilini, A.; Migliavacca, B.; Horejs-Hoeck, J.; Kaupper, T.; Roncarolo, M.G. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J. Immunol. 2006, 177, 8338–8347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirayama, Y.; Gi, M.; Yamano, S.; Tachibana, H.; Okuno, T.; Tamada, S.; Nakatani, T.; Wanibuchi, H. Anti-PD-L1 treatment enhances antitumor effect of everolimus in a mouse model of renal cell carcinoma. Cancer Sci. 2016, 107, 1736–1744. [Google Scholar] [CrossRef]
- Cao, G.; Wang, Q.; Li, G.; Meng, Z.; Liu, H.; Tong, J.; Huang, W.; Liu, Z.; Jia, Y.; Wei, J.; et al. mTOR inhibition potentiates cytotoxicity of Vgamma4 gammadelta T cells via up-regulating NKG2D and TNF-alpha. J. Leukoc. Biol. 2016, 100, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Crompton, J.G.; Sukumar, M.; Roychoudhuri, R.; Clever, D.; Gros, A.; Eil, R.L.; Tran, E.; Hanada, K.; Yu, Z.; Palmer, D.C.; et al. Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristics. Cancer Res. 2015, 75, 296–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alizadeh, D.; Wong, R.A.; Yang, X.; Wang, D.; Pecoraro, J.R.; Kuo, C.F.; Aguilar, B.; Qi, Y.; Ann, D.K.; Starr, R.; et al. IL15 Enhances CAR-T Cell Antitumor Activity by Reducing mTORC1 Activity and Preserving Their Stem Cell Memory Phenotype. Cancer Immunol. Res. 2019, 7, 759–772. [Google Scholar] [CrossRef]
- Pearce, E.L.; Walsh, M.C.; Cejas, P.J.; Harms, G.M.; Shen, H.; Wang, L.S.; Jones, R.G.; Choi, Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 2009, 460, 103–107. [Google Scholar] [CrossRef]
- Araki, K.; Turner, A.P.; Shaffer, V.O.; Gangappa, S.; Keller, S.A.; Bachmann, M.F.; Larsen, C.P.; Ahmed, R. mTOR regulates memory CD8 T-cell differentiation. Nature 2009, 460, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Pollizzi, K.N.; Patel, C.H.; Sun, I.H.; Oh, M.H.; Waickman, A.T.; Wen, J.; Delgoffe, G.M.; Powell, J.D. mTORC1 and mTORC2 selectively regulate CD8(+) T cell differentiation. J. Clin. Investig. 2015, 125, 2090–2108. [Google Scholar] [CrossRef] [Green Version]
- Turner, A.P.; Shaffer, V.O.; Araki, K.; Martens, C.; Turner, P.L.; Gangappa, S.; Ford, M.L.; Ahmed, R.; Kirk, A.D.; Larsen, C.P. Sirolimus enhances the magnitude and quality of viral-specific CD8+ T-cell responses to vaccinia virus vaccination in rhesus macaques. Am. J. Transpl. 2011, 11, 613–618. [Google Scholar] [CrossRef]
- Mannick, J.B.; Morris, M.; Hockey, H.P.; Roma, G.; Beibel, M.; Kulmatycki, K.; Watkins, M.; Shavlakadze, T.; Zhou, W.; Quinn, D.; et al. TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gammon, J.M.; Gosselin, E.A.; Tostanoski, L.H.; Chiu, Y.C.; Zeng, X.; Zeng, Q.; Jewell, C.M. Low-dose controlled release of mTOR inhibitors maintains T cell plasticity and promotes central memory T cells. J. Control. Release: Off. J. Control. Release Soc. 2017, 263, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Lin, H.W.; Sun, N.Y.; Yie, J.C.; Hung, H.C.; Chen, C.A.; Sun, W.Z.; Cheng, W.F. mTOR Inhibitors Can Enhance the Anti-Tumor Effects of DNA Vaccines through Modulating Dendritic Cell Function in the Tumor Microenvironment. Cancers 2019, 11, 617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amiel, E.; Everts, B.; Freitas, T.C.; King, I.L.; Curtis, J.D.; Pearce, E.L.; Pearce, E.J. Inhibition of mechanistic target of rapamycin promotes dendritic cell activation and enhances therapeutic autologous vaccination in mice. J. Immunol. 2012, 189, 2151–2158. [Google Scholar] [CrossRef] [Green Version]
- Figlin, R.A.; de Souza, P.; McDermott, D.; Dutcher, J.P.; Berkenblit, A.; Thiele, A.; Krygowski, M.; Strahs, A.; Feingold, J.; Boni, J.; et al. Analysis of PTEN and HIF-1alpha and correlation with efficacy in patients with advanced renal cell carcinoma treated with temsirolimus versus interferon-alpha. Cancer 2009, 115, 3651–3660. [Google Scholar] [CrossRef]
- Cho, D.; Signoretti, S.; Dabora, S.; Regan, M.; Seeley, A.; Mariotti, M.; Youmans, A.; Polivy, A.; Mandato, L.; McDermott, D.; et al. Potential histologic and molecular predictors of response to temsirolimus in patients with advanced renal cell carcinoma. Clin. Genitourin. Cancer 2007, 5, 379–385. [Google Scholar] [CrossRef]
- Janku, F.; Wheler, J.J.; Naing, A.; Stepanek, V.M.; Falchook, G.S.; Fu, S.; Garrido-Laguna, I.; Tsimberidou, A.M.; Piha-Paul, S.A.; Moulder, S.L.; et al. PIK3CA mutations in advanced cancers: Characteristics and outcomes. Oncotarget 2012, 3, 1566–1575. [Google Scholar] [CrossRef] [Green Version]
- Batsios, G.; Viswanath, P.; Subramani, E.; Najac, C.; Gillespie, A.M.; Santos, R.D.; Molloy, A.R.; Pieper, R.O.; Ronen, S.M. PI3K/mTOR inhibition of IDH1 mutant glioma leads to reduced 2HG production that is associated with increased survival. Sci. Rep. 2019, 9, 10521. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Kwok-Shing Ng, P.; Kucherlapati, M.; Chen, F.; Liu, Y.; Tsang, Y.H.; de Velasco, G.; Jeong, K.J.; Akbani, R.; Hadjipanayis, A.; et al. A Pan-Cancer Proteogenomic Atlas of PI3K/AKT/mTOR Pathway Alterations. Cancer Cell 2017, 31, 820–832. [Google Scholar] [CrossRef] [Green Version]
- Dienstmann, R.; Serpico, D.; Rodon, J.; Saura, C.; Macarulla, T.; Elez, E.; Alsina, M.; Capdevila, J.; Perez-Garcia, J.; Sanchez-Olle, G.; et al. Molecular profiling of patients with colorectal cancer and matched targeted therapy in phase I clinical trials. Mol. Cancer 2012, 11, 2062–2071. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Carreres, L.; Puyal, J.; Leal-Esteban, L.C.; Orpinell, M.; Castillo-Armengol, J.; Giralt, A.; Dergai, O.; Moret, C.; Barquissau, V.; Nasrallah, A.; et al. CDK4 regulates lysosomal function and mTORC1 activation to promote cancer cell survival. Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zibelman, M.; Wong, Y.N.; Devarajan, K.; Malizzia, L.; Corrigan, A.; Olszanski, A.J.; Denlinger, C.S.; Roethke, S.K.; Tetzlaff, C.H.; Plimack, E.R. Phase I study of the mTOR inhibitor ridaforolimus and the HDAC inhibitor vorinostat in advanced renal cell carcinoma and other solid tumors. Investig. New Drugs 2015, 33, 1040–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murugan, A.K.; Liu, R.; Xing, M. Identification and characterization of two novel oncogenic mTOR mutations. Oncogene 2019, 38, 5211–5226. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Si, L.; Li, Y.; Wu, X.; Xu, X.; Dai, J.; Tang, H.; Ma, M.; Chi, Z.; Sheng, X.; et al. Analysis of mTOR Gene Aberrations in Melanoma Patients and Evaluation of Their Sensitivity to PI3K-AKT-mTOR Pathway Inhibitors. Clin. Cancer Res. 2016, 22, 1018–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Pham, C.G.; Albanese, S.K.; Dong, Y.; Oyama, T.; Lee, C.H.; Rodrik-Outmezguine, V.; Yao, Z.; Han, S.; Chen, D.; et al. Mechanistically distinct cancer-associated mTOR activation clusters predict sensitivity to rapamycin. J. Clin. Investig. 2016, 126, 3526–3540. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Kawazu, M.; Yasuda, T.; Soda, M.; Ueno, T.; Kojima, S.; Yashiro, M.; Yoshino, I.; Ishikawa, Y.; Sai, E.; et al. Transforming somatic mutations of mammalian target of rapamycin kinase in human cancer. Cancer Sci. 2015, 106, 1687–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, T.; Nakashima, A.; Guo, L.; Coffman, K.; Tamanoi, F. Single amino-acid changes that confer constitutive activation of mTOR are discovered in human cancer. Oncogene 2010, 29, 2746–2752. [Google Scholar] [CrossRef] [Green Version]
- Verheijen, R.B.; Atrafi, F.; Schellens, J.H.M.; Beijnen, J.H.; Huitema, A.D.R.; Mathijssen, R.H.J.; Steeghs, N. Pharmacokinetic Optimization of Everolimus Dosing in Oncology: A Randomized Crossover Trial. Clin. Pharm. 2018, 57, 637–644. [Google Scholar] [CrossRef] [Green Version]
- Lunova, M.; Smolkova, B.; Lynnyk, A.; Uzhytchak, M.; Jirsa, M.; Kubinova, S.; Dejneka, A.; Lunov, O. Targeting the mTOR Signaling Pathway Utilizing Nanoparticles: A Critical Overview. Cancers 2019, 11, 82. [Google Scholar] [CrossRef] [Green Version]
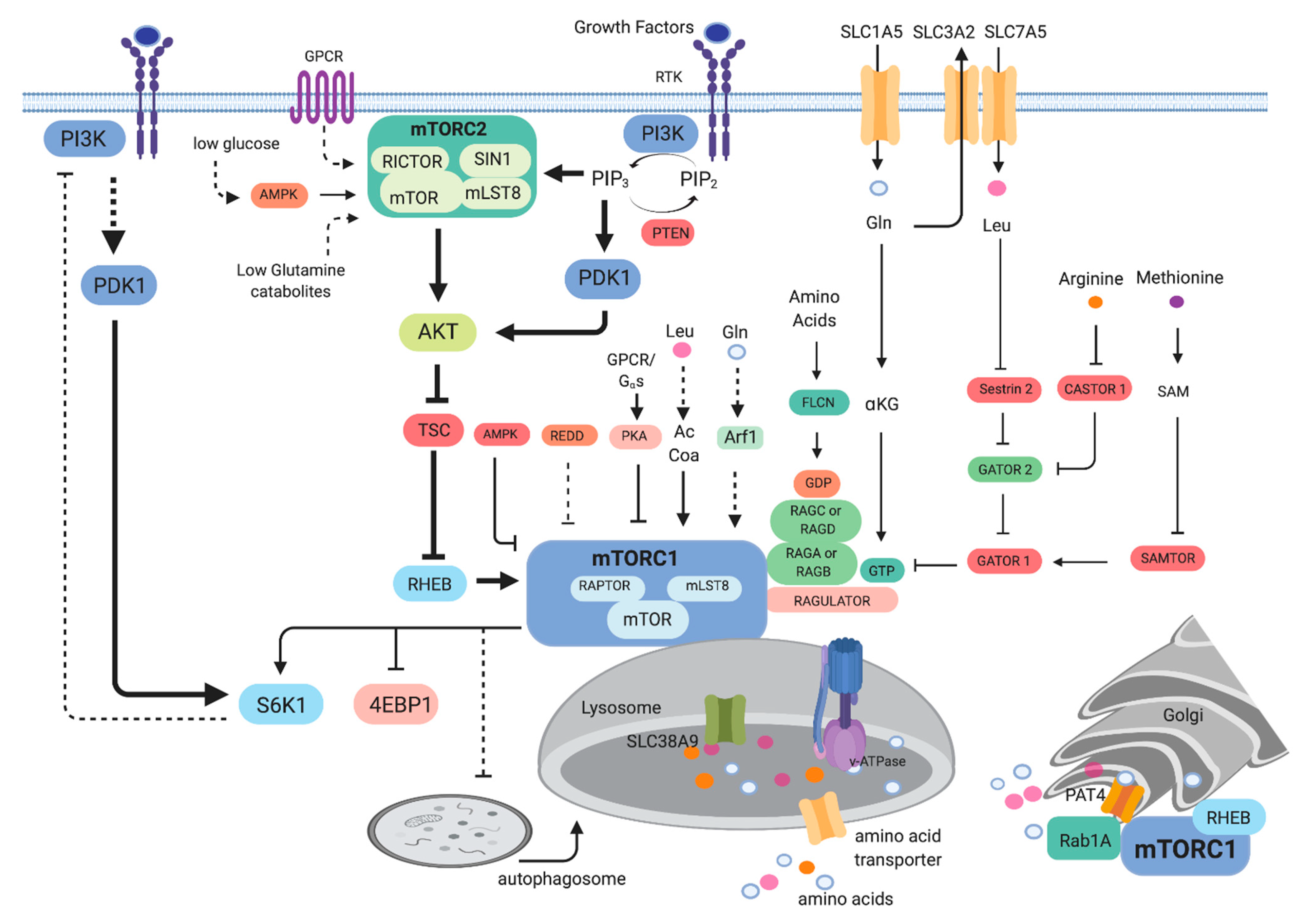
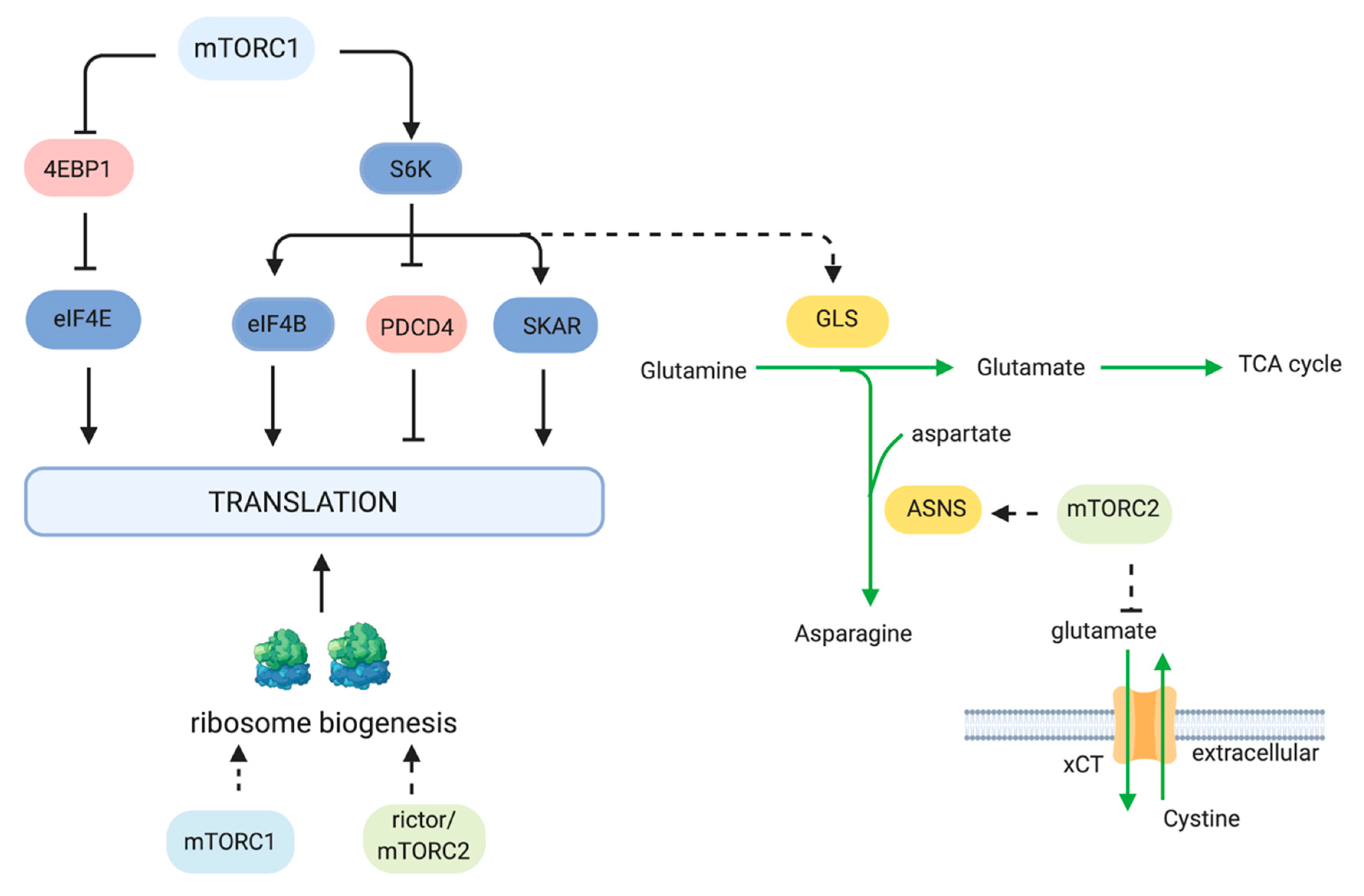





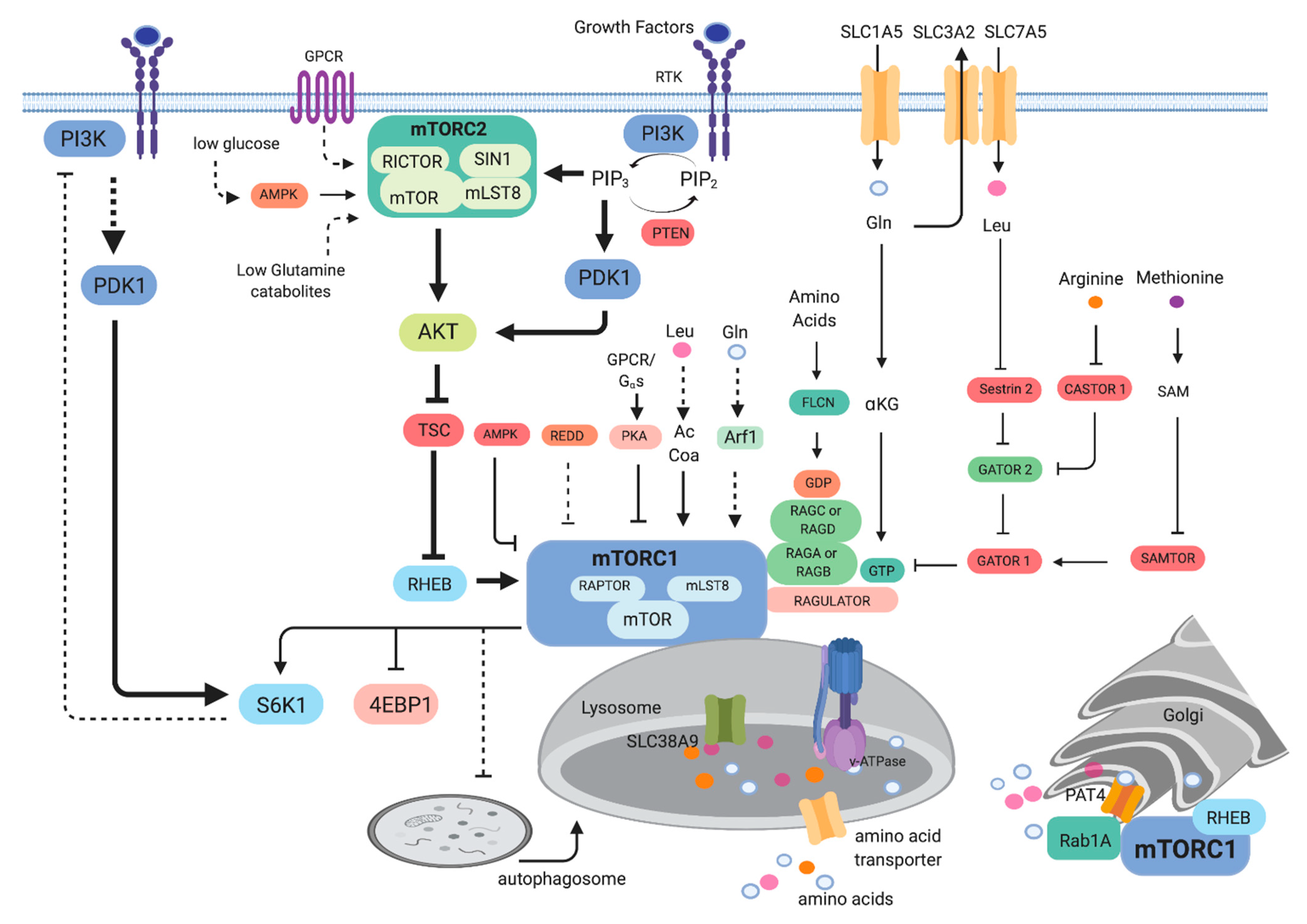{kind=link}
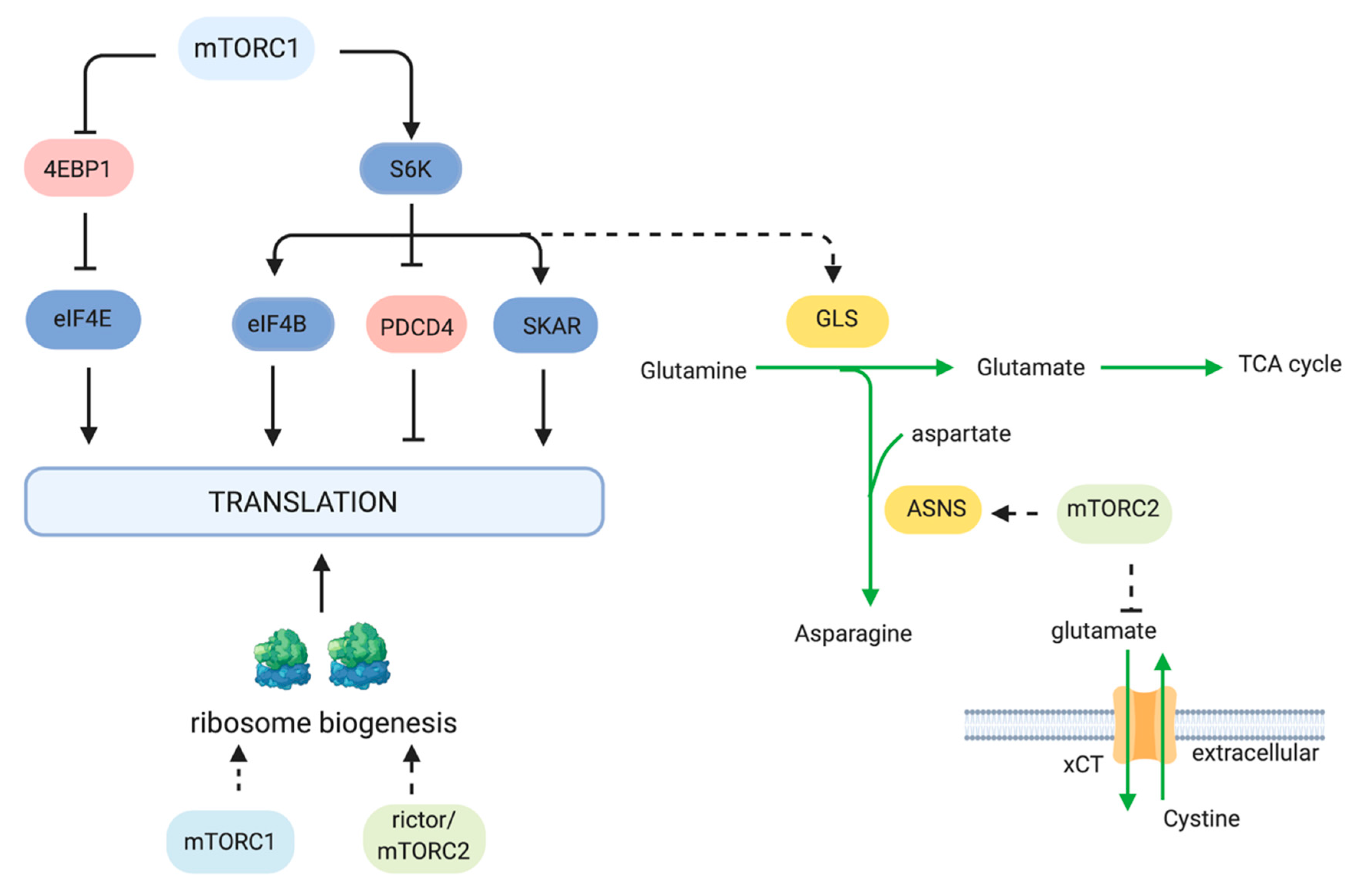{kind=link}
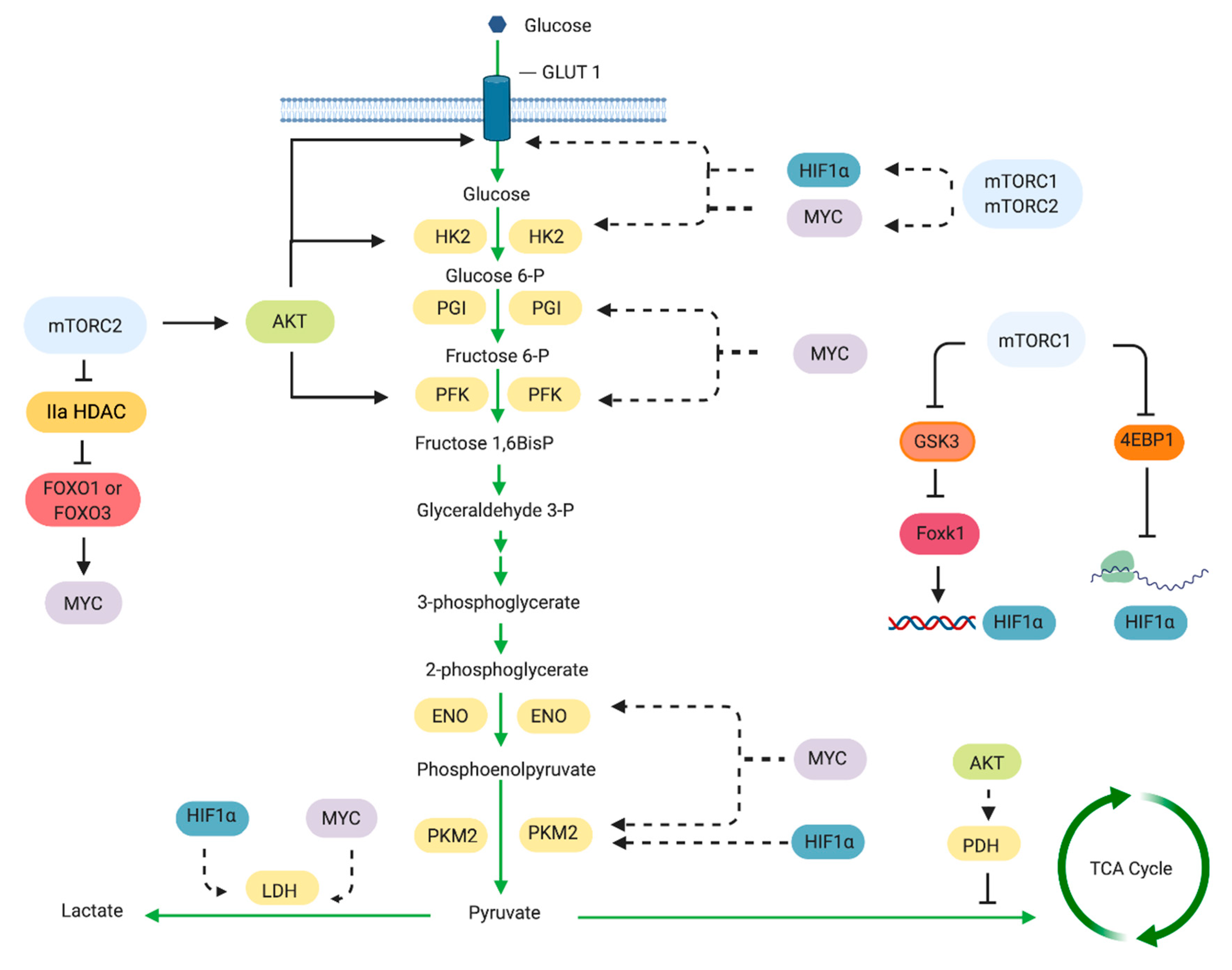{kind=link}
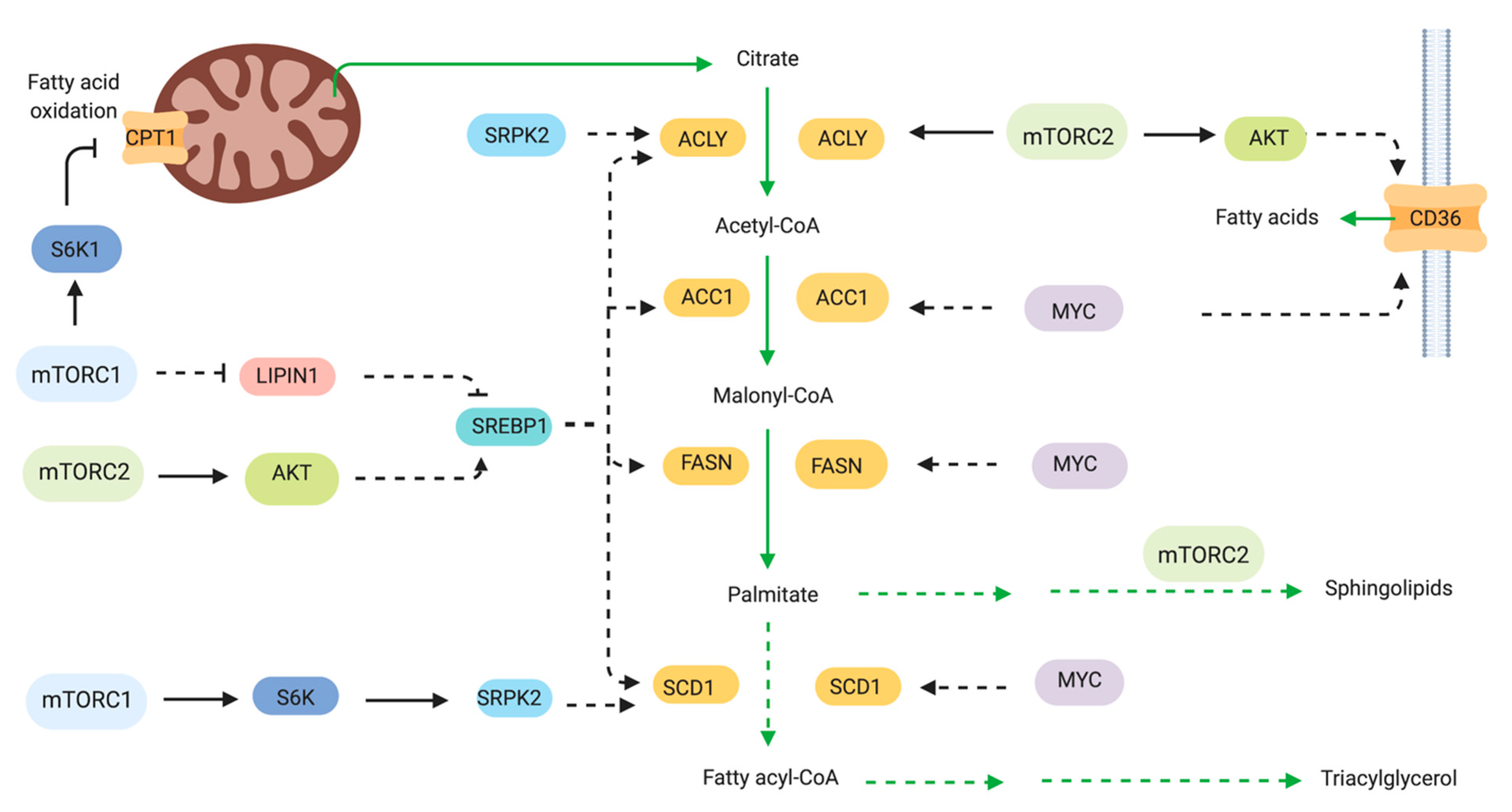{kind=link}
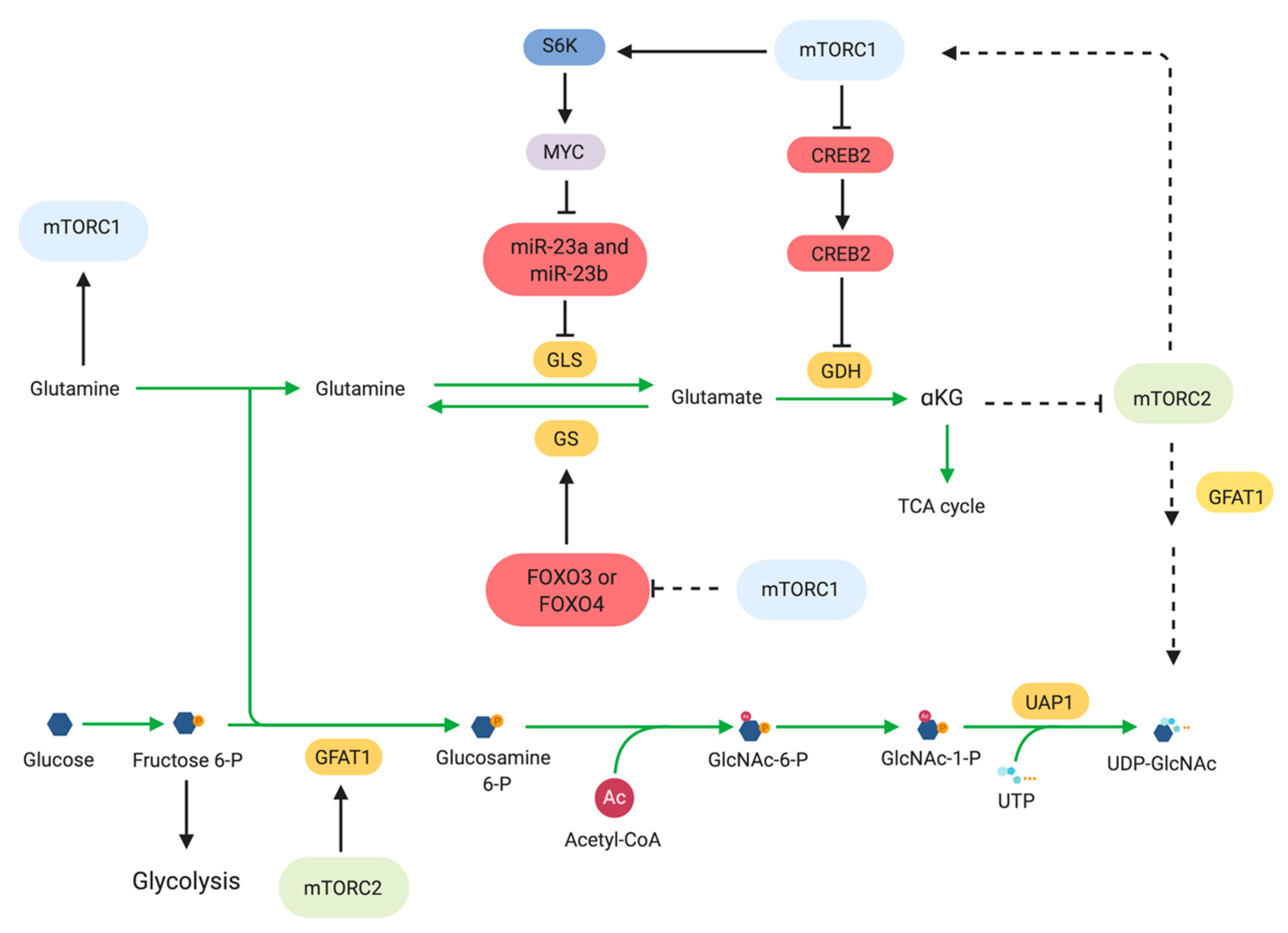{kind=link}
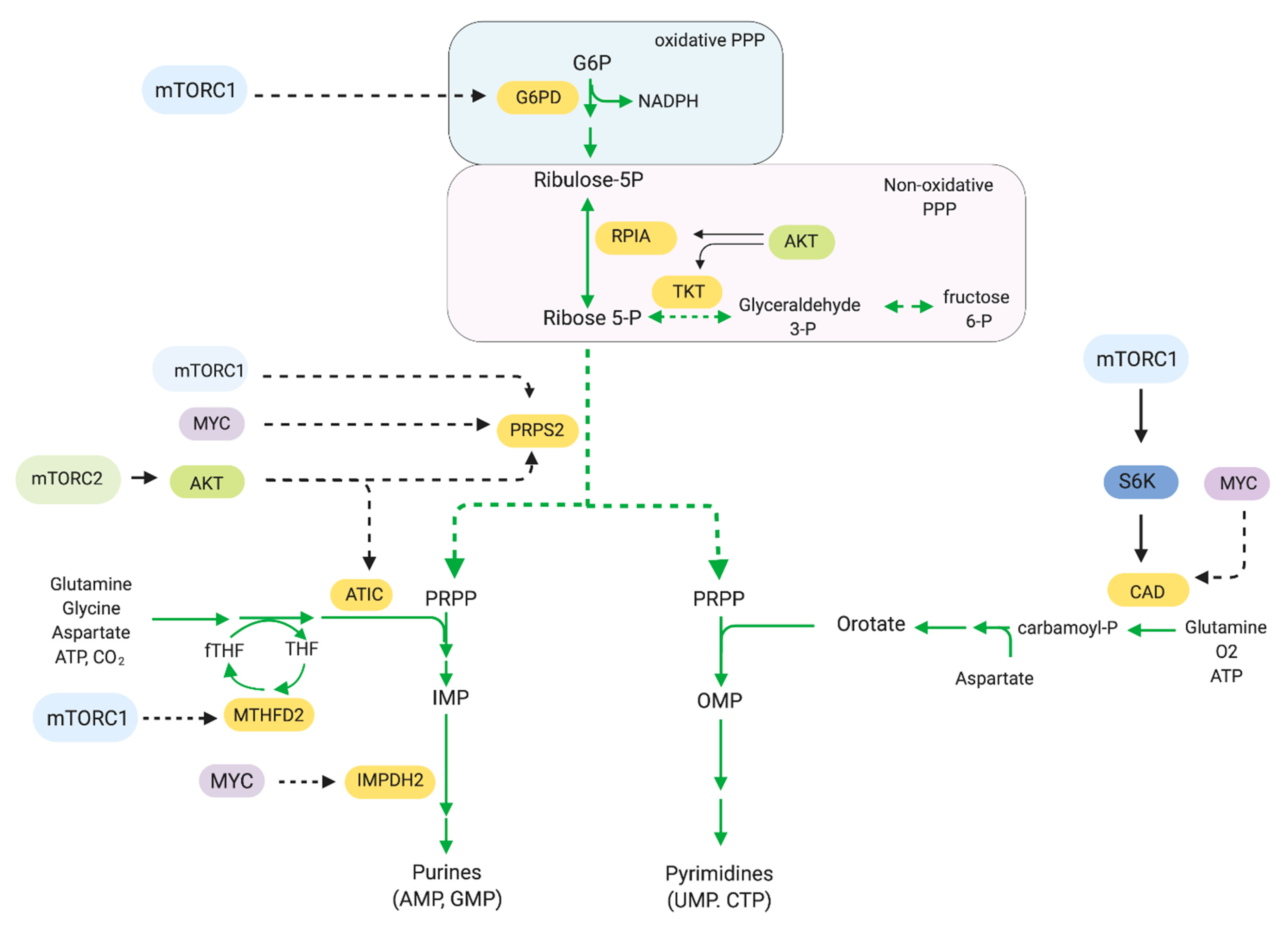{kind=link}
| Drug | Target | References |
|---|---|---|
| Rapalogs (Rapamycin Analogs) | ||
| Sirolimus | mTORC1/FKBP12 | [188,189] |
| Everolimus (Affinitor, RAD001) | mTORC1/FKBP12 | [171,172,173,174,175,176,177,181,190,191,192,193,194] |
| Temsirolimus (Torisel, CCI779) | mTORC1/FKBP12 | [178,179,180,195] |
| Ridaforolimus (deforolimus, AP23573) | mTORC1/FKBP12 | [182,183,184,185,186,187] |
| mTOR ATP-competitive inhibitors (TORKIs) | ||
| AZD8055 | mTOR | [196,197,198,199] |
| AZD2014, vistusertib | mTOR | [200,201,202,203] |
| TAK-228; INK-128; sapanisertib | mTOR | [204,205] |
| OSI-027; ASP7486 | mTOR | [206] |
| MLN0128 | mTOR | [207] |
| CC-223 | mTOR | [208] |
| Dual PI3K/mTOR inhibitors | ||
| BEZ235; dactolisib | PI3K/mTOR | [209,210,211,212,213,214,215,216,217,218,219,220] |
| PKI-587; gedatolisib | PI3K/mTOR | [221,222] |
| GDC-0980, apitolisib | PI3K/mTOR | [223,224,225] |
| SAR245409, XL765; voxtalisib | PI3K/mTOR | [226] |
| BGT226 | PI3K/mTOR | [227] |
| CMG002 | PI3K/mTOR | [228] |
| GSK2126458 | PI3K/mTOR | [229] |
| Other | ||
| JR-AB2-011 | Rictor | [230] |
| RapaLink1 | mTOR/FKBP12 | [231,232,233] |
| Drug | Target | References |
|---|---|---|
| RTK inhibitors | ||
| Trastuzumab | HER2 | [244,245] |
| Neratinib | HER2 | [246] |
| Tivantinib | c-Met | [247] |
| Cetuximab | EGFR | [248,249] |
| Bevacizumab | VEGF | [249,250] |
| Dalotuzumab | IGF-1R | [251] |
| Cixutumumab | IGF-1R | [252] |
| Pazopanib | Tyrosine Kinase | [164] |
| Lapatinib | HER2/EGFR | [253] |
| Ras/MAPK Pathway Inhibitors | ||
| Sorafenib | Raf/VEGFR | [228,254,255,256] |
| RAF265 | Pan-Raf | [257] |
| Selumetinib | MEK1/2 | [258,259] |
| MNKI-57; MNKI-4 | Mnk1/2, Mnk2 | [260] |
| Trametinib (GSK1120212) | MEK | [229,261] |
| Pimasertib | MEK | [262,263] |
| Cytotoxic Chemotherapy | ||
| Carboplatin | DNA | [264,265,266] |
| Paclitaxel | Microtubules | [266,267,268] |
| Cisplatin | DNA | [268,269] |
| Vinorelbine | Microtubules | [244,270] |
| Cyclophosphamide | DNA | [271] |
| Doxorubicin | Topoisomerase | [272] |
| Vincristine | Microtubules | [273] |
| Docetaxel | Microtubules | [274] |
| Temozolomide | DNA | [275] |
| Oxaliplatin | DNA | [276] |
| Other Targeted Therapies | ||
| Exemestane | Aromatase | [277] |
| Letrozole | Aromatase | [278] |
| Fulvestrant | Estrogen Receptor | [279] |
| Drug | Target | References |
|---|---|---|
| Metabolism Inhibitors | ||
| 5-Fluorouracil | Thymidylate Synthase | [276] |
| Capecitabine | Thymidylate Synthase | [302,303] |
| Gemcitabine | DNA | [304,305] |
| Azacitidine | DNA methyltransferase/DNA | [306] |
| Mizoribine | IMP dehydrogenase | [307] |
| L-asparaginase | Asparagine | [64] |
| Cerulenin | FASN | [308,309] |
| Metformin | Complex 1 | [310,311,312,313,314] |
| Hydroxychloroquine | Lysosomes/Autophagy | [315,316,317] |
| Other mTOR Pathway Inhibitors | ||
| NR1 | Rheb | [318] |
| Lonafarnib | Farnesyl transferase | [319] |
| Perifosine | Akt | [320] |
| MK-2206 | Akt | [321,322,323,324] |
| AZD5363 | Akt | [325] |
| GSK650394 | SGK1 | [326,327] |
| SI113 | SGK1 | [328] |
| GSK2334470 | PDK1 | [329] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magaway, C.; Kim, E.; Jacinto, E. Targeting mTOR and Metabolism in Cancer: Lessons and Innovations. Cells 2019, 8, 1584. https://doi.org/10.3390/cells8121584
Magaway C, Kim E, Jacinto E. Targeting mTOR and Metabolism in Cancer: Lessons and Innovations. Cells. 2019; 8(12):1584. https://doi.org/10.3390/cells8121584
Chicago/Turabian StyleMagaway, Cedric, Eugene Kim, and Estela Jacinto. 2019. "Targeting mTOR and Metabolism in Cancer: Lessons and Innovations" Cells 8, no. 12: 1584. https://doi.org/10.3390/cells8121584
APA StyleMagaway, C., Kim, E., & Jacinto, E. (2019). Targeting mTOR and Metabolism in Cancer: Lessons and Innovations. Cells, 8(12), 1584. https://doi.org/10.3390/cells8121584