Fluorescent TAP as a Platform for Virus-Induced Degradation of the Antigenic Peptide Transporter

 ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. DNA Constructs
2.3. Retroviral and Lentiviral Transduction
2.4. Plasmid Transfection
2.5. Generation of A TAP1/TAP2 Double Knock-Out U937 Cells for Reconstitution with Fluorescent TAP Variants
2.6. Antibodies
2.7. Flow Cytometry
2.8. Immunoblotting and Immunoprecipitation
2.9. Peptide Transport Assay
2.10. Immunofluorescence
3. Results
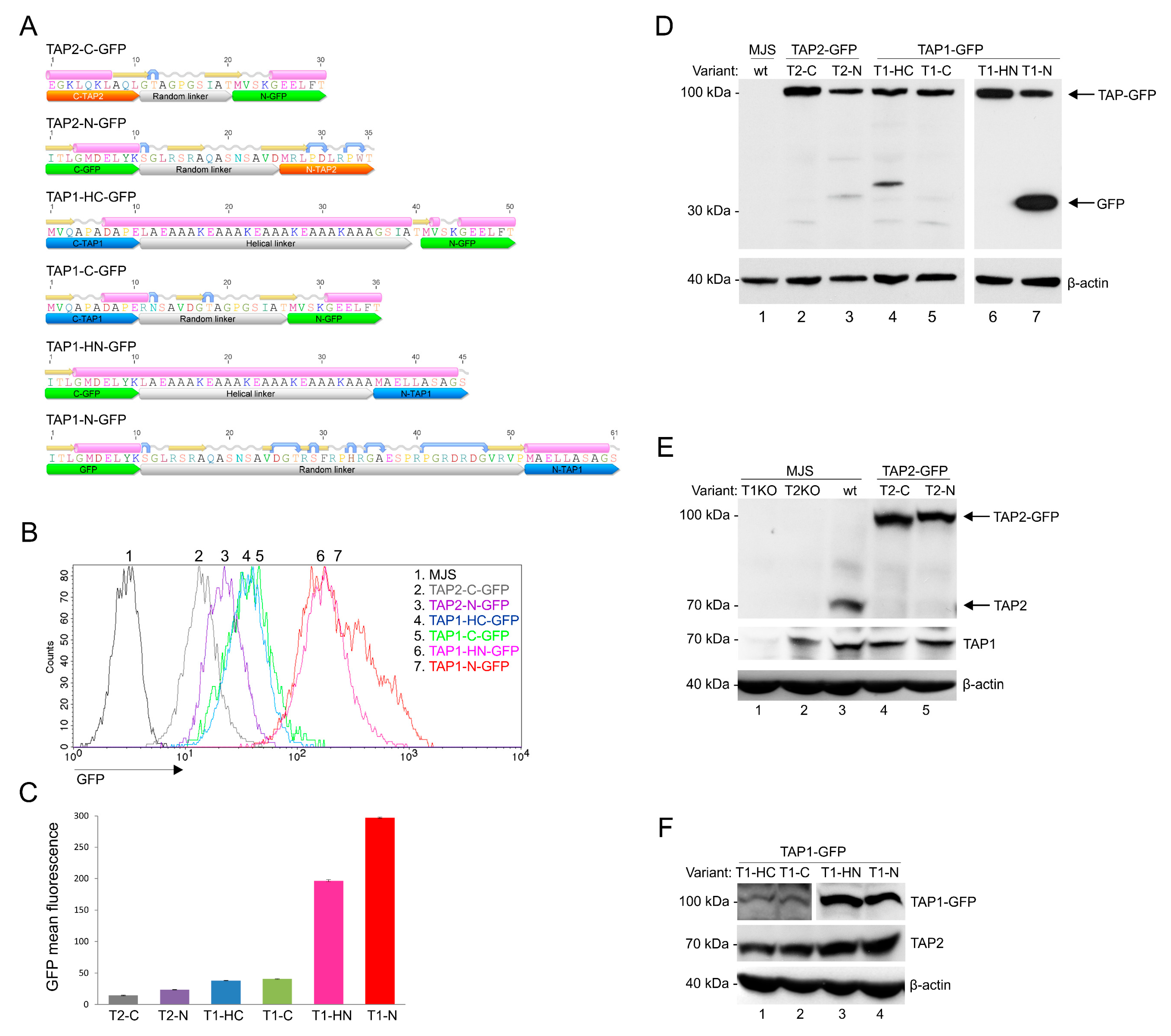
3.1. Construction and Characterization of Fluorescent TAP-GFP Variants
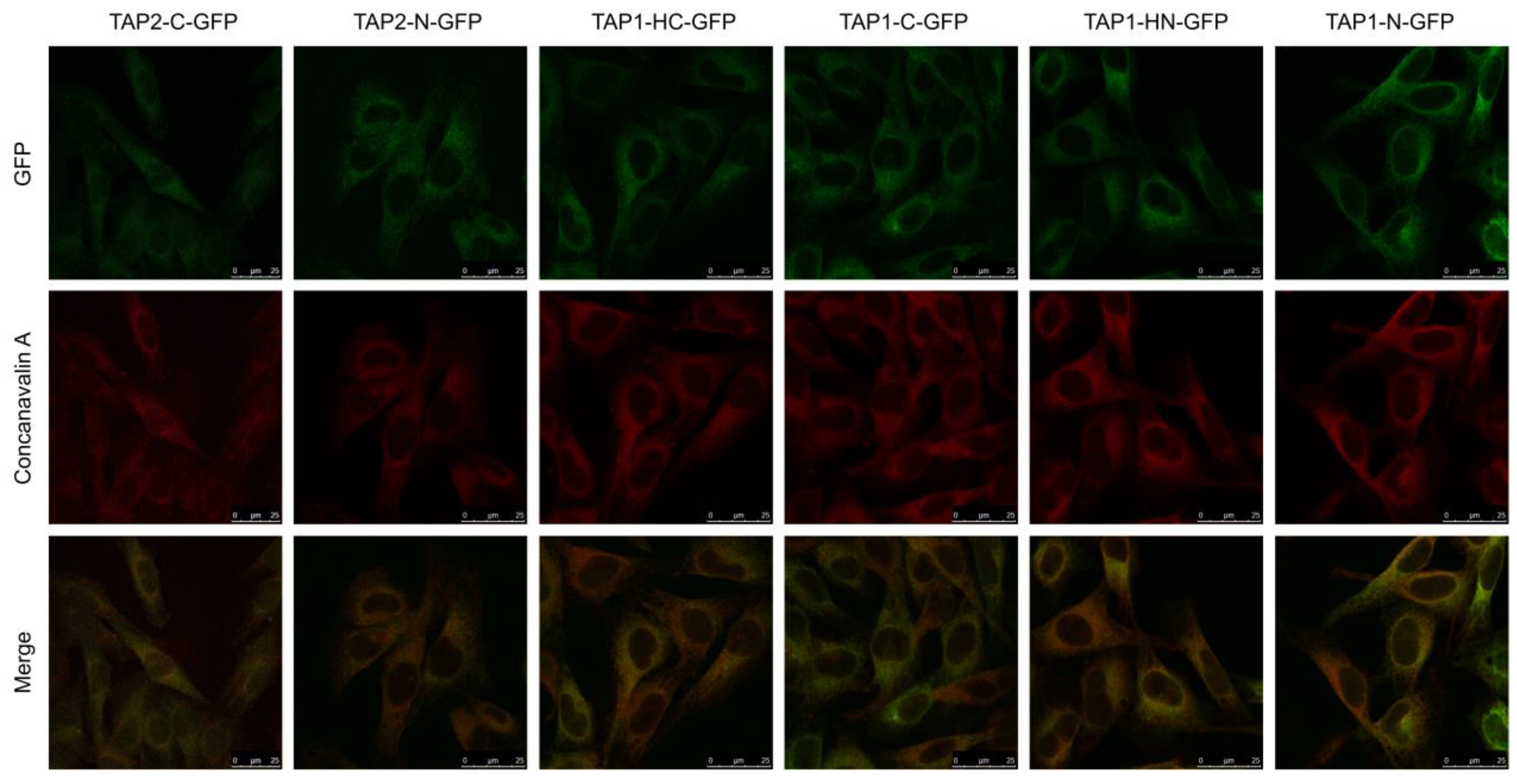
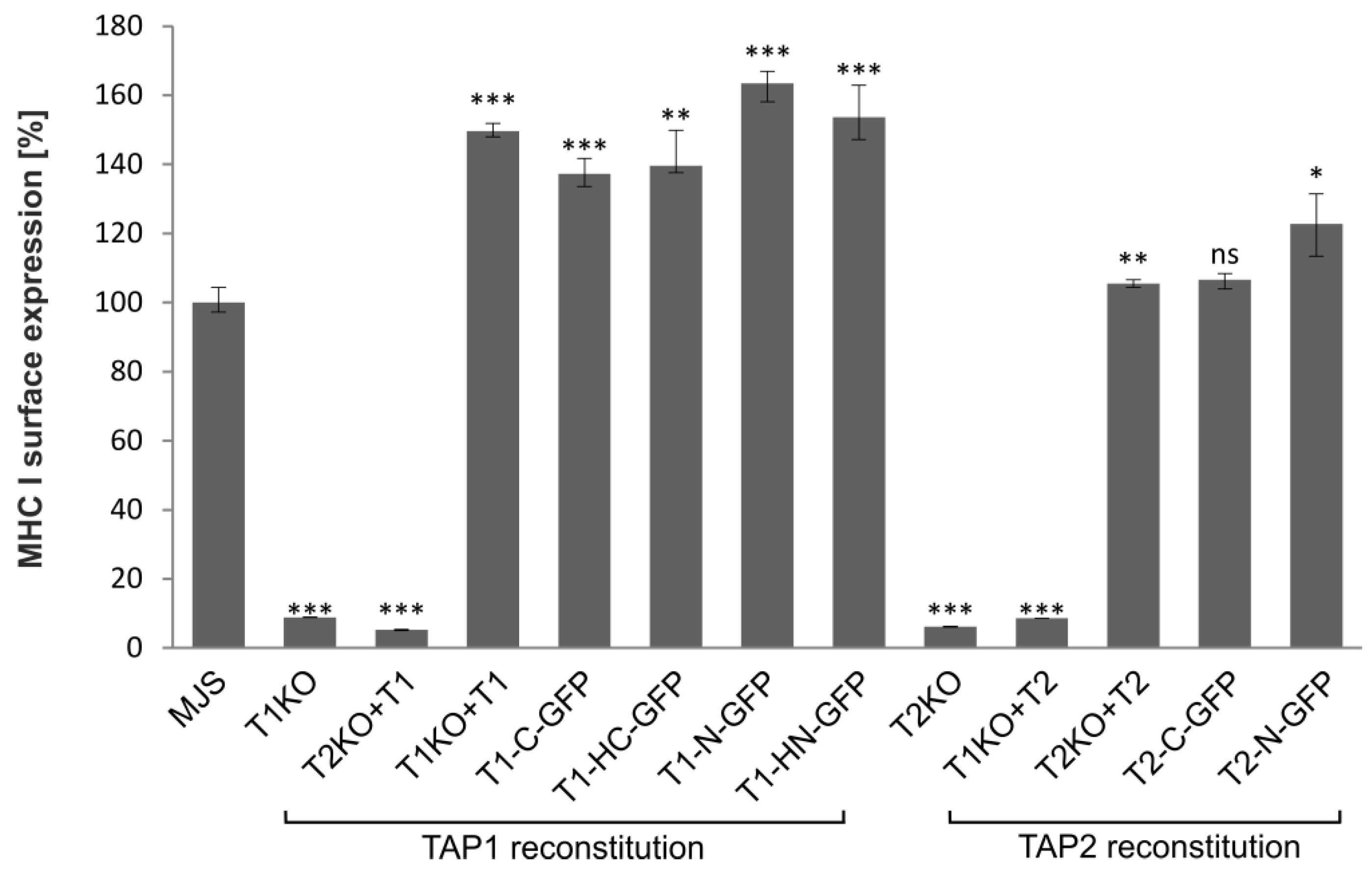
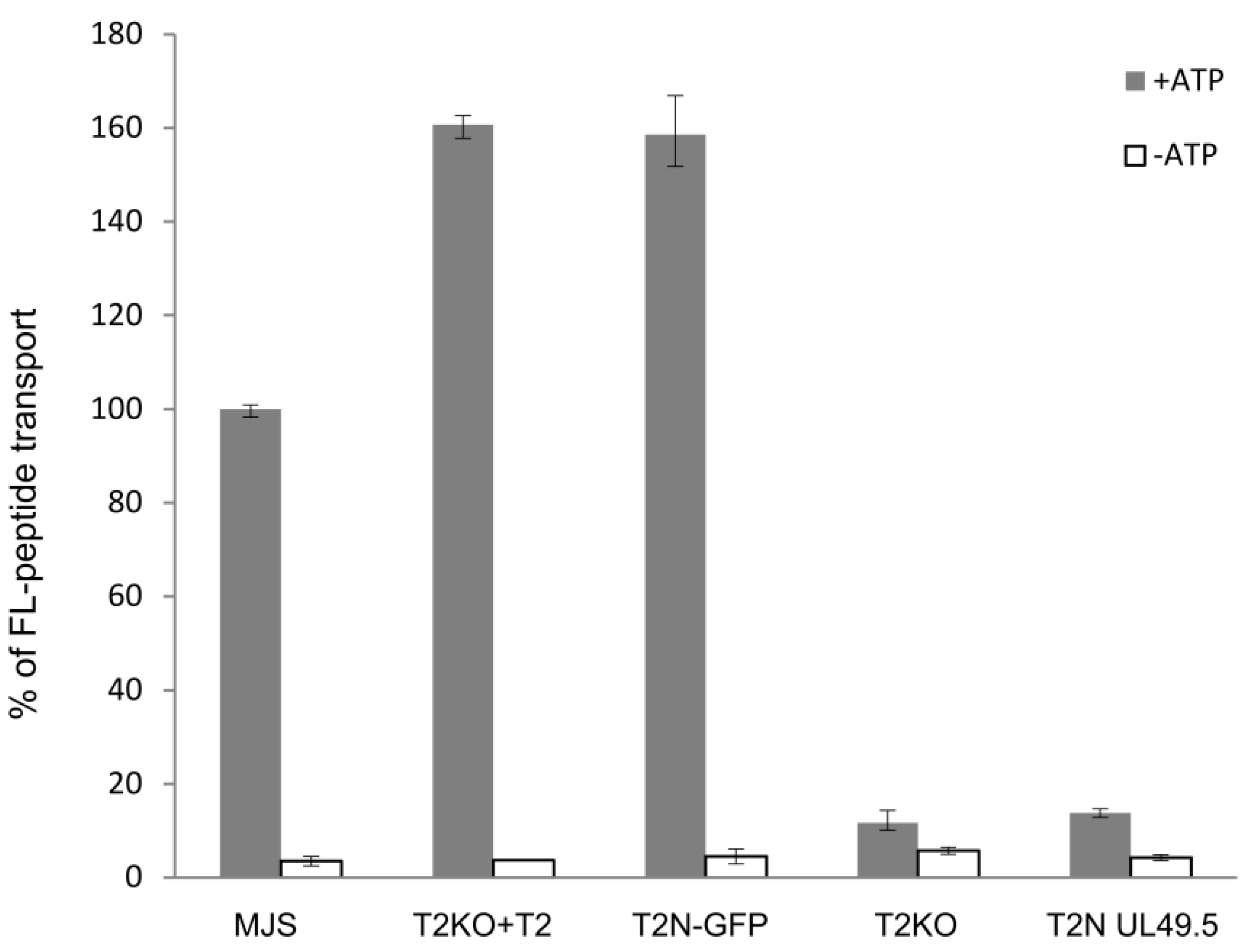
3.2. TAP-GFP Localizes in the ER and Forms a Functional Transporter
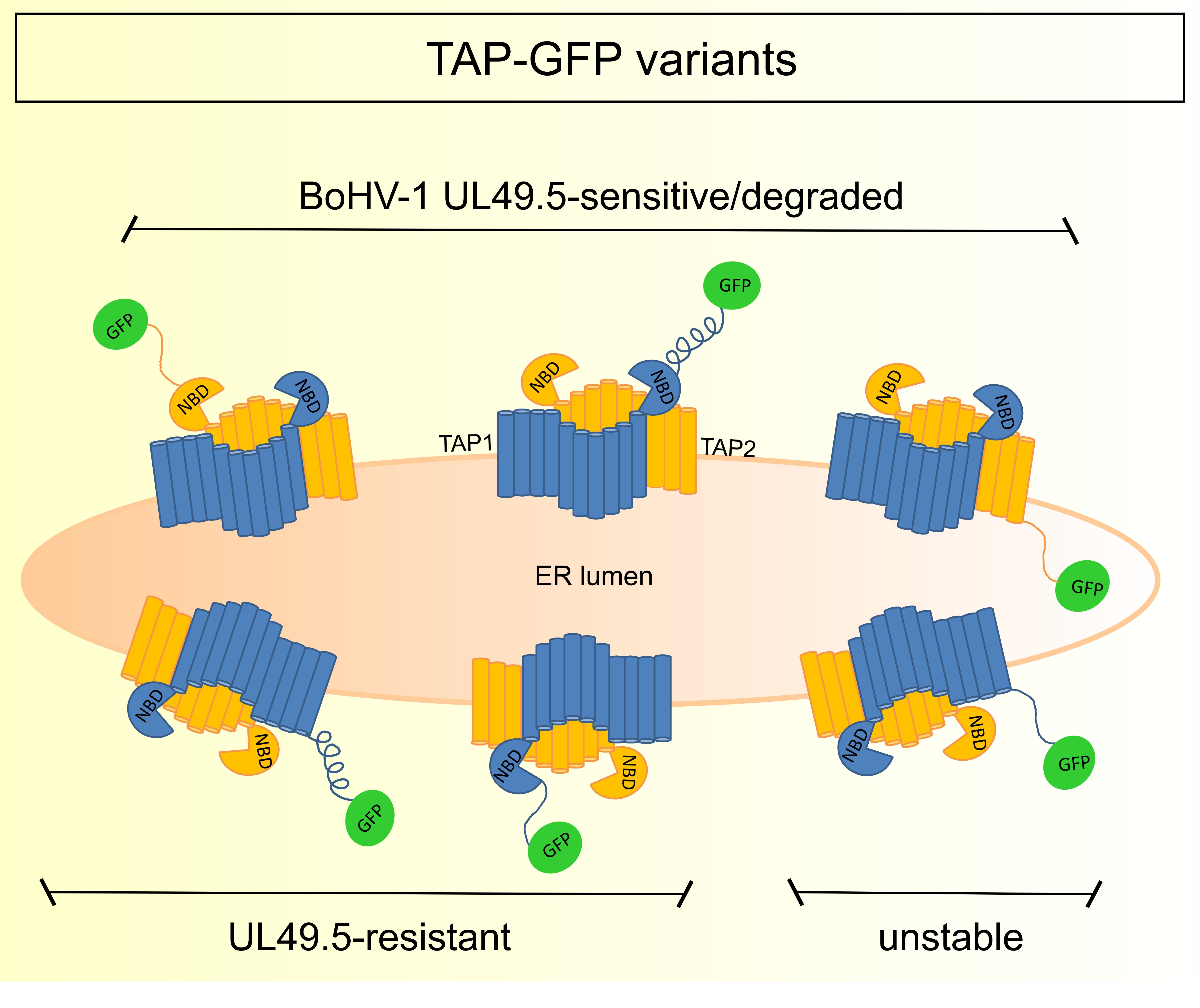
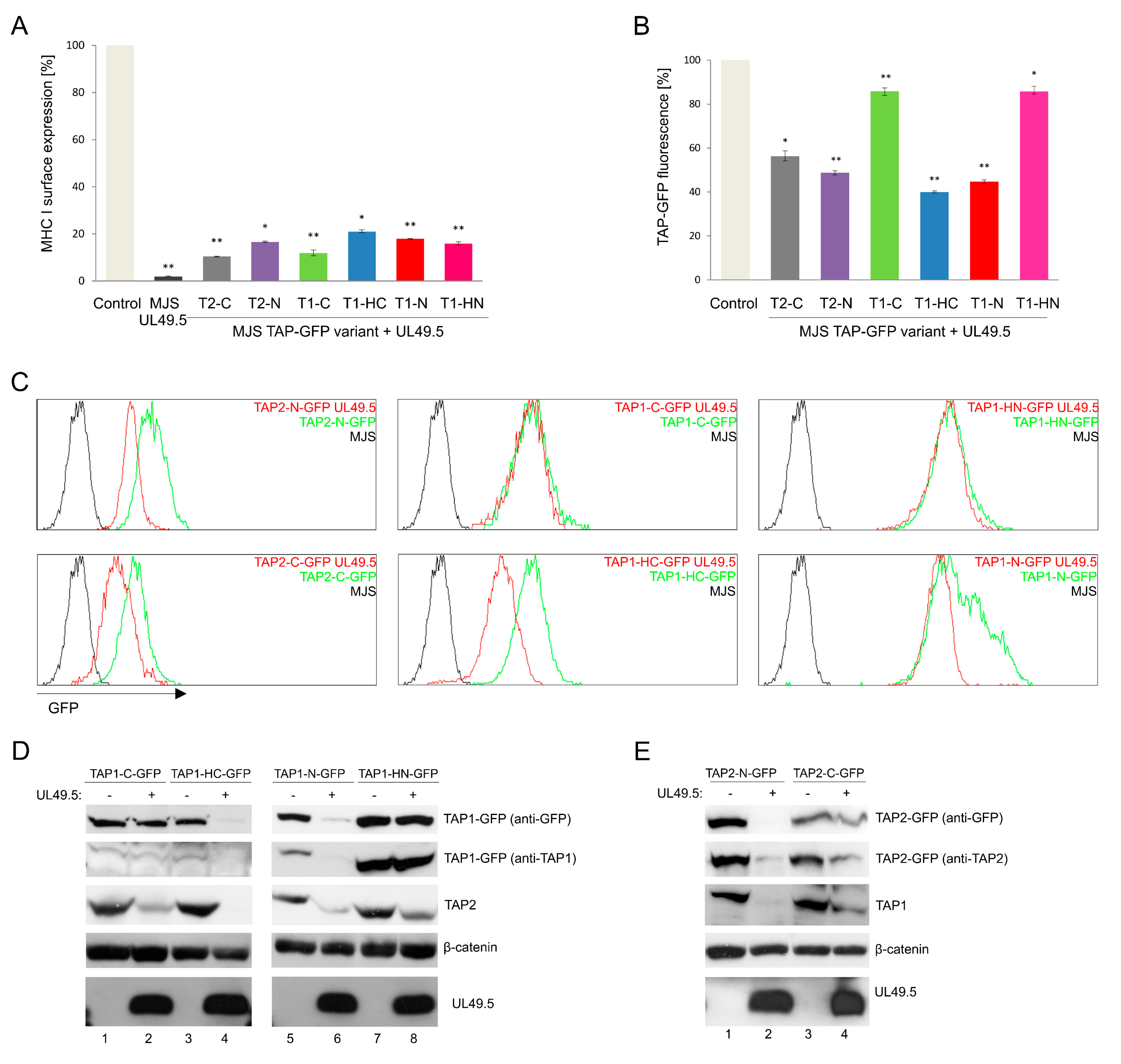
3.3. The Sensitivity of TAP-GFP Variant to UL49.5-Mediated MHC I Downregulation and TAP Degradation
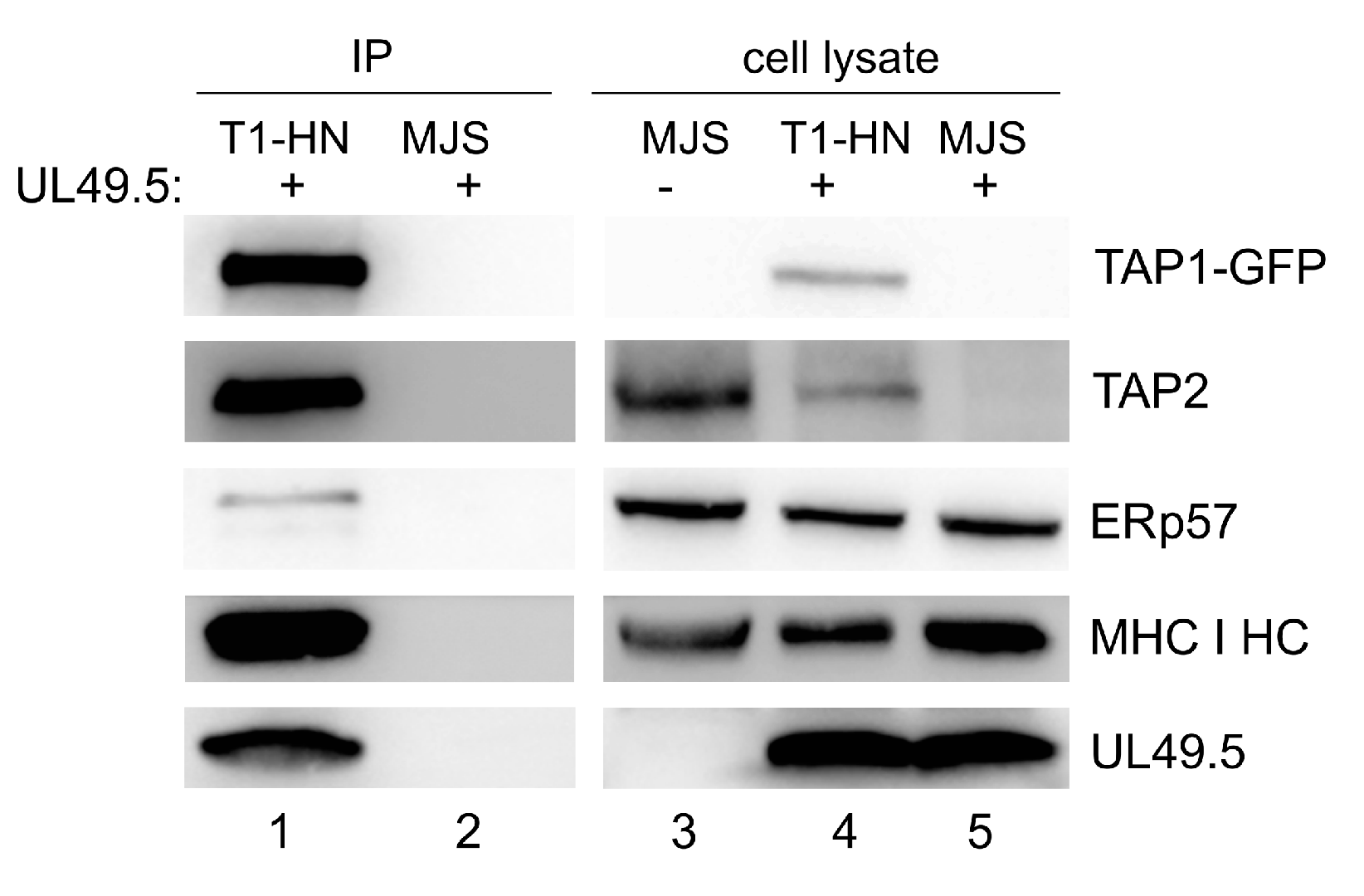
3.4. Interaction of TAP-GFP with UL49.5 and the Peptide-Loading Complex
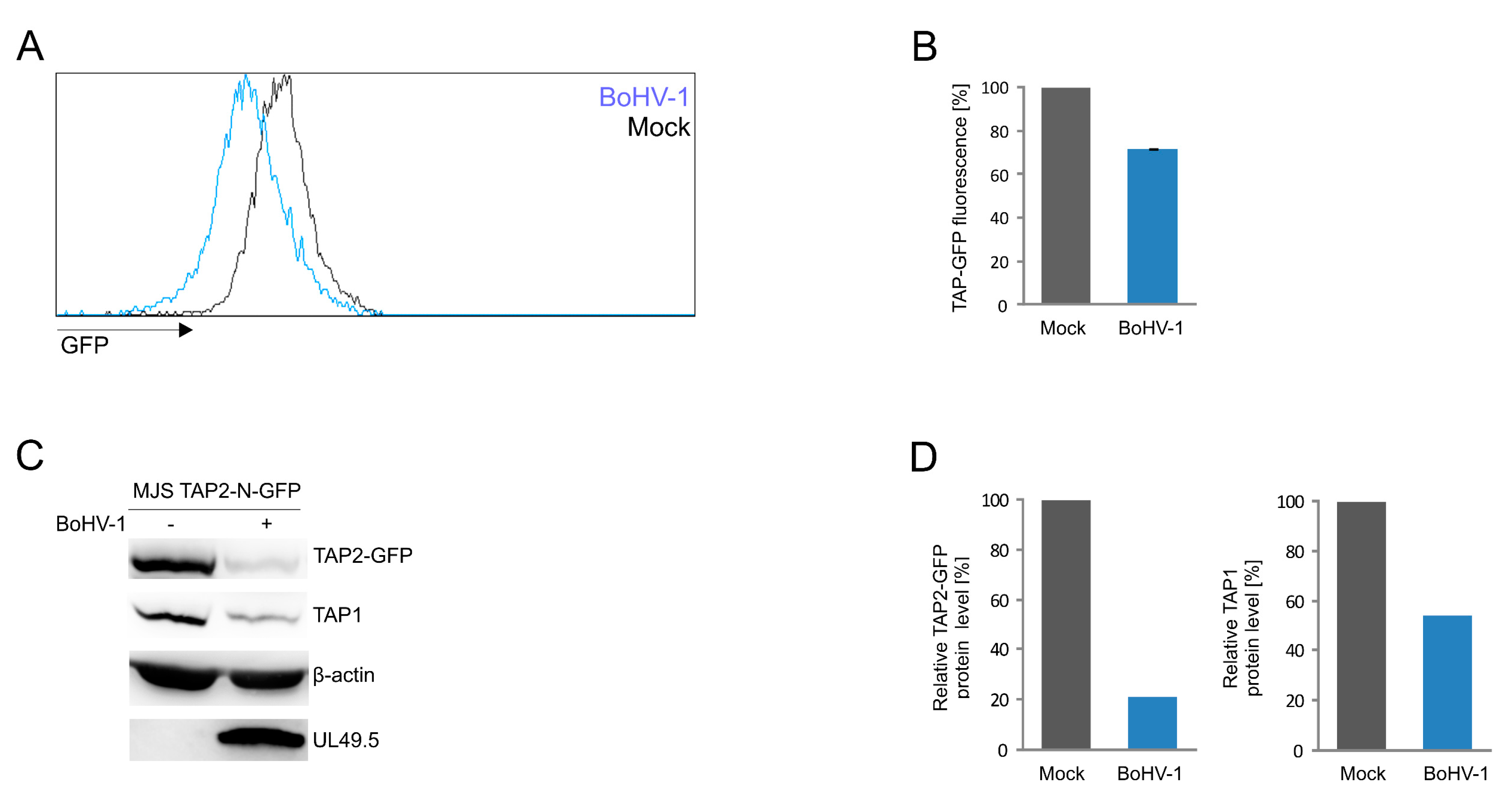
3.5. Application of the TAP2-N-GFP Variant as a Platform to Study BoHV-1 UL49.5 Activity in Virus-Infected Cells
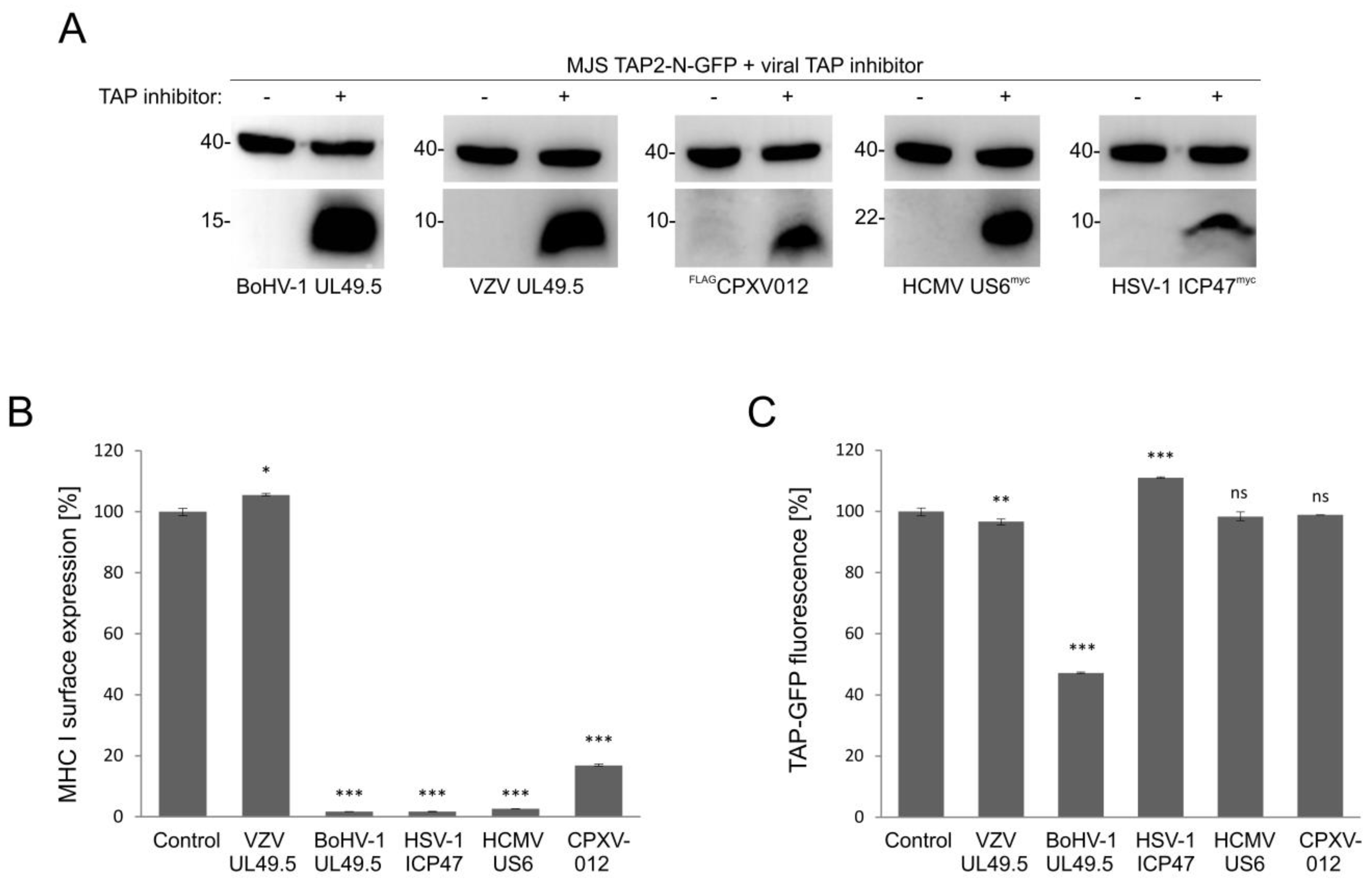
3.6. Only UL49.5 among Different Viral TAP Inhibitors Can Induce Human TAP Degradation
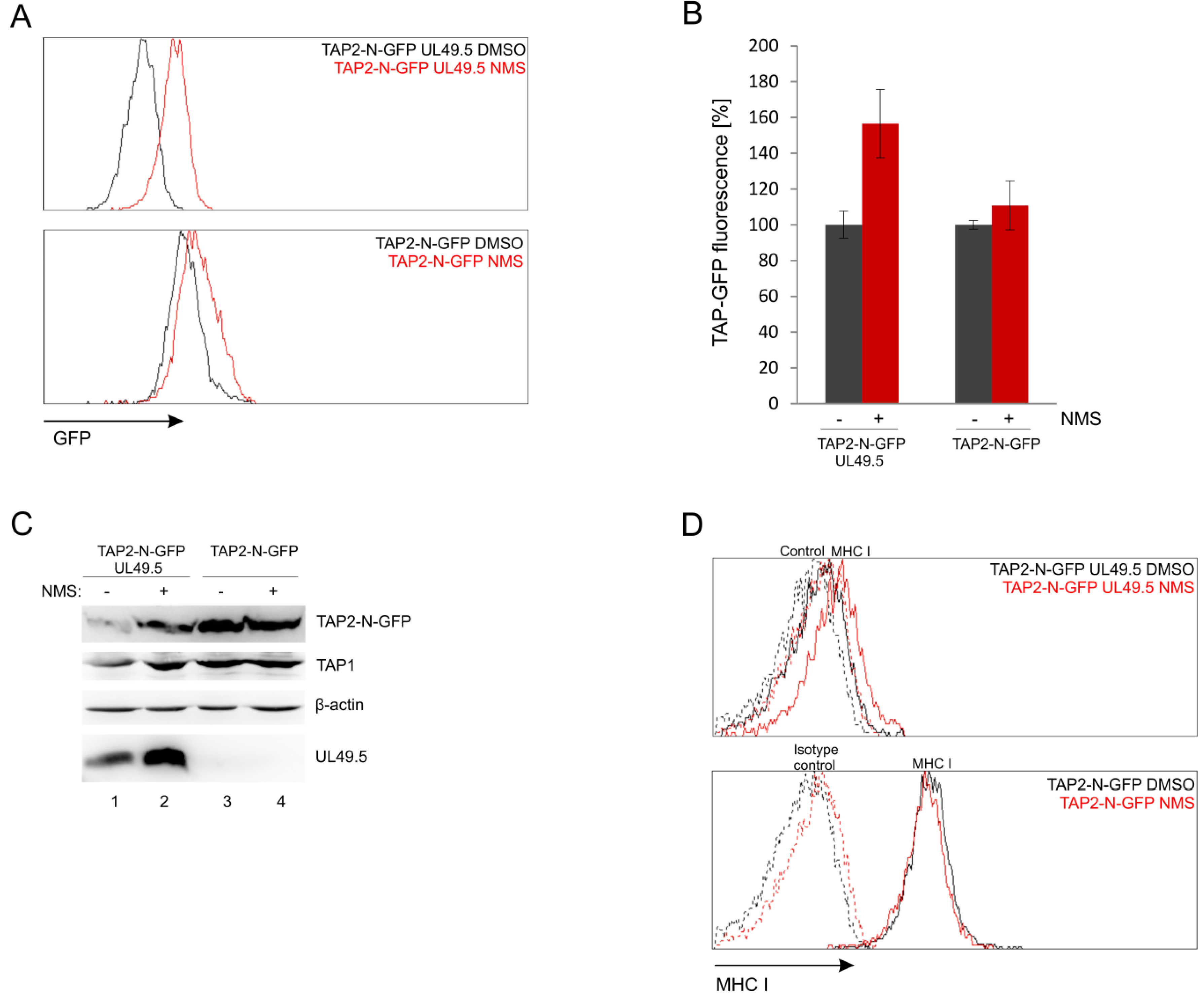
3.7. UL49.5-Induced TAP-GFP Degradation Is p97-Dependent
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jensen, P.E. Recent advances in antigen processing and presentation. Nat. Immunol. 2007, 8, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Annilo, T. Evolution of the ATP-binding cassette (ABC) transporter superfamily in vertebrates. Annu. Rev. Genom. Hum. Genet. 2005, 6, 123–142. [Google Scholar] [CrossRef] [PubMed]
- van Endert, P.M.; Tampé, R.; Meyer, T.H.; Tisch, R.; Bach, J.-F.; McDevitt, H.O. A sequential model for peptide binding and transport by the transporters associated with antigen processing. Immunity 1994, 1, 491–500. [Google Scholar] [CrossRef]
- Neefjes, J.J.; Momburg, F.; Hämmerling, G.J. Selective and ATP-dependent translocation of peptides by the MHC-encoded transporter. Science 1993, 261, 769–771. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Sivaramakrishnan, S.; Raghavan, M. Analyses of conformational states of the transporter associated with antigen processing (TAP) protein in a native cellular membrane environment. J. Biol. Chem. 2013, 288, 37039–37047. [Google Scholar] [CrossRef] [Green Version]
- Koch, J.; Guntrum, R.; Heintke, S.; Kyritsis, C.; Tampé, R. Functional dissection of the transmembrane domains of the transporter associated with antigen processing (TAP). J. Biol. Chem. 2004, 279, 10142–10147. [Google Scholar] [CrossRef] [Green Version]
- Verweij, M.C.; Horst, D.; Griffin, B.D.; Luteijn, R.D.; Davison, A.J.; Ressing, M.E.; Wiertz, E.J.H.J. Viral Inhibition of the transporter associated with antigen processing (TAP): A striking example of functional convergent evolution. PLoS Pathog. 2015, 11, e1004743. [Google Scholar] [CrossRef] [Green Version]
- Früh, K.; Ahn, K.; Djaballah, H.; Sempé, P.; Peter, M.; van Endert, P.M.; Tampé, R.; Peterson, P.A.; Yang, Y. A viral inhibitor of peptide transporters for antigen presentation. Nature 1995, 375, 415–418. [Google Scholar] [CrossRef]
- Tomazin, R.; Hill, A.B.; Jugovic, P.; York, I.; van Endert, P.; Ploegh, H.L.; Andrews, D.W.; Johnson, D.C. Stable binding of the herpes simplex virus ICP47 protein to the peptide binding site of TAP. EMBO J. 1996, 15, 3256–3266. [Google Scholar] [CrossRef]
- Oldham, M.L.; Grigorieff, N.; Chen, J. Structure of the transporter associated with antigen processing trapped by herpes simplex virus. eLife 2016, 5, e21829. [Google Scholar] [CrossRef]
- Ahn, K.; Gruhler, A.; Galocha, B.; Jones, T.R.; Wiertz, E.J.H.J.; Ploegh, H.L.; Peterson, P.A.; Yang, Y.; Früh, K. The ER-luminal domain of the HCMV glycoprotein US6 inhibits peptide translocation by TAP. Immunity 1997, 6, 613–621. [Google Scholar] [CrossRef] [Green Version]
- Kyritsis, C.; Gorbulev, S.; Hutschenreiter, S.; Pawlitschko, K.; Abele, R.; Tampé, R. Molecular mechanism and structural aspects of transporter associated with antigen processing inhibition by the cytomegalovirus protein US6. J. Biol. Chem. 2001, 276, 48031–48039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halenius, A.; Momburg, F.; Reinhard, H.; Bauer, D.; Lobigs, M.; Hengel, H. Physical and functional interactions of the cytomegalovirus US6 glycoprotein with the transporter associated with antigen processing. J. Biol. Chem. 2006, 281, 5383–5390. [Google Scholar] [CrossRef] [Green Version]
- Alzhanova, D.; Edwards, D.M.; Hammarlund, E.; Scholz, I.G.; Horst, D.; Wagner, M.J.; Upton, C.; Wiertz, E.J.; Slifka, M.K.; Früh, K. Cowpox virus inhibits the transporter associated with antigen processing to evade T cell recognition. Cell Host Microbe 2009, 6, 433–445. [Google Scholar] [CrossRef] [Green Version]
- Byun, M.; Verweij, M.C.; Pickup, D.J.; Wiertz, E.J.H.J.; Hansen, T.H.; Yokoyama, W.M. Two mechanistically distinct immune evasion proteins of cowpox virus combine to avoid antiviral CD8 T Cells. Cell Host Microbe 2009, 6, 422–432. [Google Scholar] [CrossRef] [Green Version]
- Luteijn, R.D.; Hoelen, H.; Kruse, E.; van Leeuwen, W.F.; Grootens, J.; Horst, D.; Koorengevel, M.; Drijfhout, J.W.; Kremmer, E.; Früh, K.; et al. Cowpox virus protein CPXV012 eludes CTLs by blocking ATP binding to TAP. J. Immunol. 2014, 193, 1578–1589. [Google Scholar] [CrossRef] [Green Version]
- Verweij, M.C.; Lipinska, A.D.; Koppers-Lalic, D.; van Leeuwen, W.F.; Cohen, J.I.; Kinchington, P.R.; Messaoudi, I.; Bienkowska-Szewczyk, K.; Ressing, M.E.; Rijsewijk, F.A.M.; et al. The capacity of UL49.5 proteins to inhibit TAP is widely distributed among members of the genus varicellovirus. J. Virol. 2011, 85, 2351–2363. [Google Scholar] [CrossRef] [Green Version]
- Koppers-Lalic, D.; Reits, E.A.J.; Ressing, M.E.; Lipinska, A.D.; Abele, R.; Koch, J.; Rezende, M.M.; Admiraal, P.; van Leeuwen, D.; Bienkowska-Szewczyk, K.; et al. Varicelloviruses avoid T cell recognition by UL49.5-mediated inactivation of the transporter associated with antigen processing. Proc. Natl. Acad. Sci. USA 2005, 102, 5144–5149. [Google Scholar] [CrossRef] [Green Version]
- Verweij, M.C.; Koppers-Lalic, D.; Loch, S.; Klauschies, F.; de la Salle, H.; Quinten, E.; Lehner, P.J.; Mulder, A.; Knittler, M.R.; Tampé, R.; et al. The varicellovirus UL49.5 protein blocks the transporter associated with antigen processing (TAP) by inhibiting essential conformational transitions in the 6+6 transmembrane TAP core complex. J. Immunol. 2008, 181, 4894–4907. [Google Scholar] [CrossRef] [Green Version]
- Koppers-Lalic, D.; Verweij, M.C.; Lipińska, A.D.; Wang, Y.; Quinten, E.; Reits, E.A.; Koch, J.; Loch, S.; Rezende, M.M.; Daus, F.; et al. Varicellovirus UL49.5 proteins differentially affect the function of the transporter associated with antigen processing, TAP. PLoS Pathog. 2008, 4, e1000080. [Google Scholar] [CrossRef]
- Boname, J.; May, J.; Stevenson, P. The murine gamma-herpesvirus-68 MK3 protein causes TAP degradation independent of MHC class I heavy chain degradation. Eur. J. Immunol. 2005, 35, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Herr, R.A.; Wang, X.; Loh, J.; Virgin, H.W.; Hansen, T.H. Newly discovered viral E3 ligase pK3 induces endoplasmic reticulum-associated degradation of class I major histocompatibility proteins and their membrane-bound chaperones. J. Biol. Chem. 2012, 287, 14467–14479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, I.B.; Wang, X.; Fremont, D.H. Molluscum contagiosum virus MC80 sabotages MHC-I antigen presentation by targeting tapasin for ER-associated degradation. PLoS Pathog. 2019, 15, e1007711. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zaro, J.L.; Shen, W.-C. Fusion protein linkers: Property, design and functionality. Adv. Drug Deliver. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Praest, P.; Luteijn, R.D.; Brak-Boer, I.G.J.; Lanfermeijer, J.; Hoelen, H.; Ijgosse, L.; Costa, A.I.; Gorham, R.D.; Lebbink, R.J.; Wiertz, E.J.H.J. The influence of TAP1 and TAP2 gene polymorphisms on TAP function and its inhibition by viral immune evasion proteins. Mol. Immunol. 2018, 101, 55–64. [Google Scholar] [CrossRef]
- Lipińska, A.D.; Koppers-Lalic, D.; Rychlowski, M.; Admiraal, P.; Rijsewijk, F.A.M.; Bienkowska-Szewczyk, K.; Wiertz, E.J.H.J. Bovine herpesvirus 1 UL49.5 protein inhibits the transporter associated with antigen processing despite complex formation with glycoprotein M. J. Virol. 2006, 80, 5822–5832. [Google Scholar] [CrossRef] [Green Version]
- Morgenstern, J.P.; Land, H. Advanced mammalian gene transfer: High titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acid Res. 1990, 18, 3587–3596. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Eggensperger, S.; Hank, S.; Wycisk, A.I.; Wieneke, R.; Mayerhofer, P.U.; Tampé, R. A negative feedback modulator of antigen processing evolved from a frameshift in the cowpox virus genome. PLoS Pathog. 2014, 10, e1004554. [Google Scholar] [CrossRef] [Green Version]
- Marqusee, S.; Baldwin, R.L. Helix stabilization by Glu-Lys+ salt bridges in short peptides of de novo design. Proc. Natl. Acad. Sci. USA 1987, 84, 8898–8902. [Google Scholar] [CrossRef] [Green Version]
- Arai, R.; Ueda, H.; Kitayama, A.; Kamiya, N.; Nagamune, T. Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng. Des. Sel. 2001, 14, 529–532. [Google Scholar] [CrossRef]
- Koppers-Lalic, D.; Rychlowski, M.; van Leeuwen, D.; Rijsewijk, F.A.M.; Ressing, M.E.; Neefjes, J.J.; Bienkowska-Szewczyk, K.; Wiertz, E.J.H.J. Bovine herpesvirus 1 interferes with TAP-dependent peptide transport and intracellular trafficking of MHC class I molecules in human cells. Arch. Virol. 2003, 148, 2023–2037. [Google Scholar] [CrossRef] [PubMed]
- Verweij, M.C.; Lipińska, A.D.; Koppers-Lalic, D.; Quinten, E.; Funke, J.; van Leeuwen, H.C.; Bieńkowska-Szewczyk, K.; Koch, J.; Ressing, M.E.; Wiertz, E.J.H.J. Structural and functional analysis of the TAP-inhibiting UL49.5 proteins of varicelloviruses. Mol. Immunol. 2011, 48, 2038–2051. [Google Scholar] [CrossRef] [PubMed]
- Praest, P.; de Buhr, H.; Wiertz, E.J.H.J. A flow cytometry-based approach to unravel viral interference with the MHC class I antigen processing and presentation pathway. In Antigen Processing; van Endert, P., Ed.; Springer: New York, NY, USA, 2019; Volume 1988, pp. 187–198. [Google Scholar]
- Keusekotten, K.; Leonhardt, R.M.; Ehses, S.; Knittler, M.R. Biogenesis of functional antigenic peptide transporter TAP requires assembly of pre-existing TAP1 with newly synthesized TAP2. J. Biol. Chem. 2006, 281, 17545–17551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleijmeer, M.J.; Kelly, A.; Geuze, H.J.; Slot, J.W.; Townsend, A.; Trowsdale, J. Location of MHC-encoded transporters in the endoplasmic reticulum and cis-Golgi. Nature 1992, 357, 342–344. [Google Scholar] [CrossRef] [PubMed]
- Van Kaer, L.; Ashton-Rickardt, P.G.; Ploegh, H.L.; Tonegawa, S. TAP1 mutant mice are deficient in antigen presentation, surface class I molecules, and CD4−8+ T cells. Cell 1992, 71, 1205–1214. [Google Scholar] [CrossRef]
- Antoniou, A.N.; Ford, S.; Pilley, E.S.; Blake, N.; Powis, S.J. Interactions formed by individually expressed TAP1 and TAP2 polypeptide subunits. Immunology 2002, 106, 182–189. [Google Scholar] [CrossRef]
- Xia, D.; Tang, W.K.; Ye, Y. Structure and function of the AAA+ ATPase p97/Cdc48p. Gene 2016, 583, 64–77. [Google Scholar] [CrossRef] [Green Version]
- Herbring, V.; Bäucker, A.; Trowitzsch, S.; Tampé, R. A dual inhibition mechanism of herpesviral ICP47 arresting a conformationally thermostable TAP complex. Sci. Rep. 2016, 6, 36907. [Google Scholar] [CrossRef]
- Marguet, D.; Spiliotis, E.T.; Pentcheva, T.; Lebowitz, M.; Schneck, J.; Edidin, M. Lateral diffusion of GFP-tagged H2Ld molecules and of GFP-TAP1 reports on the assembly and retention of these molecules in the endoplasmic reticulum. Immunity 1999, 11, 231–240. [Google Scholar] [CrossRef] [Green Version]
- Reits, E.A.J.; Vos, J.C.; Grommé, M.; Neefjes, J. The major substrates for TAP in vivo are derived from newly synthesized proteins. Nature 2000, 404, 774–778. [Google Scholar] [CrossRef]
- Kobayashi, A.; Maeda, T.; Maeda, M. Membrane localization of transporter associated with antigen processing (TAP)-like (ABCB9) visualized in vivo with a fluorescence protein-fusion technique. Biol. Pharm. Bull. 2004, 27, 1916–1922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghanem, E.; Fritzsche, S.; Al-Balushi, M.; Hashem, J.; Ghuneim, L.; Thomer, L.; Kalbacher, H.; van Endert, P.; Wiertz, E.; Tampe, R.; et al. The transporter associated with antigen processing (TAP) is active in a post-ER compartment. J. Cell Sci. 2010, 123, 4271–4279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karska, N.; Graul, M.; Sikorska, E.; Zhukov, I.; Ślusarz, M.J.; Kasprzykowski, F.; Lipińska, A.D.; Rodziewicz-Motowidło, S. Structure determination of UL49.5 transmembrane protein from bovine herpesvirus 1 by NMR spectroscopy and molecular dynamics. BBA Biomembr. 2019, 1861, 926–938. [Google Scholar] [CrossRef] [PubMed]
- Vos, J.C.; Spee, P.; Momburg, F.; Neefjes, J. Membrane topology and dimerization of the two subunits of the transporter associated with antigen processing reveal a three-domain structure. J. Immunol. 1999, 163, 6679–6685. [Google Scholar]
- Schrodt, S.; Koch, J.; Tampé, R. Membrane topology of the transporter associated with antigen processing (TAP1) within an assembled functional peptide-loading complex. J. Biol. Chem. 2006, 281, 6455–6462. [Google Scholar] [CrossRef] [Green Version]
- Lankat-Buttgereit, B.; Tampé, R. The transporter associated with antigen processing (TAP): A peptide transport and loading complex essential for cellular immune response. In ABC Proteins: From Bacteria to Man; Holland, B., Cole, S.P.C., Kuchler, K., Higgins, C.F., Eds.; Academic Press: Cambridge, MA, USA, 2003; pp. 533–550. [Google Scholar]
- Kageshita, T.; Hirai, S.; Ono, T.; Hicklin, D.J.; Ferrone, S. Down-regulation of HLA class I antigen-processing molecules in malignant melanoma. Am. J. Pathol. 1999, 154, 745–754. [Google Scholar] [CrossRef]
- Matschulla, T.; Berry, R.; Gerke, C.; Döring, M.; Busch, J.; Paijo, J.; Kalinke, U.; Momburg, F.; Hengel, H.; Halenius, A. A highly conserved sequence of the viral TAP inhibitor ICP47 is required for freezing of the peptide transport cycle. Sci. Rep. 2017, 7, 2933. [Google Scholar] [CrossRef] [Green Version]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wąchalska, M.; Graul, M.; Praest, P.; Luteijn, R.D.; Babnis, A.W.; Wiertz, E.J.H.J.; Bieńkowska-Szewczyk, K.; Lipińska, A.D. Fluorescent TAP as a Platform for Virus-Induced Degradation of the Antigenic Peptide Transporter. Cells 2019, 8, 1590. https://doi.org/10.3390/cells8121590
Wąchalska M, Graul M, Praest P, Luteijn RD, Babnis AW, Wiertz EJHJ, Bieńkowska-Szewczyk K, Lipińska AD. Fluorescent TAP as a Platform for Virus-Induced Degradation of the Antigenic Peptide Transporter. Cells. 2019; 8(12):1590. https://doi.org/10.3390/cells8121590
Chicago/Turabian StyleWąchalska, Magda, Małgorzata Graul, Patrique Praest, Rutger D. Luteijn, Aleksandra W. Babnis, Emmanuel J. H. J. Wiertz, Krystyna Bieńkowska-Szewczyk, and Andrea D. Lipińska. 2019. "Fluorescent TAP as a Platform for Virus-Induced Degradation of the Antigenic Peptide Transporter" Cells 8, no. 12: 1590. https://doi.org/10.3390/cells8121590
APA StyleWąchalska, M., Graul, M., Praest, P., Luteijn, R. D., Babnis, A. W., Wiertz, E. J. H. J., Bieńkowska-Szewczyk, K., & Lipińska, A. D. (2019). Fluorescent TAP as a Platform for Virus-Induced Degradation of the Antigenic Peptide Transporter. Cells, 8(12), 1590. https://doi.org/10.3390/cells8121590