1. Introduction
Oral cancer is one of the most diagnosed malignancies, with a global annual incidence of 300,373 cases [
1]. Regardless of diagnostic and therapeutic progress, from an estimated 145,353 deaths for both sexes in 2012, the oral cancer-related mortality rate continues to rise, with a predicted global mortality rate of 67.1% (n = 242,886) by 2035 because of demographic changes, thus making it one of the main causes of cancer-related deaths globally [
1,
2]. Constituting over 90% of all oral cancer histological subtypes, the oral squamous cell carcinoma (OSCC) is notably highly aggressive, often non-responsive to common anticancer therapy, and associated with early relapse and poor prognosis [
1,
2].
Currently, the treatment of choice remains surgery, radiation therapy, and/or chemotherapy, which consists of cisplatin (CDDP), docetaxel (DTX), and 5-fluorouracil (5-FU), especially for advanced-stage (III and IV) malignancies; however, this is often associated with increased risk of severe drug-induced adverse effects and toxicities, high rates of treatment failure and disease recurrence, as well as low median survival rates for OSCC patients [
3], with an estimated 5-year survival rate that is consistently below 50% over the last 30 years [
4]. In fact, despite the documented therapeutic relevance of radiotherapy for the reduction of OSCC tumor size and oral function preservation, it is common for irradiated OSCC patients to develop early loco-regional disease relapse, thus, further contributing to the dismal prognosis [
5]. Putting together, these inform the need for the discovery of novel actionable molecular target or development of novel therapeutic agents with better efficacy.
The last two decades have been characterized by accumulating evidence, implicating cancer stem cells (CSCs) in the initiation and sustenance of tumor, resistance to chemo- and/or radiation therapy, disease recurrence, and metastases [
6,
7].
Previously, we demonstrated that the direct targeting of CSCs or so-called tumor-initiating cells (TICs) in OSCC by gene loss of function [
8] or therapeutic targeting [
9] inhibited the malignant and metastatic traits of OSCC cells, eliminated their CSCs-like properties, including enhanced clonogenicity, orosphere formation, and self-renewal, as well as improved their sensitivity to chemo- and/or radiation therapy, thus, highlighting the therapeutic significance of preferentially targeting the CSCs pool as an efficacious anti-OSCC strategy for mitigating the menace of radioresistance, cancer recurrence, and metastasis, while improving survival rates in patients with OSCC. Corollary to the above implication of CSCs in cancer metastases and resistance to anticancer therapy, as well as accruing association of the aberrant expression of epithelial-to-mesenchymal transition (EMT)-inducing transcription factors with cancer stemness, the last decade has been characterized by increased documentation of existent reciprocal link between CSCs and EMT, with CSCs exhibiting EMT phenotypes, and EMT being relevant to the acquisition and maintenance of stem cell-like traits and sufficient to confer same traits on differentiated non-cancerous and cancerous cells [
10,
11,
12]. This CSCs-EMT reciprocity, mirroring a pathological positive feedback loop, plays a critical role in enhanced chemoresistance, radiotherapy resistance, disease progression, recurrence, and poor prognosis [
10,
11,
12,
13].
The ubiquitously-expressed transmembrane glycoprotein CD47, also known as integrin-associated signal transducer or protein (IAST or IAP), belongs to the immunoglobulin (Ig) superfamily and is an antigenic surface determinant protein that provides a “self” or “do not eat” signal by interacting with the N-terminus of its ligand, signal regulatory protein alpha (SIRPα) on immune cells to suppress or inhibit macrophage phagocytosis [
14]. In fact, by interacting with integrins from the β1, β2, and β3 families, CD47 induces heterotrimeric G-protein signaling, thus modulating leukocyte adhesion, cell motility, and phagocytosis. It has been recently demonstrated that CD47 is upregulated in leukemia cells and circulating hematopoietic stem cells to avoid pre-mobilization and intra-mobilization phagocytosis by macrophages, thus, lending credence to the known role of CD47 as a facilitator of immune-surveillance evasion by cancer cells [
15]. However, while the nefarious role of CD47 in tumorigenesis, therapy evasion, and poor prognosis is increasingly being demystified, the therapeutic promise of CD47-mediated CSCs-targeting remains poorly understood and largely unexplored in OSCC; thus, in the present study, we explored the probable role of loss of CD47 function in the attenuation of oral CSCs, EMT, and radiosensitivity.
In this study, we investigated and provided evidence for the feasibility of a CD47-mediated attenuation of OSCC cell viability, suppression of OSCC-SCs-like attributes and associated pluripotency, deregulation of the constitutive CSCs-EMT loop in OSCC, and enhancement of OSCC cell sensitivity to radiation therapy.
2. Materials and Methods
2.1. Patient Samples
We evaluated the prognostic relevance of CD47 expression in a cohort of OSCC patients (n = 71) aged between 29 and 72, with a median age of 48 years, who had undergone definitive treatment with curative intent in National Defense Medical Center from October 2000 to March 2013. The present study was reviewed and approved by the institutional review board (TSGHIRB 2–102-05–125), and all participants provided written informed consent. Patient distribution based on American Joint Committee on Cancer (AJCC) staging, 7th edition, was as follows - 9 stage I disease (13%), 22 stage II (31%), 13 stage III (18%), and 27 stage IV (38%), as well as normal non-tumor (2), mild dysplasia (1), moderate dysplasia (1), and severe dysplasia (3) from peritumoral tissue. Location-wise, buccal mucosa (n = 39, 51.3%) was the most common affected site, followed by tongue (n = 25, 32.9%), gingival (n = 7, 9.2%), and others (n = 5, 6.6%) [palate (n= 2), maxilla (n = 1), lip (n = 1), and tonsil (n = 1)]. None of the specimens had received radiation or a chemotherapeutic regimen. The clinicopathological characteristic of our cohort is shown in
Table 1.
2.2. Immunohistochemical (IHC) Staining
For the CD47 IHC staining analysis of OSCC (n = 71) and non-tumor oral tissues (n = 7), 1% bovine serum albumin (BSA) was used for blocking the formalin-fixed paraffin-embedded (FFPE) tissue sections before they were incubated with CD47 antibody (Santa Cruz Biotechnology Inc, Santa Cruz, CA, USA) at 4 °C overnight. The sections were then incubated with goat anti-mouse IgG (Cell Signaling Technology Inc., Danvers, MA, USA) for 1 h. Tissue staining was scored by two independent pathologists. The staining index (SI) was calculated based on the formula.
where percentage of positively stained tumor cells was graded as: 0 (0%), 1 (<10%), 2 (10–35%), 3 (35–70%), and 4 (>70%); and staining intensity graded as: 0 (no staining), 1 (weak), 2 (moderate), and 3 (strong). Positive results were defined as buffy granules in the cell membrane and cytoplasm. Scores of intensities were given 1, 2, and 3 depending on the color intensity as weak, moderate, strong staining. The quick score (Q score) is defined as the percentage of staining cells (%) multiplied by a score of intensity. The membrane and cytoplasm scores were calculated in each. The total Q score was given as membrane score plus cytoplasm score.
2.3. Reagents
Gibco® RPMI 1640, trypsin/EDTA, dimethyl sulfoxide (DMSO), phosphate-buffered saline (PBS), sulforhodamine B (SRB) medium, acetic acid, and TRIS base were also purchased from Sigma Aldrich Co. (St. Louis, MO, USA).
2.4. Cell Lines and Culture
The human OSCC cell lines HSC-3 and FaDu were obtained from the American Type Culture Collection (ATCC. Manassas, VA., USA), while the SAS and TW2.6 cells were kindly provided by Prof. Hsiao Michael (Genomic Research Center, Academia Sinica). Both cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen Life Technologies, Carlsbad, CA, USA), supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Invitrogen, Life Technologies, Carlsbad, CA, USA) and incubated at 37 °C in 5% humidified CO2 incubator. The OSCC cells were passaged at 98% confluence, and the medium was changed every 72 h before exposure to 5 Gy–15 Gy of radiotherapy.
2.5. shRNA Transfection of OSCC Cell Lines
The SAS, TW2.6, HSC-3, or FaDu cells were then transfected with shRNA specifically targeting CD47 or control/scramble shRNA purchased from Shanghai GenePharma Co., Ltd (Shanghai, China). The shRNA sequences were as follows: shCD47-1 - 5’-CGTCACAGGCAGGACCCACTGCCCA-3’; shCD47-2 - 5’-CCACAGATG TACAAGGGATGACCACAGTGTCATT-3’; and scramble shCD47 - 5’-CGTGACAGCCACGACCGACTGCGCA-3’. The OSCC cells were transfected with packed and harvested shRNA-coding lentiviruses following the manufacturer’s instruction. Briefly, 5 × 104 SAS or TW2.6 cells plated in 24-well plates were primed with 8 μg/mL hexadimethrine bromide, and viral particles were added to the culture medium at a multiplicity of infection (MOI) of 6. After transfection for 12 h, the virus-containing medium was replaced with fresh culture medium. The selection of viable OSCC cells stably transfected with shCD47 was performed with 2 μg/mL puromycin the next day, and the surviving colonies were expanded for further experiments.
2.6. Orosphere Formation and Self-Renewal Assay
For generation of orospheres, the OSCC cells were seeded at 5 × 104 cells/well in 6-well non-adherent plates (Corning Inc., Corning, NY, USA) in serum-free DMEM/F12 medium (11330057; Thermo Fisher Scientific Inc, Bartlesville, OK, USA.) supplemented with basic fibroblast growth factor (bFGF) (20 ng/mL, Invitrogen, Carlsbad, CA, USA), B27 supplement (Invitrogen, Carlsbad, CA, USA), 5 μg/mL insulin (91077C; Sigma Aldrich Co. (St. Louis, MO, USA)), and epidermal growth factor (EGF) (20 ng/mL, Millipore, Bedford, MA, USA). The cells were then incubated at 37 °C in a 5% humidified CO2 incubator for 12–15 days, and the formed orospheres were observed and counted using an inverted phase-contrast microscope. After 12 days of culture, primary orospheres consisting of ≥50 μm were counted, and images were taken under the microscope. Secondary orospheres were generated by dissociating primary orospheres using the trypsinization method, and the dissociated primary orospheres were then pipetted through a 22G needle to obtain a single-cell suspension (Thermo Fisher Scientific Inc, Bartlesville, OK, USA). After dissociation of the primary orospheres, cell seeding was done as for primary tumorspheres. After 12–15 days of culture, secondary orospheres consisting of ≥50 µm were counted, and the images were taken under the microscope.
2.7. Colony Formation Assay
WT or shCD47 SAS, TW2.6, HSC-3, or FaDu cells were plated in triplicate at 2 × 104 cells per well in 6-well plates and cultured for 12 days. After the colonies formed attained the cluster size of ≥50 cells, they were washed with PBS two times, fixed with 95% methanol for 15 min, and then stained with crystal violet at room temperature for 15 min. This was followed by colony visualization and counting under the microscope. The size and number of colonies formed were estimated with the ChemiDoc-XRS imager from the QuantityOne software package (Bio-Rad, Hercules, CA, USA).
2.8. Radiation and Cell Viability Assay
3 × 104 WT or shCD47 SAS, TW2.6, HSC-3, or FaDu cells were seeded and cultivated per plate in triplicate in 96-well plates. After 24 h, both adherent and floating cells were harvested, baseline cell-count was carried out, then triplicate wells were irradiated with 5 Gy–15 Gy or left without irradiation for another 24 h. The irradiated or unexposed OSCC cells were then harvested, fixed with 10% trichloroacetic acid (TCA), washed with ddH2O, and stained with 0.4% SRB (w/v) in 1% acetic acid. The unbound SRB dye was carefully washed off with 1% acetic acid several times, and then plated air-dried. The bound SRB dye was solubilized in 10 mM Trizma base, and absorbance was read using a microplate reader at 570 nm wavelength. Variation in cell numbers was relative to the baseline number of viable cells before irradiation.
2.9. Western Blot Analysis
For western blotting, cells were lysed using RIPA buffer (Cell Signaling Technology Inc., Danvers, MA, USA) with a cocktail of protease inhibitors (Sigma, St. Louis, MO, USA). Ten micrograms of protein samples were separated using 10% SDS-PAGE electrophoresis and transferred to polyvinylidene fluoride (PVDF) membranes using the Bio-Rad Mini-Protein electro-transfer system (Bio-Rad Laboratories Inc, CA, USA). Then, non-specific binding was blocked by incubating the membranes in 5% skimmed milk in Tris-buffered saline with 0.1% Tween 20 (TBST) for 1 h at room temperature, and then the membranes were incubated overnight at 4 °C with the antibodies against CD47 (1:1000, B6H12, sc-12730, Santa Cruz Biotechnology Inc, Santa Cruz, CA, USA), OCT4 (1:1000, C-10, sc-5279, Santa Cruz), SOX2 (1:1000, A-5, sc-365964, Santa Cruz), CD133 (1:1000, MAB4399-1, EMD Millipore, Temecula, CA, USA), vimentin (1:1000, D21H3, #5741, Cell Signaling Technology Inc., Danvers, MA, USA), Slug (1:1000, C19G7, #9585, Cell Signaling Technology Inc., Danvers, MA, USA), Snail (1:1000, C15D3, #3879, Cell Signaling Technology Inc., Danvers, MA, USA), N-cadherin (1:1000, D4R1H, #13116, Cell Signaling Technology Inc., Danvers, MA, USA), E-cadherin (1:1000, 24E10, #3195, Cell Signaling Technology Inc., Danvers, MA, USA), and GAPDH (1:500, G-9, sc-365062, Santa Cruz), listed in
Supplementary Table S1. The membranes were later incubated with the corresponding horseradish peroxidase (HRP)-conjugated anti-rabbit IgG or anti-mouse IgG secondary antibodies (Cell Signaling Technology Inc., Danvers, MA, USA) for 1 h at room temperature and washed with PBS four times. Protein band detection was done by the enhanced chemiluminescence (ECL) detection system (Thermo Fisher Scientific Inc., Waltham, MA, USA), and protein band quantification was done using ImageJ v. 1.46 (
https://imagej.nih.gov/ij/). Target protein expression was normalized to that of GAPDH, and assays were repeated four times in triplicates.
2.10. Wound Healing Migration Assay
For the wound-healing migration assay, OSCC cells were seeded in 6-well plates and cultured to 100% confluence. At full confluence, a 1 mm wound was made along the median axis of the well using a yellow pipette tip. Cell migration into the wound area was then observed at 0, 3, 6, and 12 h time-points under the microscope. The assay was performed three times in triplicates.
2.11. Matrigel Invasion and Migration Assay
Cell invasion assays were performed in Boyden chambers (pore size = 8 μm) with the upper side of the filter covered with 0.2% Matrigel diluted in DMEM. The 1 × 104 irradiated or un-irradiated WT, shCD47-1, or shCD47-2 OSCC cells in serum-free culture medium were plated in the upper chambers, while the lower chambers contained complete culture medium with 10% FBS as a chemoattractant. After overnight incubation, the un-invaded OSCC cells on the upper side of the filter were carefully removed with a cotton bud, while the invaded cells on the lower side of the membrane were washed, fixed in 95% ethanol and then stained with 10% Giemsa dye. The number of invaded cells was counted in 4 randomly selected fields of each membrane and averaged to obtain a representative number of the invaded cells. For the Boyden chamber migration assay, the porous membranes without the Matrigel were used.
2.12. Immunofluorescence Staining
For the immunofluorescence analysis, the OSCC cells were plated in 6-well chamber slides (Nunc™, Thermo Fisher Scientific Inc., Waltham, MA, USA) for 24 h, fixed in 2% paraformaldehyde at room temperature for 10 min, permeabilized with 0.1% Triton X-100 in 0.01 M PBS (pH 7.4) containing 0.2% BSA, air-dried, and rehydrated in PBS. The cells were then incubated with antibody against CD47, Oct4, Sox2, c-Myc, vimentin, or E-cadherin diluted 1:200 in PBS containing 3% normal goat serum at room temperature for 2 h, followed by incubation with anti-rabbit IgG fluorescein isothiocyanate (FITC)-conjugated secondary antibody (Jackson ImmunoResearch Lab. Inc., West Grove, PA, USA). The cells were allowed to rest at room temperature for 1 h, washed in PBS, and mounted using Vectashield mounting medium while counterstaining with 4′,6-diamidino-2-phenylindole (DAPI, D3571, Molecular Probes, Life Technologies Co., Carlsbad, CA, USA). Images were captured using a Zeiss Axiophot (Carl Zeiss Co. Ltd., Hsinchu City, Taiwan) fluorescence microscope, and the microphotographs were analyzed using ImageJ software v. 1.46 (
https://imagej.nih.gov/ij/).
2.13. Statistical Analysis
Each experiment was performed at least 3 times in triplicates. All statistical analyses were carried out using IBM SPSS Statistics for Windows, Version 25.0 (Released 2017; Armonk, NY: IBM Corp. USA). All data represent means ± standard deviation (SD). Comparison between two groups was estimated using the 2-sided Student’s t-test, while the one-way analysis of variance (ANOVA) was used for comparison between 3 or more groups. The association between the differential expression of CD47 and overall survival (OS) in patients with OSCC was determined using univariate Cox proportional regression of covariates, including the age, gender, AJCC stage, pathological grade, local recurrence, and lymph node involvement. Variables for which p < 0.05 were identified as significantly associated with prognosis, and Cox multivariate analysis was subsequently performed for these variables. Hazard ratios (HRs) and 95% confidence intervals (CIs) for multivariate analyses were computed using the Cox proportional hazards regression. p-value <0.05 was considered statistically significant.
4. Discussion
Despite advances made in the diagnostic approach and therapeutic strategies, OSCC remains a clinical challenge with great financial and emotional implications for patients and caregivers alike. The failure of conventional anticancer therapy in OSCC is increasingly being associated with the presence and activities of CSCs, where these OSCC-SCs are constitutively resistant to chemotherapeutic agents and radiation therapy. The last two decades have been characterized by heightened interest in and increased exploration of the role of CSCs in the resistance or reduced sensitivity of OSCC cells to standard chemotherapeutics or radiation therapy, with most research focused on the discovery of new therapeutic targets and/or development of novel anticancer drugs that preferentially target CSCs-enrichment in oral cancer cells [
5,
6,
7,
8,
9,
21].
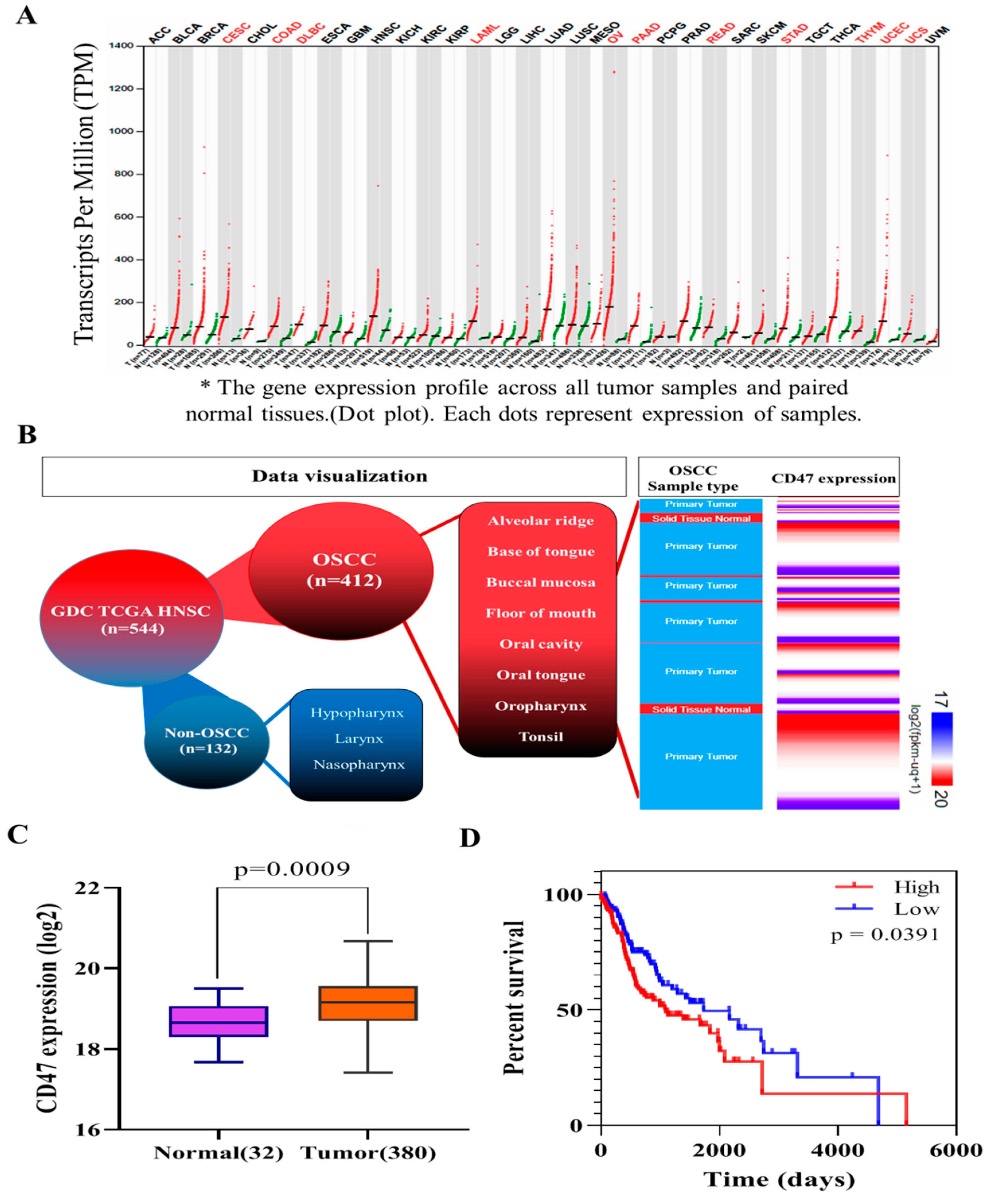
In this present study, we hypothesized and provided evidence that the ”don’t eat me” signal—CD47—modulated radiosensitivity by CSCs regulation and EMT deactivation in OSCC cells. We demonstrated that CD47 was aberrantly expressed in human OSCC and influenced the survival rate of patients with OSCC (
Figure 1 and
Figure 2;
Table 2). This finding corroborated the recently published work of Ye X., et al., who showed the overexpression of CD47 in Tca8113, Cal-27, and SCC-9 OSCC cell lines and week expression of same in normal oral keratinocytes. Consistent with our data showing that high CD47 expression conferred ~20% survival disadvantage and positively correlated with disease stage progression, they also suggested that CD47 might serve as a reliable predictive biomarker for oral pre-cancer and cancer progression, thus hinting on its probable role as an important molecular target for designing novel anticancer therapeutics for OSCC patients [
22]. Also demonstrated to be ectopically expressed in several other cancer types, strong evidence abounds implicating CD47 in the pathogenesis and progression of malignancies by its inherent ability to inhibit the phagocytosis of transformed or malignant cells [
23,
24,
25]. This anti-phagocytosis potential aligning with the cancer cells’ escape from immune surveillance, evasion of cell death, and sustenance of chronic proliferation sequel to enhanced mitogenic and pluripotency signals is evocative of the Hanahan and Weinberg’s hallmarks of cancer [
26] and reminiscent of our current understanding of CSCs biology and activities [
27]. Our understanding is that cancer cells are neither homogeneous nor solitary bio-entities; they interact with their microenvironment with the emission of inhibitory and activating cues, such as the mitogenic and angiogenic signals, respectively, to facilitate tumor progress [
26,
27]. The heterogeneous cell phenotype, tumor-initiating potential, perpetuity of OSCC cells, and the poorly chartered territory of OSCC-SCs eradication is now at the forefront of oral cancer research. The current paradigm is that a small subset of cancer cells, which in the context of this present work is termed OSCC-SCs, confers the unique self-renewal and cancer regeneration abilities on OSCC cells [
27].
In search of OSCC-relevant actionable molecular targets, our work for the first time, to the best of our knowledge, demonstrated that CD47 modulated the CSCs-like phenotype in OSCC, as evidenced by concomitant upregulation of known pluripotency transcription factors and stemness markers with CD47 expression, and co-suppression of SOX2, OCT4, c-MYC, and CD133 protein expression following shRNA-mediated downregulation of CD47 expression in wild-type adherent and anchorage-independent orospheres (
Figure 3). As already alluded above, CSCs within the OSCC tumor nests co-express OCT4, SOX2, NANOG, phosphorylated STAT3 (pSTAT3), CD133, CD44, and c-MYC [
18]. Our result is of clinical relevance since, in the CSCs model, the net loss of cancer cells due to programmed or spontaneous cell death elicits a short-term reduction in population size, but over the long-term, counter-intuitively promotes tumor growth, as previously quiescent OSCC-SCs are reactivated, re-enter the cell cycle, proliferate, and repopulate the tumor bulk. Thus, the significance of efficient therapeutic targeting of OSCC-SCs is by the genetic ablation of CD47 expression and/or activity, as demonstrated in our work. Our findings are corroborated by recently demonstrated induction of macrophage phagocytosis of lung cancer cells and lung CSCs, as well as the inhibition of lung CSCs-induced tumor growth in immune-deficient mice xenograft models by blocking CD47 function with anti-CD47 antibodies [
28]. Similarly, enhanced phagocytosis of CD47-rich CD133+ ovarian TICs, which are relatively resistant to current anticancer treatment, is triggered by treatment with anti-CD47 mAb or CD47 knockdown [
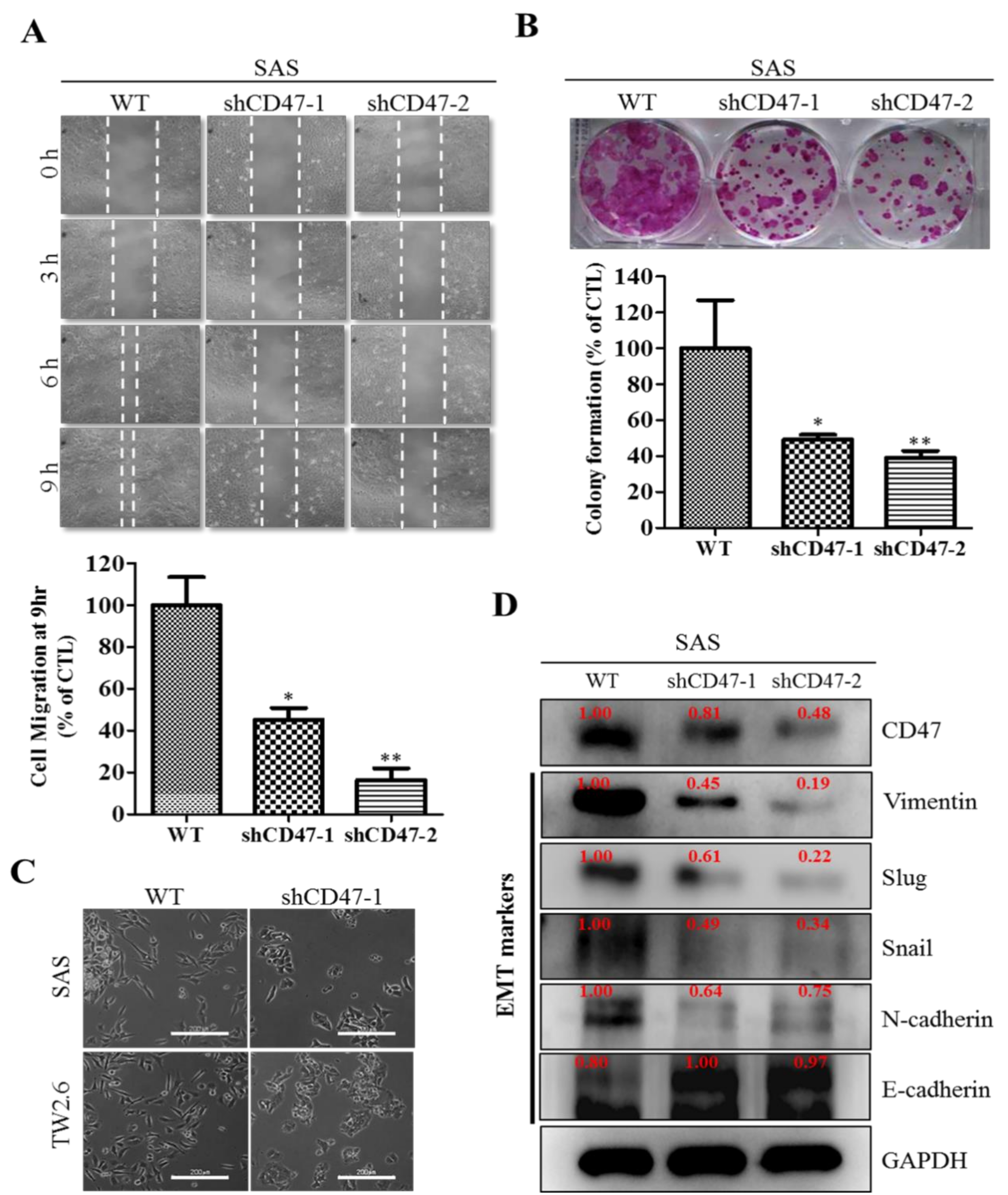
29]. We also demonstrated that the downregulation of CD47 induced the loss of the mesenchymal (fibroblast-like) phenotype and acquisition of an epithelial (ovoid-shaped) phenotype, attenuating the EMT and migration capacity of OSCC cells (
Figure 4). We posit that by facilitating cytoskeleton remodeling, cell elongation, and loss of cell-cell/cell-basal membrane adhesion, CD47 induces enhanced migration and invasion, resistance to chemoradiotherapy, and evasion of cell death, which are characteristics of EMT in OSCC; upon completion of the transition, the cells degrade the underlying extracellular matrix (ECM) and commence dispersion to secondary anatomic sites. The disruption of this CD47-induced phenotypic transition would be consistent with our demonstrated shCD47-induced reversal of cadherin switching with resultant E-cadherin/N-cadherin ratio greater than 1, suppression of E-cadherin master regulators--Snail and Slug [
30], as well as the intermediate filament, vimentin, which is associated with enhanced invasiveness, higher prevalence of lymph node involvement, disease recurrence, and poor prognosis [
30,
31]. Aside from demonstrating that CD47 modulates the expression and subcellular localization of mesenchymal and epithelial factors in OSCC, we also provided evidence, at least in part, that CD47 was a master regulator of stem cell development, differentiation, and maintenance, as well as positive regulator of cell migration and cell motility (
Figure 5). These findings are particularly insightful and therapeutically-relevant, especially as the “migrating, self-renewing and symmetrically-dividing CSCs shape the primary tumor, and are also exclusively capable of distant seeding, whereas the majority of non-stem cancer cells (that can be frequently detected as circulating tumor cells) are intrinsically only able to form dormant micrometastases” [
27]. To the best of our understanding of the OSCC SC-EMT, while it is difficult to delineate clearly whether it is the undifferentiated or differentiated OSCC cells responsible for observed EMT phenotype in our study, we would allude to the recent comprehensive overview of related theme, in which Kim et al. suggested that “early, undifferentiated cells with mesenchymal phenotype are characterized by a shift from E-cadherin expression to N-cadherin expression along with the expression of Snails, vimentin and metalloproteases”, and that these “early undifferentiated cells with a mesenchymal phenotype retain the expression of several totipotent transcription factors (e.g., Oct4 and Nanog), which indicates that these cells can adopt a mesenchymal phenotype without losing their pluripotency” [
32]. In light of this, we posit a regulatory role for CD47 signaling at the interphase between the CSCs and EMT phenotype of OSCC cells.
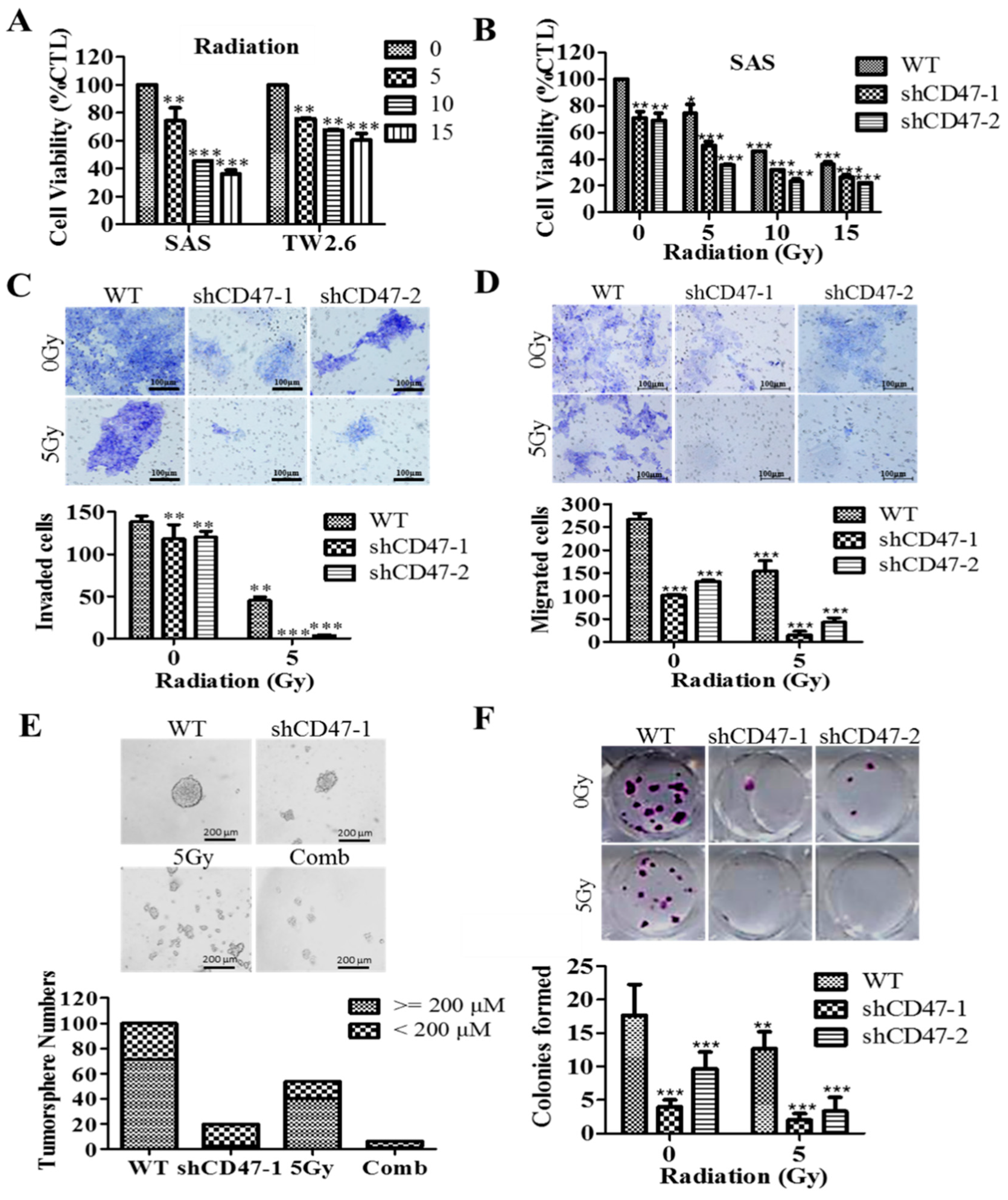
By inference, extrinsic perturbations, such as CD47 blockage/knockdown and cytotoxic treatments, including radiation therapy, may mold oral tumor by selective targeting of the aggressive OSCC cells, including OSCC-SCs, which subsequently facilitate malignant growth; thus, any efficacious therapy must eradicate OSCC-SCs; however, there is mounting evidence, showing these cells are intrinsically less or in-sensitive to current OSCC anticancer therapy. Thus, interestingly, having shown the existence of a positive correlation of CD47 expression with CSC, EMT, and metastatic phenotypes, as well as an inverse correlation with OSCC patients’ overall survival, cell death; we finally demonstrated that the suppression of CD47 expression enhanced the sensitivity of OSCC-SCs to radiation therapy, as evidenced by the remarkable synergistic effect of concurrent CD47 knockdown and radiotherapy on cell viability, migration, invasiveness, clonogenic, and orospheric survival (
Figure 6). This has clinical significance since radiotherapy is a common and very vital component of the multidisciplinary treatment for patients with OSCC, especially those with unresectable oral cavity tumors, cases where surgery is technically improbable for the early-stage disease with high risk of cosmetic or functional defect, high operative risk secondary to co-morbidity or subpar performance status [
33,
34].
Human papillomavirus (HPV) infection has been implicated in over 25% of HNSCCs. In fact, many of the HPV positive HNSCCs are oropharynx cancer, including tongue base and tonsil [
35,
36]. Clinically, the HPV status of patients is being touted as a predictor of treatment response and survival rate [
3,
37,
38]. It has been suggested that the better prognosis of HPV(+) HNSCCs involves the host immune system, especially as immune cells, which were originally suppressed in the tumor microenvironment, have been shown to be re-activated through the crosstalk of immunogenic signals between tumorous cells and immune cells by exosomes [
38]. Tumors release exosomes containing a lot of antigens. In contrast to HPV(−) tumor, exosomes released by HPV (+) tumors do exhibit prolong immunogenic interaction with immune cells due to the regulation of the expression and/or activity of CD47 on the membrane of cancerous cells. The membrane protein CD47 functions as a “don’t eat me” signal and thus limits the clearance of cancerous cells by circulating monocytes [
38,
39]. Radiation has been shown to induce anti-tumor immune response [
40] and associated with a dose-dependent decrease in the surface expression of CD47 on HPV(+) HNSCC cells, resulting in improved clearance of tumorous cell, in vitro and in vivo [
39].
Our initial analysis of the TCGA-HNSCC cohort (n = 604) gene expression profile dataset showed it contained limited number of the HPV positive HNSCC as sorted by FISH or p16 testing; most of the tumor sites were located at the tongue base and oropharynx, which were excluded in our original TCGA analysis since our study focus was oral cancer instead. In addition, the expression level of CD47 in these samples was low or null. These findings suggested that the therapeutic potential of targeting CD47 was likely independent of the HPV status of the patients with OSCC. The mechanism underlying the response of CD47 to radiation in OSCC remained relatively underexplored, thus, necessitating a further investigation of the use of CD47 as a potential therapeutic target in anti-OSCC treatment.
As with most studies of this nature, this present study had several limitations within which the findings presented herein should be interpreted carefully. This study was a retrospective, single-center investigation of the selected biomarker in a relatively small cohort of Taiwanese OSCC patients (n = 71), limiting the ‘generalizability’ of the findings reported, especially to other ethno-geographics and raises the question of probable predisposition to selection biases.
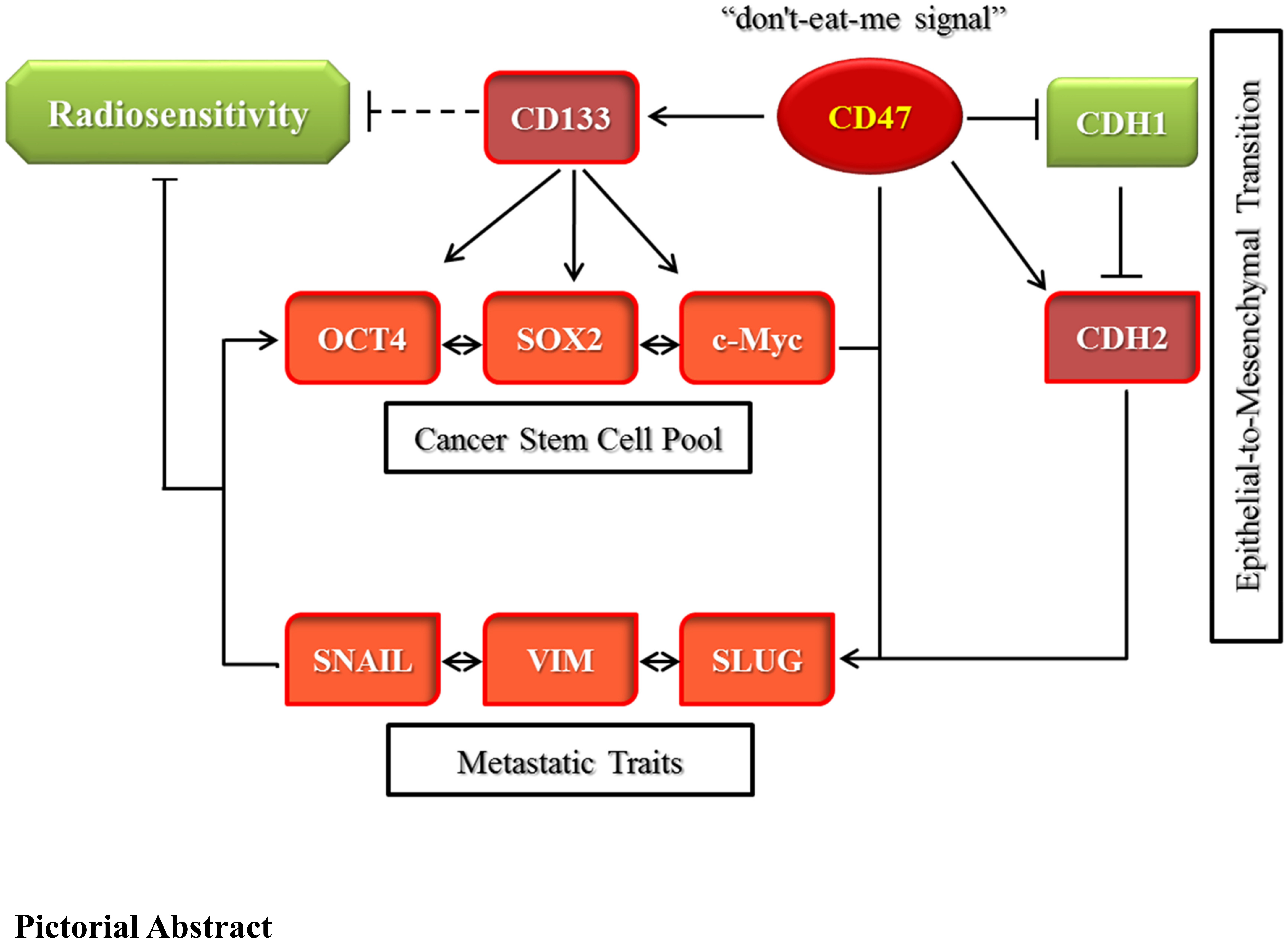
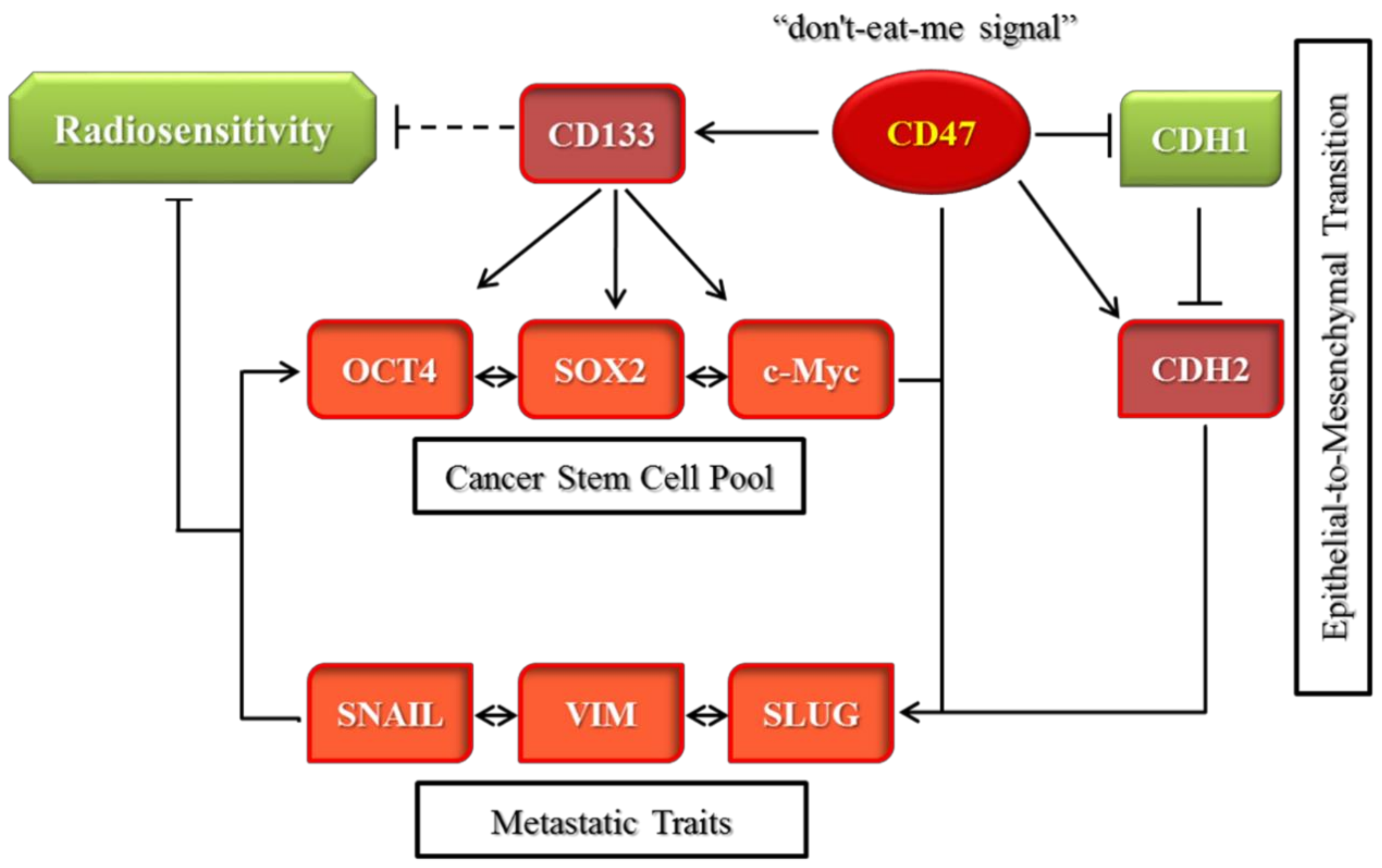
In conclusion, as depicted in
Figure 7, we demonstrated that CD47 knockdown alone or combined with radiation therapy significantly inhibited the survival and/or proliferation of OSCC-SCs. The CD47 knockdown of shCD47 also suppressed orosphere formation, reduced colony formation, and enhanced radiosensitivity in OSCC by dysregulation of our proposed CD47-CSCs-EMT signaling loop. The findings of our present study lent credence to the role of CD47 as a putative actionable therapeutic target with high anticancer efficacy and laid another brick on the foundation for further exploration of the clinical feasibility and therapeutic application of ‘CD47 ablation’ as a novel strategy for improving therapeutic outcome in oral cancer clinics.

 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}