Therapeutic Modulation of Autophagy in Leukaemia and Lymphoma

 ,
,  and
and 
Abstract
:
1. Introduction
Macroautophagy—Janus-Faced Role in Cancer
2. Haematopoiesis Development and Autophagy
2.1. Haematopoiesis
2.2. Autophagy in HSCs
2.3. Autophagy in Development and Differentiation of Lymphocytes
2.3.1. T Lymphocytes
2.3.2. B Lymphocytes
2.3.3. NK Cells
2.4. Autophagy in Development and Differentiation of Erythrocytes
2.5. Autophagy in Development and Differentiation of Macrophages, Neutrophils and Megakaryocytes
2.5.1. Macrophages
2.5.2. Neutrophils
2.5.3. Megakaryocytes
3. Autophagy and Lymphoid Tumours
3.1. Aberrant Autophagy in Lymphomas and Lymphoid Leukaemia
3.1.1. Lymphomas
3.1.2. Acute Lymphoid Leukaemia
3.2. Autophagy-Based Treatment Strategies in Lymphomas and Lymphoid Leukaemia
3.2.1. Lymphomas
3.2.2. Acute Lymphoid Leukaemia
4. Autophagy and Myeloid Tumours
4.1. Aberrant Autophagy in Myeloid Dysplastic Syndromes and Myeloid Leukaemia
4.1.1. Myelodysplastic Syndrome (MDS)
4.1.2. Acute Myeloid Leukaemia
4.1.3. Chronic Myeloid Leukaemia
4.2. Autophagy-Based Treatment Strategies in Myeloid Dysplastic Syndromes and Myeloid Leukaemia
4.2.1. Myelodysplastic Syndrome (MDS)
4.2.2. Acute Myeloid Leukaemia
4.3. Chronic Myeloid Leukaemia
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Klionsky, D.J.; Codogno, P. The mechanism and physiological function of macroautophagy. J. Innate Immun. 2013, 5, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the Integrated Stress Response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Komatsu, M.; Finley, K.; Simonsen, A. Autophagy: More Than a Nonselective Pathway. Int. J. Cell Biol. 2012, 2012, e219625. [Google Scholar] [CrossRef] [PubMed]
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, Metabolism, and Cancer. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.K.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The Role of Atg Proteins in Autophagosome Formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Proikas-Cezanne, T.; Takacs, Z.; Dönnes, P.; Kohlbacher, O. WIPI proteins: Essential PtdIns3P effectors at the nascent autophagosome. J. Cell Sci. 2015, 128, 207–217. [Google Scholar] [CrossRef]
- Khaminets, A.; Behl, C.; Dikic, I. Ubiquitin-Dependent and Independent Signals in Selective Autophagy. Trends Cell Biol. 2016, 26, 6–16. [Google Scholar] [CrossRef]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef]
- Mochida, K.; Oikawa, Y.; Kimura, Y.; Kirisako, H.; Hirano, H.; Ohsumi, Y.; Nakatogawa, H. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 2015, 522, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Löhr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Cecconi, F.; Levine, B. The role of autophagy in mammalian development: Cell makeover rather than cell death. Dev. Cell 2008, 15, 344–357. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-Y.; White, E. Role of Autophagy in Cancer Prevention. Cancer Prev. Res. 2011, 4, 973–983. [Google Scholar] [CrossRef]
- Strohecker, A.M.; White, E. Autophagy promotes BrafV600E-driven lung tumorigenesis by preserving mitochondrial metabolism. Autophagy 2014, 10, 384–385. [Google Scholar] [CrossRef]
- Guo, J.Y.; White, E. Autophagy is required for mitochondrial function, lipid metabolism, growth, and fate of KRAS(G12D)-driven lung tumors. Autophagy 2013, 9, 1636–1638. [Google Scholar] [CrossRef]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef]
- Guo, J.Y.; Chen, H.-Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.S.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef]
- Thorburn, A.; Thamm, D.H.; Gustafson, D.L. Autophagy and cancer therapy. Mol. Pharmacol. 2014, 85, 830–838. [Google Scholar] [CrossRef]
- Orsini, M.; Morceau, F.; Dicato, M.; Diederich, M. Autophagy as a pharmacological target in hematopoiesis and hematological disorders. Biochem. Pharmacol. 2018, 152, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Doulatov, S.; Notta, F.; Laurenti, E.; Dick, J.E. Hematopoiesis: A human perspective. Cell Stem Cell 2012, 10, 120–136. [Google Scholar] [CrossRef] [PubMed]
- Orkin, S.H.; Zon, L.I. Hematopoiesis: An evolving paradigm for stem cell biology. Cell 2008, 132, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Kohli, L.; Passegué, E. Surviving change: The metabolic journey of hematopoietic stem cells. Trends Cell Biol. 2014, 24, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.J.; Seita, J.; Czechowicz, A.; Bhattacharya, D.; Bryder, D.; Weissman, I.L. Hematopoietic stem cell quiescence attenuates DNA damage response and permits DNA damage accumulation during aging. Cell Cycle 2007, 6, 2371–2376. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, M. Differentiation and proliferation of hematopoietic stem cells. Blood 1993, 81, 2844–2853. [Google Scholar] [PubMed]
- Van Zant, G.; Liang, Y. The role of stem cells in aging. Exp. Hematol. 2003, 31, 659–672. [Google Scholar] [CrossRef]
- Morrison, S.J.; Wandycz, A.M.; Akashi, K.; Globerson, A.; Weissman, I.L. The aging of hematopoietic stem cells. Nat. Med. 1996, 2, 1011–1016. [Google Scholar] [CrossRef]
- Sudo, K.; Ema, H.; Morita, Y.; Nakauchi, H. Age-associated characteristics of murine hematopoietic stem cells. J. Exp. Med. 2000, 192, 1273–1280. [Google Scholar] [CrossRef]
- Rossi, D.J.; Bryder, D.; Zahn, J.M.; Ahlenius, H.; Sonu, R.; Wagers, A.J.; Weissman, I.L. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc. Natl. Acad. Sci. USA 2005, 102, 9194–9199. [Google Scholar] [CrossRef]
- Chambers, S.M.; Shaw, C.A.; Gatza, C.; Fisk, C.J.; Donehower, L.A.; Goodell, M.A. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 2007, 5, e201. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, M.; Soilleux, E.J.; Djordjevic, G.; Tripp, R.; Lutteropp, M.; Sadighi-Akha, E.; Stranks, A.J.; Glanville, J.; Knight, S.; Jacobsen, S.-E.W.; et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J. Exp. Med. 2011, 208, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lee, J.Y.; Wei, H.; Tanabe, O.; Engel, J.D.; Morrison, S.J.; Guan, J.-L. FIP200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood 2010, 116, 4806–4814. [Google Scholar] [CrossRef] [PubMed]
- Warr, M.R.; Binnewies, M.; Flach, J.; Reynaud, D.; Garg, T.; Malhotra, R.; Debnath, J.; Passegué, E. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 2013, 494, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Cai, J.; Zhang, S.; Yuan, N.; Fang, Y.; Wang, Z.; Li, X.; Cao, D.; Xu, F.; Lin, W.; et al. Autophagy Sustains Hematopoiesis Through Targeting Notch. Stem Cells Dev. 2015, 24, 2660–2673. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Fang, Y.; Cai, J.; Li, X.; Xu, F.; Yuan, N.; Zhang, S.; Wang, J. ROS functions as an upstream trigger for autophagy to drive hematopoietic stem cell differentiation. Hematology 2016, 21, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lansinger, O.M.; Flach, J.; Verovskaya, E.V.; Figueroa, M.E.; Passegué, E. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017, 543, 205–210. [Google Scholar] [CrossRef]
- Cooper, M.D. The early history of B cells. Nat. Rev. Immunol. 2015, 15, 191–197. [Google Scholar] [CrossRef]
- Gascoigne, N.R.J.; Rybakin, V.; Acuto, O.; Brzostek, J. TCR Signal Strength and T Cell Development. Annu. Rev. Cell Dev. Biol. 2016, 32, 327–348. [Google Scholar] [CrossRef]
- Bhandoola, A.; von Boehmer, H.; Petrie, H.T.; Zúñiga-Pflücker, J.C. Commitment and developmental potential of extrathymic and intrathymic T cell precursors: Plenty to choose from. Immunity 2007, 26, 678–689. [Google Scholar] [CrossRef]
- Pua, H.H.; Dzhagalov, I.; Chuck, M.; Mizushima, N.; He, Y.-W. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J. Exp. Med. 2007, 204, 25–31. [Google Scholar] [CrossRef]
- Stephenson, L.M.; Miller, B.C.; Ng, A.; Eisenberg, J.; Zhao, Z.; Cadwell, K.; Graham, D.B.; Mizushima, N.N.; Xavier, R.; Virgin, H.W.; et al. Identification of Atg5-dependent transcriptional changes and increases in mitochondrial mass in Atg5-deficient T lymphocytes. Autophagy 2009, 5, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Capan, E.; Zhao, Y.; Zhao, J.; Stolz, D.; Watkins, S.C.; Jin, S.; Lu, B. Autophagy is induced in CD4+ T cells and important for the growth factor-withdrawal cell death. J. Immunol. 2006, 177, 5163–5168. [Google Scholar] [CrossRef]
- Paul, S.; Kashyap, A.K.; Jia, W.; He, Y.-W.; Schaefer, B.C. Selective autophagy of the adaptor protein Bcl10 modulates T cell receptor activation of NF-κB. Immunity 2012, 36, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Bell, B.D.; Leverrier, S.; Weist, B.M.; Newton, R.H.; Arechiga, A.F.; Luhrs, K.A.; Morrissette, N.S.; Walsh, C.M. FADD and caspase-8 control the outcome of autophagic signaling in proliferating T cells. Proc. Natl. Acad. Sci. USA 2008, 105, 16677–16682. [Google Scholar] [CrossRef]
- Lünemann, J.D.; Münz, C. Autophagy in CD4+ T-cell immunity and tolerance. Cell Death Differ. 2009, 16, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Rivera Vargas, T.; Cai, Z.; Shen, Y.; Dosset, M.; Benoit-Lizon, I.; Martin, T.; Roussey, A.; Flavell, R.A.; Ghiringhelli, F.; Apetoh, L. Selective degradation of PU.1 during autophagy represses the differentiation and antitumour activity of TH9 cells. Nat. Commun. 2017, 8, 559. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Long, L.; Yang, K.; Guy, C.; Shrestha, S.; Chen, Z.; Wu, C.; Vogel, P.; Neale, G.; Green, D.R.; et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat. Immunol. 2016, 17, 277–285. [Google Scholar] [CrossRef]
- O’Sullivan, D.; van der Windt, G.J.W.; Huang, S.C.-C.; Curtis, J.D.; Chang, C.-H.; Buck, M.D.; Qiu, J.; Smith, A.M.; Lam, W.Y.; DiPlato, L.M.; et al. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity 2014, 41, 75–88. [Google Scholar] [CrossRef]
- Zhu, L.; Xie, X.; Zhang, L.; Wang, H.; Jie, Z.; Zhou, X.; Shi, J.; Zhao, S.; Zhang, B.; Cheng, X.; et al. TBK-binding protein 1 regulates IL-15-induced autophagy and NKT cell survival. Nat. Commun. 2018, 9, 2812. [Google Scholar] [CrossRef]
- Pieper, K.; Grimbacher, B.; Eibel, H. B-cell biology and development. J. Allergy Clin. Immunol. 2013, 131, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.C.; Zhao, Z.; Stephenson, L.M.; Cadwell, K.; Pua, H.H.; Lee, H.K.; Mizushima, N.N.; Iwasaki, A.; He, Y.-W.; Swat, W.; et al. The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy 2008, 4, 309–314. [Google Scholar] [CrossRef]
- Arnold, J.; Murera, D.; Arbogast, F.; Fauny, J.-D.; Muller, S.; Gros, F. Autophagy is dispensable for B-cell development but essential for humoral autoimmune responses. Cell Death Differ. 2016, 23, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Hong, M.J.; Sun, H.; Wang, L.; Shi, X.; Gilbert, B.E.; Corry, D.B.; Kheradmand, F.; Wang, J. Essential role for autophagy in the maintenance of immunological memory against influenza infection. Nat. Med. 2014, 20, 503–510. [Google Scholar] [CrossRef]
- Chen, M.; Kodali, S.; Jang, A.; Kuai, L.; Wang, J. Requirement for autophagy in the long-term persistence but not initial formation of memory B cells. J. Immunol. 2015, 194, 2607–2615. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Martin, N.; Maldonado, P.; Gasparrini, F.; Frederico, B.; Aggarwal, S.; Gaya, M.; Tsui, C.; Burbage, M.; Keppler, S.J.; Montaner, B.; et al. A switch from canonical to noncanonical autophagy shapes B cell responses. Science 2017, 355, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Pengo, N.; Scolari, M.; Oliva, L.; Milan, E.; Mainoldi, F.; Raimondi, A.; Fagioli, C.; Merlini, A.; Mariani, E.; Pasqualetto, E.; et al. Plasma cells require autophagy for sustainable immunoglobulin production. Nat. Immunol. 2013, 14, 298–305. [Google Scholar] [CrossRef]
- Wang, S.; Xia, P.; Huang, G.; Zhu, P.; Liu, J.; Ye, B.; Du, Y.; Fan, Z. FoxO1-mediated autophagy is required for NK cell development and innate immunity. Nat. Commun. 2016, 7, 11023. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, T.E.; Geary, C.D.; Weizman, O.-E.; Geiger, T.L.; Rapp, M.; Dorn, G.W.; Overholtzer, M.; Sun, J.C. Atg5 Is Essential for the Development and Survival of Innate Lymphocytes. Cell Rep. 2016, 15, 1910–1919. [Google Scholar] [CrossRef] [PubMed]
- Morvan, M.G.; Lanier, L.L. NK cells and cancer: You can teach innate cells new tricks. Nat. Rev. Cancer 2016, 16, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Berchem, G.; Janji, B. Targeting autophagy blocks melanoma growth by bringing natural killer cells to the tumor battlefield. Autophagy 2018, 14, 730–732. [Google Scholar] [CrossRef] [PubMed]
- Baginska, J.; Viry, E.; Berchem, G.; Poli, A.; Noman, M.Z.; van Moer, K.; Medves, S.; Zimmer, J.; Oudin, A.; Niclou, S.P.; et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc. Natl. Acad. Sci. USA 2013, 2013, 04790. [Google Scholar] [CrossRef] [PubMed]
- Takano-Ohmuro, H.; Mukaida, M.; Kominami, E.; Morioka, K. Autophagy in embryonic erythroid cells: Its role in maturation. Eur. J. Cell Biol. 2000, 79, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Fader, C.M.; Salassa, B.N.; Grosso, R.A.; Vergara, A.N.; Colombo, M.I. Hemin induces mitophagy in a leukemic erythroblast cell line. Biol. Cell 2016, 108, 77–95. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.-A.; Sanalkumar, R.; O’Geen, H.; Linnemann, A.K.; Chang, C.-J.; Bouhassira, E.E.; Farnham, P.J.; Keles, S.; Bresnick, E.H. Autophagy driven by a master regulator of hematopoiesis. Mol. Cell. Biol. 2012, 32, 226–239. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Cai, J.; Li, X.; Yuan, N.; Zhang, S. Autophagy governs erythroid differentiation both in vitro and in vivo. Hematology 2016, 21, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Aerbajinai, W.; Giattina, M.; Lee, Y.T.; Raffeld, M.; Miller, J.L. The proapoptotic factor Nix is coexpressed with Bcl-xL during terminal erythroid differentiation. Blood 2003, 102, 712–717. [Google Scholar] [CrossRef]
- Chen, M.; Sandoval, H.; Wang, J. Selective mitochondrial autophagy during erythroid maturation. Autophagy 2008, 4, 926–928. [Google Scholar] [CrossRef]
- Sandoval, H.; Thiagarajan, P.; Dasgupta, S.K.; Schumacher, A.; Prchal, J.T.; Chen, M.; Wang, J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008, 454, 232–235. [Google Scholar] [CrossRef]
- Schwarten, M.; Mohrlüder, J.; Ma, P.; Stoldt, M.; Thielmann, Y.; Stangler, T.; Hersch, N.; Hoffmann, B.; Merkel, R.; Willbold, D. Nix directly binds to GABARAP: A possible crosstalk between apoptosis and autophagy. Autophagy 2009, 5, 690–698. [Google Scholar] [CrossRef]
- Betin, V.M.S.; Singleton, B.K.; Parsons, S.F.; Anstee, D.J.; Lane, J.D. Autophagy facilitates organelle clearance during differentiation of human erythroblasts: Evidence for a role for ATG4 paralogs during autophagosome maturation. Autophagy 2013, 9, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Li-Harms, X.; Milasta, S.; Lynch, J.; Wright, C.; Joshi, A.; Iyengar, R.; Neale, G.; Wang, X.; Wang, Y.-D.; Prolla, T.A.; et al. Mito-protective autophagy is impaired in erythroid cells of aged mtDNA-mutator mice. Blood 2015, 125, 162–174. [Google Scholar] [CrossRef]
- Kundu, M.; Lindsten, T.; Yang, C.-Y.; Wu, J.; Zhao, F.; Zhang, J.; Selak, M.A.; Ney, P.A.; Thompson, C.B. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 2008, 112, 1493–1502. [Google Scholar] [CrossRef] [PubMed]
- Joo, J.H.; Dorsey, F.C.; Joshi, A.; Hennessy-Walters, K.M.; Rose, K.L.; McCastlain, K.; Zhang, J.; Iyengar, R.; Jung, C.H.; Suen, D.-F.; et al. Hsp90-Cdc37 chaperone complex regulates Ulk1- and Atg13-mediated mitophagy. Mol. Cell 2011, 43, 572–585. [Google Scholar] [CrossRef] [PubMed]
- Orsini, M.; Chateauvieux, S.; Rhim, J.; Gaigneaux, A.; Cheillan, D.; Christov, C.; Dicato, M.; Morceau, F.; Diederich, M. Sphingolipid-mediated inflammatory signaling leading to autophagy inhibition converts erythropoiesis to myelopoiesis in human hematopoietic stem/progenitor cells. Cell Death Differ. 2018. [Google Scholar] [CrossRef] [PubMed]
- Mancias, J.D.; Pontano Vaites, L.; Nissim, S.; Biancur, D.E.; Kim, A.J.; Wang, X.; Liu, Y.; Goessling, W.; Kimmelman, A.C.; Harper, J.W. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Ginhoux, F.; Jung, S. Monocytes and macrophages: Developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014, 14, 392–404. [Google Scholar] [CrossRef]
- Jacquel, A.; Obba, S.; Boyer, L.; Dufies, M.; Robert, G.; Gounon, P.; Lemichez, E.; Luciano, F.; Solary, E.; Auberger, P. Autophagy is required for CSF-1-induced macrophagic differentiation and acquisition of phagocytic functions. Blood 2012, 119, 4527–4531. [Google Scholar] [CrossRef]
- Zhang, Y.; Morgan, M.J.; Chen, K.; Choksi, S.; Liu, Z. Induction of autophagy is essential for monocyte-macrophage differentiation. Blood 2012, 119, 2895–2905. [Google Scholar] [CrossRef]
- Obba, S.; Hizir, Z.; Boyer, L.; Selimoglu-Buet, D.; Pfeifer, A.; Michel, G.; Hamouda, M.-A.; Gonçalvès, D.; Cerezo, M.; Marchetti, S.; et al. The PRKAA1/AMPKα1 pathway triggers autophagy during CSF1-induced human monocyte differentiation and is a potential target in CMML. Autophagy 2015, 11, 1114–1129. [Google Scholar] [CrossRef] [PubMed]
- Stranks, A.J.; Hansen, A.L.; Panse, I.; Mortensen, M.; Ferguson, D.J.P.; Puleston, D.J.; Shenderov, K.; Watson, A.S.; Veldhoen, M.; Phadwal, K.; et al. Autophagy Controls Acquisition of Aging Features in Macrophages. J. Innate Immun. 2015, 7, 375–391. [Google Scholar] [CrossRef] [PubMed]
- Shan, M.; Qin, J.; Jin, F.; Han, X.; Guan, H.; Li, X.; Zhang, J.; Zhang, H.; Wang, Y. Autophagy suppresses isoprenaline-induced M2 macrophage polarization via the ROS/ERK and mTOR signaling pathway. Free Radic. Biol. Med. 2017, 110, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Rizo, V.; Martínez-Guzmán, M.A.; Iñiguez-Gutierrez, L.; García-Orozco, A.; Alvarado-Navarro, A.; Fafutis-Morris, M. Neutrophil Extracellular Traps and Its Implications in Inflammation: An Overview. Front. Immunol. 2017, 8, 81. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, M.J.; Radic, M. Neutrophil extracellular traps: Double-edged swords of innate immunity. J. Immunol. 2012, 189, 2689–2695. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-K.; Park, M.J.; Lee, H.W.; Lee, H.S.; Choi, S.R.; Song, Y.R.; Kim, H.J.; Park, H.-C.; Kim, S.G. The relationship between autophagy, increased neutrophil extracellular traps formation and endothelial dysfunction in chronic kidney disease. Clin. Immunol. 2018, 197, 189–197. [Google Scholar] [CrossRef]
- Remijsen, Q.; Berghe, T.V.; Wirawan, E.; Asselbergh, B.; Parthoens, E.; De Rycke, R.; Noppen, S.; Delforge, M.; Willems, J.; Vandenabeele, P. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011, 21, 290–304. [Google Scholar] [CrossRef]
- Kajiume, T.; Kobayashi, M. Human granulocytes undergo cell death via autophagy. Cell Death Discov. 2018, 4, 111. [Google Scholar] [CrossRef]
- Satake, S.; Hirai, H.; Hayashi, Y.; Shime, N.; Tamura, A.; Yao, H.; Yoshioka, S.; Miura, Y.; Inaba, T.; Fujita, N.; et al. C/EBPβ is involved in the amplification of early granulocyte precursors during candidemia-induced “emergency” granulopoiesis. J. Immunol. 2012, 189, 4546–4555. [Google Scholar] [CrossRef]
- Riffelmacher, T.; Clarke, A.; Richter, F.C.; Stranks, A.; Pandey, S.; Danielli, S.; Hublitz, P.; Yu, Z.; Johnson, E.; Schwerd, T.; et al. Autophagy-Dependent Generation of Free Fatty Acids Is Critical for Normal Neutrophil Differentiation. Immunity 2017, 47, 466–480. [Google Scholar] [CrossRef]
- Ocana, A.; Nieto-Jiménez, C.; Pandiella, A.; Templeton, A.J. Neutrophils in cancer: Prognostic role and therapeutic strategies. Mol. Cancer 2017, 16, 137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shi, H.; Yuan, X.; Jiang, P.; Qian, H.; Xu, W. Tumor-derived exosomes induce N2 polarization of neutrophils to promote gastric cancer cell migration. Mol. Cancer 2018, 17, 146. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, N.; Capobianco, A.; Rovere-Querini, P.; Ramirez, G.A.; Tombetti, E.; Valle, P.D.; Monno, A.; D’Alberti, V.; Gasparri, A.M.; Franchini, S.; et al. Platelet microparticles sustain autophagy-associated activation of neutrophils in systemic sclerosis. Sci. Transl. Med. 2018, 10, eaao3089. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, M.; Carniato, F.; Tonello, S.; Migliario, M.; Invernizzi, M.; Rocchetti, V.; Marchese, L.; Renò, F. Charged molecular silica trigger in vitro NETosis in human granulocytes via both oxidative and autophagic pathways. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 7058–7068. [Google Scholar] [PubMed]
- Park, S.Y.; Shrestha, S.; Youn, Y.-J.; Kim, J.-K.; Kim, S.-Y.; Kim, H.J.; Park, S.-H.; Ahn, W.-G.; Kim, S.; Lee, M.G.; et al. Autophagy Primes Neutrophils for Neutrophil Extracellular Trap Formation during Sepsis. Am. J. Respir. Crit. Care Med. 2017, 196, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Li, T.; Cao, M.; Si, Y.; Wu, X.; Zhao, L.; Yao, Z.; Zhang, Y.; Fang, S.; Deng, R.; et al. Extracellular DNA traps released by acute promyelocytic leukemia cells through autophagy. Cell Death Dis. 2016, 7, e2283. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Wei, Q.; Shin, J.N.; Abdel Fattah, E.; Bonilla, D.L.; Xiang, Q.; Eissa, N.T. Autophagy Is Required for Neutrophil-Mediated Inflammation. Cell Rep. 2015, 12, 1731–1739. [Google Scholar] [CrossRef]
- Iula, L.; Keitelman, I.A.; Sabbione, F.; Fuentes, F.; Guzman, M.; Galletti, J.G.; Gerber, P.P.; Ostrowski, M.; Geffner, J.R.; Jancic, C.C.; et al. Autophagy Mediates Interleukin-1β Secretion in Human Neutrophils. Front. Immunol. 2018, 9, 269. [Google Scholar] [CrossRef]
- Machlus, K.R.; Italiano, J.E. The incredible journey: From megakaryocyte development to platelet formation. J. Cell Biol. 2013, 201, 785–796. [Google Scholar] [CrossRef]
- Cao, Y.; Cai, J.; Zhang, S.; Yuan, N.; Li, X.; Fang, Y.; Song, L.; Shang, M.; Liu, S.; Zhao, W.; et al. Loss of autophagy leads to failure in megakaryopoiesis, megakaryocyte differentiation, and thrombopoiesis in mice. Exp. Hematol. 2015, 43, 488–494. [Google Scholar] [CrossRef]
- Feng, W.; Chang, C.; Luo, D.; Su, H.; Yu, S.; Hua, W.; Chen, Z.; Hu, H.; Liu, W. Dissection of autophagy in human platelets. Autophagy 2014, 10, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, J.W. How I treat double-hit lymphoma. Blood 2017, 130, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Nicotra, G.; Mercalli, F.; Peracchio, C.; Castino, R.; Follo, C.; Valente, G.; Isidoro, C. Autophagy-active beclin-1 correlates with favourable clinical outcome in non-Hodgkin lymphomas. Mod. Pathol. 2010, 23, 937–950. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-J.; Zhu, Y.-J.; Lin, T.-Y.; Jiang, W.-Q.; Huang, H.-Q.; Li, Z.-M. Beclin 1 expression predicts favorable clinical outcome in patients with diffuse large B-cell lymphoma treated with R-CHOP. Hum. Pathol. 2011, 42, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.-L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Invest. 2003, 112, 1809–1820. [Google Scholar] [CrossRef]
- Bertolo, C.; Roa, S.; Sagardoy, A.; Mena-Varas, M.; Robles, E.F.; Martinez-Ferrandis, J.I.; Sagaert, X.; Tousseyn, T.; Orta, A.; Lossos, I.S.; et al. LITAF, a BCL6 target gene, regulates autophagy in mature B-cell lymphomas. Br. J. Haematol. 2013, 162, 621–630. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, X.; Zhang, Y.; Yang, J.; Xu, Y.; Zhao, Y.; Wang, X. CUL4B regulates autophagy via JNK signaling in diffuse large B-cell lymphoma. Cell Cycle 2019. [Google Scholar] [CrossRef]
- El-Khoury, V.; Pierson, S.; Szwarcbart, E.; Brons, N.H.C.; Roland, O.; Cherrier-De Wilde, S.; Plawny, L.; Van Dyck, E.; Berchem, G. Disruption of autophagy by the histone deacetylase inhibitor MGCD0103 and its therapeutic implication in B-cell chronic lymphocytic leukemia. Leukemia 2014, 28, 1636–1646. [Google Scholar] [CrossRef]
- Hanihara-Tatsuzawa, F.; Miura, H.; Kobayashi, S.; Isagawa, T.; Okuma, A.; Manabe, I.; MaruYama, T. Control of Toll-like receptor-mediated T cell-independent type 1 antibody responses by the inducible nuclear protein IκB-ζ. J. Biol. Chem. 2014, 289, 30925–30936. [Google Scholar] [CrossRef] [PubMed]
- Fonte, E.; Vilia, M.G.; Reverberi, D.; Sana, I.; Scarfò, L.; Ranghetti, P.; Orfanelli, U.; Cenci, S.; Cutrona, G.; Ghia, P.; et al. Toll-like receptor 9 stimulation can induce IκBζ expression and IgM secretion in chronic lymphocytic leukemia cells. Haematologica 2017, 102, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, L.; Kristensen, T.; Abildgaard, N.; Thomassen, M.; Frederiksen, M.; Mourits-Andersen, T.; Møller, M.B. High expression of PI3K core complex genes is associated with poor prognosis in chronic lymphocytic leukemia. Leuk. Res. 2015, 39, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Narayan, G.; Arias-Pulido, H.; Koul, S.; Vargas, H.; Zhang, F.F.; Villella, J.; Schneider, A.; Terry, M.B.; Mansukhani, M.; Murty, V.V. Frequent promoter methylation of CDH1, DAPK, RARB, and HIC1 genes in carcinoma of cervix uteri: Its relationship to clinical outcome. Mol. Cancer 2003, 2, 24. [Google Scholar] [CrossRef] [PubMed]
- Raval, A.; Tanner, S.M.; Byrd, J.C.; Angerman, E.B.; Perko, J.D.; Chen, S.-S.; Hackanson, B.; Grever, M.R.; Lucas, D.M.; Matkovic, J.J.; et al. Downregulation of death-associated protein kinase 1 (DAPK1) in chronic lymphocytic leukemia. Cell 2007, 129, 879–890. [Google Scholar] [CrossRef] [PubMed]
- Gade, P.; Kimball, A.S.; DiNardo, A.C.; Gangwal, P.; Ross, D.D.; Boswell, H.S.; Keay, S.K.; Kalvakolanu, D.V. Death-associated Protein Kinase-1 Expression and Autophagy in Chronic Lymphocytic Leukemia Are Dependent on Activating Transcription Factor-6 and CCAAT/Enhancer-binding Protein-β. J. Biol. Chem. 2016, 291, 22030–22042. [Google Scholar] [CrossRef] [PubMed]
- Bologna, C.; Buonincontri, R.; Serra, S.; Vaisitti, T.; Audrito, V.; Brusa, D.; Pagnani, A.; Coscia, M.; D’Arena, G.; Mereu, E.; et al. SLAMF1 regulation of chemotaxis and autophagy determines CLL patient response. J. Clin. Investig. 2016, 126, 181–194. [Google Scholar] [CrossRef]
- Fisher, R.I.; Bernstein, S.H.; Kahl, B.S.; Djulbegovic, B.; Robertson, M.J.; De Vos, S.; Epner, E.; Krishnan, A.; Leonard, J.P.; Lonial, S.; et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J. Clin. Oncol. 2006, 24, 4867–4874. [Google Scholar] [CrossRef]
- Coiffier, B.; Ribrag, V. Exploring mammalian target of rapamycin (mTOR) inhibition for treatment of mantle cell lymphoma and other hematologic malignancies. Leuk. Lymphoma 2009, 50, 1916–1930. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, Z.; Miranda, R.N.; Medeiros, L.J.; McCarty, N. TG2 and NF-κB Signaling Coordinates the Survival of Mantle Cell Lymphoma Cells via IL6-Mediated Autophagy. Cancer Res. 2016, 76, 6410–6423. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhao, J.; Al-Zoghaibi, F.; Zhou, A.; Wiedmer, T.; Silverman, R.H.; Sims, P.J. Transcriptional control of the human plasma membrane phospholipid scramblase 1 gene is mediated by interferon-alpha. Blood 2000, 95, 2593–2599. [Google Scholar] [PubMed]
- Zhao, K.-W.; Li, X.; Zhao, Q.; Huang, Y.; Li, D.; Peng, Z.-G.; Shen, W.-Z.; Zhao, J.; Zhou, Q.; Chen, Z.; et al. Protein kinase Cdelta mediates retinoic acid and phorbol myristate acetate-induced phospholipid scramblase 1 gene expression: Its role in leukemic cell differentiation. Blood 2004, 104, 3731–3738. [Google Scholar] [CrossRef] [PubMed]
- Mastorci, K.; Montico, B.; Faè, D.A.; Sigalotti, L.; Ponzoni, M.; Inghirami, G.; Dolcetti, R.; Dal Col, J. Phospholipid scramblase 1 as a critical node at the crossroad between autophagy and apoptosis in mantle cell lymphoma. Oncotarget 2016, 7, 41913–41928. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, Z.; Neelapu, S.S.; Romaguera, J.; McCarty, N. Hedgehog inhibitors selectively target cell migration and adhesion of mantle cell lymphoma in bone marrow microenvironment. Oncotarget 2016, 7, 14350–14365. [Google Scholar] [CrossRef] [PubMed]
- Maclean, K.H.; Dorsey, F.C.; Cleveland, J.L.; Kastan, M.B. Targeting lysosomal degradation induces p53-dependent cell death and prevents cancer in mouse models of lymphomagenesis. J. Clin. Investig. 2008, 118, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Kuehl, W.M.; Bergsagel, P.L. Molecular pathogenesis of multiple myeloma and its premalignant precursor. J. Clin. Invest. 2012, 122, 3456–3463. [Google Scholar] [CrossRef]
- Alexanian, R.; Bonnet, J.; Gehan, E.; Haut, A.; Hewlett, J.; Lane, M.; Monto, R.; Wilson, H. Combination chemotherapy for multiple myeloma. Cancer 1972, 30, 382–389. [Google Scholar] [CrossRef]
- Mcelwain, T.J.; Powles, R.L. High-Dose Intravenous Melphalan For Plasma-Cell Leukaemia And Myeloma. Lancet 1983, 322, 822–824. [Google Scholar] [CrossRef]
- Ma, M.H.; Yang, H.H.; Parker, K.; Manyak, S.; Friedman, J.M.; Altamirano, C.; Wu, Z.; Borad, M.J.; Frantzen, M.; Roussos, E.; et al. The proteasome inhibitor PS-341 markedly enhances sensitivity of multiple myeloma tumor cells to chemotherapeutic agents. Clin. Cancer Res. 2003, 9, 1136–1144. [Google Scholar]
- Jelinek, T.; Hajek, R. PD-1/PD-L1 inhibitors in multiple myeloma: The present and the future. OncoImmunology 2016, 5, e1254856. [Google Scholar] [CrossRef]
- Allegra, A.; Sant’Antonio, E.; Penna, G.; Alonci, A.; D’Angelo, A.; Russo, S.; Cannavò, A.; Gerace, D.; Musolino, C. Novel therapeutic strategies in multiple myeloma: Role of the heat shock protein inhibitors. Eur. J. Haematol. 2011, 86, 93–110. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.S.; Li, N.; Weinhold, N.; Försti, A.; Ali, M.; van Duin, M.; Thorleifsson, G.; Johnson, D.C.; Chen, B.; Halvarsson, B.-M.; et al. Genome-wide association study identifies multiple susceptibility loci for multiple myeloma. Nat. Commun. 2016, 7, 12050. [Google Scholar] [CrossRef] [PubMed]
- Went, M.; Sud, A.; Försti, A.; Halvarsson, B.-M.; Weinhold, N.; Kimber, S.; van Duin, M.; Thorleifsson, G.; Holroyd, A.; Johnson, D.C.; et al. Identification of multiple risk loci and regulatory mechanisms influencing susceptibility to multiple myeloma. Nat. Commun. 2018, 9, 3707. [Google Scholar] [CrossRef] [PubMed]
- Jung, G.; Roh, J.; Lee, H.; Gil, M.; Yoon, D.H.; Suh, C.; Jang, S.; Park, C.-J.; Huh, J.; Park, C.-S. Autophagic Markers BECLIN 1 and LC3 are Associated with Prognosis of Multiple Myeloma. Acta Haematol. 2015, 134, 17–24. [Google Scholar] [CrossRef] [PubMed]
- De Veirman, K.; Menu, E.; Maes, K.; De Beule, N.; De Smedt, E.; Maes, A.; Vlummens, P.; Fostier, K.; Kassambara, A.; Moreaux, J.; et al. Myeloid-derived suppressor cells induce multiple myeloma cell survival by activating the AMPK pathway. Cancer Lett. 2019, 442, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Lv, A.-E.; Li, H.-P.; Han, D.-H.; Zhang, Y.-P. LncRNA MALAT-1 Elevates HMGB1 to Promote Autophagy Resulting in Inhibition of Tumor Cell Apoptosis in Multiple Myeloma. J. Cell. Biochem. 2017, 118, 3341–3348. [Google Scholar] [CrossRef] [PubMed]
- Desantis, A.; Bruno, T.; Catena, V.; De Nicola, F.; Goeman, F.; Iezzi, S.; Sorino, C.; Ponzoni, M.; Bossi, G.; Federico, V.; et al. Che-1-induced inhibition of mTOR pathway enables stress-induced autophagy. EMBO J. 2015, 34, 1214–1230. [Google Scholar] [CrossRef] [PubMed]
- Hoang, B.; Benavides, A.; Shi, Y.; Frost, P.; Lichtenstein, A. Effect of autophagy on multiple myeloma cell viability. Mol. Cancer Ther. 2009, 8, 1974–1984. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhou, P.; Chen, X.; Zhao, L.; Tan, J.; Yang, Y.; Fang, Y.; Zhou, J. The novel autophagy inhibitor elaiophylin exerts antitumor activity against multiple myeloma with mutant TP53 in part through endoplasmic reticulum stress-induced apoptosis. Cancer Biol. Ther. 2017, 18, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Lamy, L.; Ngo, V.N.; Emre, N.C.T.; Shaffer, A.L.; Yang, Y.; Tian, E.; Nair, V.; Kruhlak, M.J.; Zingone, A.; Landgren, O.; et al. Control of autophagic cell death by caspase-10 in multiple myeloma. Cancer Cell 2013, 23, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Luo, W.; Zeng, C.; Zhang, Y.; Wang, L.; Yao, W.; Nie, C. PP2A mediates apoptosis or autophagic cell death in multiple myeloma cell lines. Oncotarget 2017, 8, 80770–80789. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E. Gammaherpesviruses and lymphoproliferative disorders. Annu. Rev. Pathol. 2014, 9, 349–372. [Google Scholar] [CrossRef] [PubMed]
- Simonelli, C.; Spina, M.; Cinelli, R.; Talamini, R.; Tedeschi, R.; Gloghini, A.; Vaccher, E.; Carbone, A.; Tirelli, U. Clinical features and outcome of primary effusion lymphoma in HIV-infected patients: A single-institution study. J. Clin. Oncol. 2003, 21, 3948–3954. [Google Scholar] [CrossRef] [PubMed]
- Masud Alam, M.; Kariya, R.; Kawaguchi, A.; Matsuda, K.; Kudo, E.; Okada, S. Inhibition of autophagy by chloroquine induces apoptosis in primary effusion lymphoma in vitro and in vivo through induction of endoplasmic reticulum stress. Apoptosis 2016, 21, 1191–1201. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-Y.; Chen, C.-Y.; Chiou, Y.-H.; Shyu, H.-W.; Lin, K.-H.; Chou, M.-C.; Huang, M.-H.; Wang, Y.-F. Epigallocatechin-3-Gallate Suppresses Human Herpesvirus 8 Replication and Induces ROS Leading to Apoptosis and Autophagy in Primary Effusion Lymphoma Cells. Int. J. Mol. Sci. 2018, 19, 16. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.D.; Lamant, L.; Kenner, L.; Brugières, L. Anaplastic large cell lymphoma in paediatric and young adult patients. Br. J. Haematol. 2016, 173, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Lowe, E.J.; Gross, T.G. Anaplastic large cell lymphoma in children and adolescents. Pediatr. Hematol. Oncol. 2013, 30, 509–519. [Google Scholar] [CrossRef]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non- Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef]
- Chiarle, R.; Voena, C.; Ambrogio, C.; Piva, R.; Inghirami, G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat. Rev. Cancer 2008, 8, 11–23. [Google Scholar] [CrossRef]
- Roskoski, R. Anaplastic lymphoma kinase (ALK): Structure, oncogenic activation, and pharmacological inhibition. Pharmacol. Res. 2013, 68, 68–94. [Google Scholar] [CrossRef]
- Christensen, J.G.; Zou, H.Y.; Arango, M.E.; Li, Q.; Lee, J.H.; McDonnell, S.R.; Yamazaki, S.; Alton, G.R.; Mroczkowski, B.; Los, G. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol. Cancer Ther 2007, 6, 3314–3322. [Google Scholar] [CrossRef] [PubMed]
- Mologni, L. Current and future treatment of anaplastic lymphoma kinase-rearranged cancer. World J. Clin. Oncol. 2015, 6, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Crescenzo, R.; Inghirami, G. Anaplastic lymphoma kinase inhibitors. Curr. Opin. Pharmacol. 2015, 23, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Gambacorti Passerini, C.; Farina, F.; Stasia, A.; Redaelli, S.; Ceccon, M.; Mologni, L.; Messa, C.; Guerra, L.; Giudici, G.; Sala, E.; et al. Crizotinib in advanced, chemoresistant anaplastic lymphoma kinase-positive lymphoma patients. J. Natl. Cancer Inst. 2014, 106, djt378. [Google Scholar] [CrossRef] [PubMed]
- Mosse, Y.P.; Lim, M.S.; Voss, S.D.; Wilner, K.; Ruffner, K.; Laliberte, J.; Rolland, D.; Balis, F.M.; Maris, J.M.; Weigel, B.J.; et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: A Children’s Oncology Group phase 1 consortium study. Lancet Oncol. 2013, 14, 472–480. [Google Scholar] [CrossRef]
- Lai, R.; Ingham, R.J. The pathobiology of the oncogenic tyrosine kinase NPM-ALK: A brief update. Ther. Adv. Hematol. 2013, 4, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Mastini, C.; Martinengo, C.; Inghirami, G.; Chiarle, R. Anaplastic lymphoma kinase: An oncogene for tumor vaccination. J. Mol. Med. 2009, 87, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Mitou, G.; Frentzel, J.; Desquesnes, A.; Le Gonidec, S.; AlSaati, T.; Beau, I.; Lamant, L.; Meggetto, F.; Espinos, E.; Codogno, P.; et al. Targeting autophagy enhances the anti-tumoral action of crizotinib in ALK-positive anaplastic large cell lymphoma. Oncotarget 2015, 6, 30149–30164. [Google Scholar] [CrossRef]
- Fisher, R.I.; LeBlanc, M.; Press, O.W.; Maloney, D.G.; Unger, J.M.; Miller, T.P. New Treatment Options Have Changed the Survival of Patients with Follicular Lymphoma. JCO 2005, 23, 8447–8452. [Google Scholar] [CrossRef]
- Hiddemann, W.; Kneba, M.; Dreyling, M.; Schmitz, N.; Lengfelder, E.; Schmits, R.; Reiser, M.; Metzner, B.; Harder, H.; Hegewisch-Becker, S.; et al. Frontline therapy with rituximab added to the combination of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) significantly improves the outcome for patients with advanced-stage follicular lymphoma compared with therapy with CHOP alone: Results of a prospective randomized study of the German Low-Grade Lymphoma Study Group. Blood 2005, 106, 3725–3732. [Google Scholar] [PubMed]
- Martinelli, G.; Schmitz, S.-F.H.; Utiger, U.; Cerny, T.; Hess, U.; Bassi, S.; Okkinga, E.; Stupp, R.; Stahel, R.; Heizmann, M.; et al. Long-term follow-up of patients with follicular lymphoma receiving single-agent rituximab at two different schedules in trial SAKK 35/98. J. Clin. Oncol. 2010, 28, 4480–4484. [Google Scholar] [CrossRef] [PubMed]
- Kridel, R.; Sehn, L.H.; Gascoyne, R.D. Pathogenesis of follicular lymphoma. J. Clin. Investig. 2012, 122, 3424–3431. [Google Scholar] [CrossRef] [PubMed]
- Gravelle, P.; Do, C.; Franchet, C.; Mueller, S.; Oberic, L.; Ysebaert, L.; Larocca, L.M.; Hohaus, S.; Calmels, M.-N.; Frenois, F.-X.; et al. Impaired functional responses in follicular lymphoma CD8+TIM-3+ T lymphocytes following TCR engagement. Oncoimmunology 2016, 5, e1224044. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, A.; Marzec, J.; Clear, A.; Petty, R.D.; Coutinho, R.; Matthews, J.; Wilson, A.; Iqbal, S.; Calaminici, M.; Gribben, J.G.; et al. Dysregulation of autophagy in human follicular lymphoma is independent of overexpression of BCL-2. Oncotarget 2014, 5, 11653–11668. [Google Scholar] [CrossRef] [PubMed]
- Giatromanolaki, A.; Koukourakis, M.I.; Pouliliou, S.; Gatter, K.C.; Pezzella, F.; Harris, A.L.; Sivridis, E. Overexpression of LC3A autophagy protein in follicular and diffuse large B-cell lymphomas. Hematol. Oncol. Stem Cell Ther. 2013, 6, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G.; Goorha, S.; Radtke, I.; Miller, C.B.; Coustan-Smith, E.; Dalton, J.D.; Girtman, K.; Mathew, S.; Ma, J.; Pounds, S.B.; et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007, 446, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Holleman, A.; Cheok, M.H.; den Boer, M.L.; Yang, W.; Veerman, A.J.P.; Kazemier, K.M.; Pei, D.; Cheng, C.; Pui, C.-H.; Relling, M.V.; et al. Gene-expression patterns in drug-resistant acute lymphoblastic leukemia cells and response to treatment. N. Engl. J. Med. 2004, 351, 533–542. [Google Scholar] [CrossRef]
- Andersson, A.K.; Ma, J.; Wang, J.; Chen, X.; Gedman, A.L.; Dang, J.; Nakitandwe, J.; Holmfeldt, L.; Parker, M.; Easton, J.; et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat. Genet. 2015, 47, 330–337. [Google Scholar] [CrossRef]
- Ottmann, O.G.; Wassmann, B.; Pfeifer, H.; Giagounidis, A.; Stelljes, M.; Duhrsen, U.; Schmalzing, M.; Wunderle, L.; Binckebanck, A.; Hoelzer, D. Imatinib compared with chemotherapy as front-line treatment of elderly patients with Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ALL). Cancer 2007, 109, 2068–2076. [Google Scholar] [CrossRef]
- Kantarjian, H.; Thomas, D.; Wayne, A.S.; O’Brien, S. Monoclonal antibody-based therapies: A new dawn in the treatment of acute lymphoblastic leukemia. J. Clin. Oncol. 2012, 30, 3876–3883. [Google Scholar] [CrossRef]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.-N.; Liu, Q.-Q.; Zhang, S.-P.; Yuan, N.; Cao, Y.; Cai, J.-Y.; Lin, W.-W.; Xu, F.; Wang, Z.-J.; Chen, B.; et al. Alternative messenger RNA splicing of autophagic gene Beclin 1 in human B-cell acute lymphoblastic leukemia cells. Asian Pac. J. Cancer Prev. 2014, 15, 2153–2158. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xu, F.; Yuan, N.; Niu, Y.; Lin, W.; Cao, Y.; Cai, J.; Song, L.; Li, X.; Fang, Y.; et al. Rapamycin inhibits pre-B acute lymphoblastic leukemia cells by downregulating DNA and RNA polymerases. Leuk. Res. 2014, 38, 940–947. [Google Scholar] [CrossRef] [PubMed]
- Simioni, C.; Cani, A.; Martelli, A.M.; Zauli, G.; Tabellini, G.; McCubrey, J.; Capitani, S.; Neri, L.M. Activity of the novel mTOR inhibitor Torin-2 in B-precursor acute lymphoblastic leukemia and its therapeutic potential to prevent Akt reactivation. Oncotarget 2014, 5, 10034–10047. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Gopinathan, G.; Sukumar, J.T.; Gribben, J.G. Blocking autophagy prevents bortezomib-induced NF-κB activation by reducing I-κBα degradation in lymphoma cells. PLoS ONE 2012, 7, e32584. [Google Scholar] [CrossRef] [PubMed]
- Zang, C.; Eucker, J.; Liu, H.; Coordes, A.; Lenarz, M.; Possinger, K.; Scholz, C.W. Inhibition of pan-class I phosphatidyl-inositol-3-kinase by NVP-BKM120 effectively blocks proliferation and induces cell death in diffuse large B-cell lymphoma. Leuk. Lymphoma 2014, 55, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; He, M.; Cheng, F.; Bai, R.; da Silva, S.R.; Aguiar, R.C.T.; Gao, S.-J. Tenovin-6 inhibits proliferation and survival of diffuse large B-cell lymphoma cells by blocking autophagy. Oncotarget 2017, 8, 14912–14924. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Wang, T.; Song, Z.; Peng, L.; Gao, M.; Hermine, O.; Rousseaux, S.; Khochbin, S.; Mi, J.-Q.; Wang, J. Induction of autophagy and autophagy-dependent apoptosis in diffuse large B-cell lymphoma by a new antimalarial artemisinin derivative, SM1044. Cancer Med. 2018, 7, 380–396. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-J.; Chai, Y.; Guo, X.-J.; Chu, S.-L.; Zhang, L.-S. The effects of the long non-coding RNA MALAT-1 regulated autophagy-related signaling pathway on chemotherapy resistance in diffuse large B-cell lymphoma. Biomed. Pharmacother. 2017, 89, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Hou, W.; Goldstein, L.A.; Lu, C.; Stolz, D.B.; Yin, X.M.; Rabinowich, H. Involvement of protective autophagy in TRAIL resistance of apoptosis-defective tumor cells. J. Biol. Chem. 2008, 283, 19665–19677. [Google Scholar] [CrossRef] [PubMed]
- Amrein, L.; Soulières, D.; Johnston, J.B.; Aloyz, R. P53 and autophagy contribute to dasatinib resistance in primary CLL lymphocytes. Leuk. Res. 2011, 35, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Kovaleva, V.; Mora, R.; Park, Y.J.; Plass, C.; Chiramel, A.I.; Bartenschlager, R.; Doḧner, H.; Stilgenbauer, S.; Pscherer, A.; Lichter, P.; et al. miRNA-130a targets ATG2B and DICER1 to inhibit autophagy and trigger killing of chronic lymphocytic leukemia cells. Cancer Res. 2012, 72, 1763–1772. [Google Scholar] [CrossRef] [PubMed]
- Stellrecht, C.M.; Chen, L.S.; Ayres, M.L.; Dennison, J.B.; Shentu, S.; Chen, Y.; Keating, M.J.; Wierda, W.G.; Gandhi, V. Chlorinated adenosine analogue induces AMPK and autophagy in chronic lymphocytic leukaemia cells during therapy. Br. J. Haematol. 2017, 179, 266–271. [Google Scholar] [CrossRef] [PubMed]
- El-Khoury, V.; Moussay, E.; Janji, B.; Palissot, V.; Aouali, N.; Brons, N.H.C.; Van Moer, K.; Pierson, S.; Van Dyck, E.; Berchem, G. The histone deacetylase inhibitor MGCD0103 induces apoptosis in B-cell chronic lymphocytic leukemia cells through a mitochondria-mediated caspase activation cascade. Mol. Cancer Ther. 2010, 9, 1349–1360. [Google Scholar] [CrossRef] [PubMed]
- MacCallum, S.F.; Groves, M.J.; James, J.; Murray, K.; Appleyard, V.; Prescott, A.R.; Drbal, A.A.; Nicolaou, A.; Cunningham, J.; Haydock, S.; et al. Dysregulation of autophagy in chronic lymphocytic leukemia with the small-molecule Sirtuin inhibitor Tenovin-6. Sci. Rep. 2013, 3, 1275. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Tan, B.; Gao, S.-J. Tenovin-6 impairs autophagy by inhibiting autophagic flux. Cell Death Dis. 2017, 8, e2608. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Zhang, W.; Yang, L.; Pelicano, H.; Zhou, K.; Yin, R.; Huang, R.; Zeng, J. Targeting the autophagy in bone marrow stromal cells overcomes resistance to vorinostat in chronic lymphocytic leukemia. Onco Targets Ther. 2018, 11, 5151–5170. [Google Scholar] [CrossRef]
- Chen, Z.; Teo, A.E.; McCarty, N. ROS-induced CXCR4 signaling regulates mantle cell lymphoma (MCL) cell survival and drug resistance in the bone marrow microenvironment via autophagy. Clin. Cancer Res. 2016, 22, 187–199. [Google Scholar] [CrossRef]
- Xiao, Y.; Guan, J. 17-AAG enhances the cytotoxicity of flavopiridol in mantle cell lymphoma via autophagy suppression. Neoplasma 2015, 62, 391–397. [Google Scholar] [CrossRef]
- Alinari, L.; Baiocchi, R.A.; Prætorius-Ibba, M. FTY720-induced blockage of autophagy enhances anticancer efficacy of milatuzumab in mantle cell lymphoma: Is FTY720 the next autophagy-blocking agent in lymphoma treatment? Autophagy 2012, 8, 416–417. [Google Scholar] [CrossRef]
- Alinari, L.; Yu, B.; Christian, B.A.; Yan, F.; Shin, J.; Lapalombella, R.; Hertlein, E.; Lustberg, M.E.; Quinion, C.; Zhang, X.; et al. Combination anti-CD74 (milatuzumab) and anti-CD20 (rituximab) monoclonal antibody therapy has in vitro and in vivo activity in mantle cell lymphoma. Blood 2011, 117, 4530–4541. [Google Scholar] [CrossRef] [PubMed]
- Heine, S.; Kleih, M.; Giménez, N.; Böpple, K.; Ott, G.; Colomer, D.; Aulitzky, W.E.; van der Kuip, H.; Silkenstedt, E. Cyclin D1-CDK4 activity drives sensitivity to bortezomib in mantle cell lymphoma by blocking autophagy-mediated proteolysis of NOXA. J. Hematol. Oncol. 2018, 11, 112. [Google Scholar] [CrossRef] [PubMed]
- Rosich, L.; Xargay-Torrent, S.; López-Guerra, M.; Campo, E.; Colomer, D.; Roué, G. Counteracting autophagy overcomes resistance to everolimus in mantle cell lymphoma. Clin. Cancer Res. 2012, 18, 5278–5289. [Google Scholar] [CrossRef] [PubMed]
- Nahimana, A.; Attinger, A.; Aubry, D.; Greaney, P.; Ireson, C.; Thougaard, A.V.; Tjrnelund, J.; Dawson, K.M.; Dupuis, M.; Duchosal, M.A. The NAD biosynthesis inhibitor APO866 has potent antitumor activity against hematologic malignancies. Blood 2009, 113, 3276–3286. [Google Scholar] [CrossRef] [PubMed]
- Ginet, V.; Puyal, J.; Rummel, C.; Aubry, D.; Breton, C.; Cloux, A.J.; Majjigapu, S.R.; Sordat, B.; Vogel, P.; Bruzzone, S.; et al. A critical role of autophagy in antileukemia/lymphoma effects of APO866, an inhibitor of NAD biosynthesis. Autophagy 2014, 10, 603–617. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.K.; Yu, D.; Lum, J.J.; Bui, T.; Christophorou, M.A.; Evan, G.I.; Thomas-Tikhonenko, A.; Thompson, C.B. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J. Clin. Investig. 2007, 117, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Hart, L.S.; Cunningham, J.T.; Datta, T.; Dey, S.; Tameire, F.; Lehman, S.L.; Qiu, B.; Zhang, H.; Cerniglia, G.; Bi, M.; et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J. Clin. Investig. 2012, 122, 4621–4634. [Google Scholar] [CrossRef]
- Ni, Z.; Dai, X.; Wang, B.; Ding, W.; Cheng, P.; Xu, L.; Lian, J.; He, F. Natural Bcl-2 inhibitor (−)-gossypol induces protective autophagy via reactive oxygen species-high mobility group box 1 pathway in Burkitt lymphoma. Leuk. Lymphoma 2013, 54, 2263–2268. [Google Scholar] [CrossRef]
- Zeng, X.; Li, Y.; Fan, J.; Zhao, H.; Xian, Z.; Sun, Y.; Wang, Z.; Wang, S.; Zhang, G.; Ju, D. Recombinant human arginase induced caspase-dependent apoptosis and autophagy in non-Hodgkin’s lymphoma cells. Cell Death Dis. 2013, 4, e840. [Google Scholar] [CrossRef]
- Métayer, L.E.; Brown, R.D.; Carlebur, S.; Burke, G.A.A.; Brown, G.C. Mechanisms of cell death induced by arginase and asparaginase in precursor B-cell lymphoblasts. Apoptosis 2018. [Google Scholar] [CrossRef]
- Fan, J.; Zeng, X.; Li, Y.; Wang, S.; Yang, P.; Cao, Z.; Wang, Z.; Song, P.; Mei, X.; Ju, D. A novel therapeutic approach against B-cell non-Hodgkin’s lymphoma through co-inhibition of Hedgehog signaling pathway and autophagy. Tumour Biol. 2016, 37, 7305–7314. [Google Scholar] [CrossRef] [PubMed]
- Pujals, A.; Favre, L.; Pioche-Durieu, C.; Robert, A.; Meurice, G.; Le Gentil, M.; Chelouah, S.; Martin-Garcia, N.; Le Cam, E.; Guettier, C.; et al. Constitutive autophagy contributes to resistance to TP53-mediated apoptosis in Epstein-Barr virus-positive latency III B-cell lymphoproliferations. Autophagy 2015, 11, 2275–2287. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Zeng, X.; Li, Y.; Wang, S.; Wang, Z.; Sun, Y.; Gao, H.; Zhang, G.; Feng, M.; Ju, D. Autophagy Plays a Critical Role in ChLym-1-Induced Cytotoxicity of Non-Hodgkin’s Lymphoma Cells. PLoS ONE 2013, 8, e72478. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; Rizzello, C.; Romeo, M.A.; Yadav, S.; Santarelli, R.; D’Orazi, G.; Faggioni, A.; Cirone, M. Concomitant reduction of c-Myc expression and PI3K/AKT/mTOR signaling by quercetin induces a strong cytotoxic effect against Burkitt’s lymphoma. Int. J. Biochem. Cell Biol. 2016, 79, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-L.; Wei, H.-L.; Chen, J.; Wang, B.; Xie, B.; Fan, L.-L.; Li, L.-J. Arsenic trioxide induces autophagy and antitumor effects in Burkitt’s lymphoma Raji cells. Oncol. Rep. 2014, 32, 1557–1563. [Google Scholar] [CrossRef]
- Dong, L.H.; Cheng, S.; Zheng, Z.; Wang, L.; Shen, Y.; Shen, Z.X.; Chen, S.J.; Zhao, W.L. Histone deacetylase inhibitor potentiated the ability of MTOR inhibitor to induce autophagic cell death in Burkitt leukemia/lymphoma. J. Hematol. Oncol. 2013, 6, 1–11. [Google Scholar] [CrossRef]
- Ono, K.; Sato, T.; Iyama, S.; Tatekoshi, A.; Hashimoto, A.; Kamihara, Y.; Horiguchi, H.; Kikuchi, S.; Kawano, Y.; Takada, K.; et al. A novel strategy inducing autophagic cell death in Burkitt’s lymphoma cells with anti-CD19-targeted liposomal rapamycin. Blood Cancer J. 2014, 4, e180. [Google Scholar] [CrossRef]
- Turzanski, J.; Daniels, I.; Haynes, A.P. Involvement of macroautophagy in the caspase-independent killing of Burkitt lymphoma cell lines by rituximab. Br. J. Haematol. 2009, 145, 137–140. [Google Scholar] [CrossRef]
- Oliva, L.; Cenci, S. Autophagy in plasma cell pathophysiology. Front. Immunol. 2014, 5, 103. [Google Scholar] [CrossRef]
- Pan, Y.; Gao, Y.; Chen, L.; Gao, G.; Dong, H.; Yang, Y.; Dong, B.; Chen, X. Targeting autophagy augments in vitro and in vivo antimyeloma activity of DNA-damaging chemotherapy. Clin. Cancer Res. 2011, 17, 3248–3258. [Google Scholar] [CrossRef]
- Guo, X.; He, D.; Zhang, E.; Chen, J.; Chen, Q.; Li, Y.; Yang, L.; Yang, Y.; Zhao, Y.; Wang, G.; et al. HMGB1 knockdown increases MM cell vulnerability by regulating autophagy and DNA damage repair. J. Exp. Clin. Cancer Res. 2018, 37, 205. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, Y.; Zhou, L.; Leng, Y.; Lin, H.; Kmieciak, M.; Pei, X.Y.; Jones, R.; Orlowski, R.Z.; Dai, Y.; et al. A Bim-targeting strategy overcomes adaptive bortezomib resistance in myeloma through a novel link between autophagy and apoptosis. Blood 2014, 124, 2687–2697. [Google Scholar] [CrossRef] [PubMed]
- Kaliszczak, M.; van Hechanova, E.; Li, Y.; Alsadah, H.; Parzych, K.; Auner, H.W.; Aboagye, E.O. The HDAC6 inhibitor C1A modulates autophagy substrates in diverse cancer cells and induces cell death. Br. J. Cancer 2018, 119, 1278–1287. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Zhang, Y.; Wang, W.; Wu, J.; Yang, Q.; Xu, W.; Jiang, S.; Han, Y.; Yu, K.; Zhang, S. Inhibition of autophagy enhances the antitumour activity of tigecycline in multiple myeloma. J. Cell. Mol. Med. 2018, 22, 5955–5963. [Google Scholar] [CrossRef]
- Zhao, X.; Fang, Y.; Yang, Y.; Qin, Y.; Wu, P.; Wang, T.; Lai, H.; Meng, L.; Wang, D.; Zheng, Z.; et al. Elaiophylin, a novel autophagy inhibitor, exerts antitumor activity as a single agent in ovarian cancer cells. Autophagy 2015, 11, 1849–1863. [Google Scholar] [CrossRef] [PubMed]
- Jagannathan, S.; Abdel-Malek, M.A.Y.; Malek, E.; Vad, N.; Latif, T.; Anderson, K.C.; Driscoll, J.J. Pharmacologic screens reveal metformin that suppresses GRP78-dependent autophagy to enhance the anti-myeloma effect of bortezomib. Leukemia 2015, 29, 2184–2191. [Google Scholar] [CrossRef] [PubMed]
- Jarauta, V.; Jaime, P.; Gonzalo, O.; de Miguel, D.; Ramírez-Labrada, A.; Martínez-Lostao, L.; Anel, A.; Pardo, J.; Marzo, I.; Naval, J. Inhibition of autophagy with chloroquine potentiates carfilzomib-induced apoptosis in myeloma cells in vitro and in vivo. Cancer Lett. 2016, 382, 1–10. [Google Scholar] [CrossRef]
- Baranowska, K.; Misund, K.; Starheim, K.K.; Holien, T.; Johansson, I.; Darvekar, S.; Buene, G.; Waage, A.; Bjørkøy, G.; Sundan, A. Hydroxychloroquine potentiates carfilzomib toxicity towards myeloma cells. Oncotarget 2016, 7, 70845–70856. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, Y.; Xu, H.; Shi, C.; Jin, F.; Li, W. Profilin 1 induces drug resistance through Beclin1 complex-mediated autophagy in multiple myeloma. Cancer Sci. 2018, 109, 2706–2716. [Google Scholar] [CrossRef]
- Zhang, H.; Pang, Y.; Ma, C.; Li, J.; Wang, H.; Shao, Z. ClC5 Decreases the Sensitivity of Multiple Myeloma Cells to Bortezomib via Promoting Prosurvival Autophagy. Oncol. Res. 2018, 26, 421–429. [Google Scholar] [CrossRef]
- Lu, D.; Yang, C.; Zhang, Z.; Cong, Y.; Xiao, M. Knockdown of Linc00515 Inhibits Multiple Myeloma Autophagy and Chemoresistance by Upregulating miR-140-5p and Downregulating ATG14. Cell. Physiol. Biochem. 2018, 48, 2517–2527. [Google Scholar] [CrossRef]
- Milan, E.; Perini, T.; Resnati, M.; Orfanelli, U.; Oliva, L.; Raimondi, A.; Cascio, P.; Bachi, A.; Marcatti, M.; Ciceri, F.; et al. A plastic SQSTM1/p62-dependent autophagic reserve maintains proteostasis and determines proteasome inhibitor susceptibility in multiple myeloma cells. Autophagy 2015, 11, 1161–1178. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Xie, W.; Kuhn, D.J.; Voorhees, P.M.; Lopez-Girona, A.; Mendy, D.; Corral, L.G.; Krenitsky, V.P.; Xu, W.; Moutouh-de Parseval, L.; et al. Targeting the p27 E3 ligase SCF(Skp2) results in p27- and Skp2-mediated cell-cycle arrest and activation of autophagy. Blood 2008, 111, 4690–4699. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.-F.; Liu, X.; Gao, M.; Zhang, Y.-N.; Liu, J. Endoplasmic reticulum stress induces autophagy and apoptosis while inhibiting proliferation and drug resistance in multiple myeloma through the PI3K/Akt/mTOR signaling pathway. Oncotarget 2017, 8, 61093–61106. [Google Scholar] [CrossRef] [PubMed]
- Desantis, V.; Saltarella, I.; Lamanuzzi, A.; Mariggiò, M.A.; Racanelli, V.; Vacca, A.; Frassanito, M.A. Autophagy: A New Mechanism of Prosurvival and Drug Resistance in Multiple Myeloma. Transl. Oncol. 2018, 11, 1350–1357. [Google Scholar] [CrossRef]
- Yun, Z.; Zhichao, J.; Hao, Y.; Ou, J.; Ran, Y.; Wen, D.; Qun, S. Targeting autophagy in multiple myeloma. Leuk. Res. 2017, 59, 97–104. [Google Scholar] [CrossRef]
- Sommermann, T.; O’Neill, K.; Plas, D.R.; Cahir-McFarland, E. IKKβ and NF-κB transcription govern lymphoma cell survival through AKT-induced plasma membrane trafficking of GLUT1. Cancer Res. 2011, 7291–7300. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; Santarelli, R.; Lotti, L.V.; Di Renzo, L.; Gonnella, R.; Garufi, A.; Trivedi, P.; Frati, L.; D’Orazi, G.; Faggioni, A.; et al. JNK and macroautophagy activation by bortezomib has a pro-survival effect in primary effusion lymphoma cells. PLoS ONE 2013, 8, e75965. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; Rizzello, C.; Gilardini Montani, M.S.; Cuomo, L.; Vitillo, M.; Santarelli, R.; Gonnella, R.; D’Orazi, G.; Faggioni, A.; Cirone, M. Quercetin induces apoptosis and autophagy in primary effusion lymphoma cells by inhibiting PI3K/AKT/mTOR and STAT3 signaling pathways. J. Nutr. Biochem. 2017, 41, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Torossian, A.; Broin, N.; Frentzel, J.; Daugrois, C.; Gandarillas, S.; AlSaati, T.; Lamant, L.; Brousset, P.; Giuriato, S.; Espinos, E. Blockade of crizotinib-induced BCL-2 elevation in ALK-positive ALCL triggers autophagy associated with cell death. Haematologica 2019. [Google Scholar] [CrossRef]
- Brem, E.A.; Thudium, K.; Khubchandani, S.; Tsai, P.C.; Olejniczak, S.H.; Bhat, S.; Riaz, W.; Gu, J.; Iqbal, A.; Campagna, R.; et al. Distinct cellular and therapeutic effects of obatoclax in rituximab-sensitive and -resistant lymphomas. Br. J. Haematol. 2011, 153, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Leseux, L.; Laurent, G.; Laurent, C.; Rigo, M.; Blanc, A.; Olive, D.; Bezombes, C. PKC zeta mTOR pathway: A new target for rituximab therapy in follicular lymphoma. Blood 2008, 111, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Laane, E.; Tamm, K.P.; Buentke, E.; Ito, K.; Kharaziha, P.; Oscarsson, J.; Corcoran, M.; Björklund, A.-C.; Hultenby, K.; Lundin, J.; et al. Cell death induced by dexamethasone in lymphoid leukemia is mediated through initiation of autophagy. Cell Death Differ. 2009, 16, 1018–1029. [Google Scholar] [CrossRef] [PubMed]
- Grander, D.; Kharaziha, P.; Laane, E.; Pokrovskaja, K.; Panaretakis, T. Autophagy as the main means of cytotoxicity by glucocorticoids in hematological malignancies. Autophagy 2009, 5, 1198–1200. [Google Scholar] [CrossRef]
- Polak, A.; Kiliszek, P.; Sewastianik, T.; Szydłowski, M.; Jabłońska, E.; Białopiotrowicz, E.; Górniak, P.; Markowicz, S.; Nowak, E.; Grygorowicz, M.A.; et al. MEK Inhibition Sensitizes Precursor B-Cell Acute Lymphoblastic Leukemia (B-ALL) Cells to Dexamethasone through Modulation of mTOR Activity and Stimulation of Autophagy. PLoS ONE 2016, 11, e0155893. [Google Scholar] [CrossRef]
- Ristic, B.; Bosnjak, M.; Arsikin, K.; Mircic, A.; Suzin-Zivkovic, V.; Bogdanovic, A.; Perovic, V.; Martinovic, T.; Kravic-Stevovic, T.; Bumbasirevic, V.; et al. Idarubicin induces mTOR-dependent cytotoxic autophagy in leukemic cells. Exp. Cell Res. 2014, 326, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Neri, L.M.; Cani, A.; Martelli, A.M.; Simioni, C.; Junghanss, C.; Tabellini, G.; Ricci, F.; Tazzari, P.L.; Pagliaro, P.; McCubrey, J.A.; et al. Targeting the PI3K/Akt/mTOR signaling pathway in B-precursor acute lymphoblastic leukemia and its therapeutic potential. Leukemia 2014, 28, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Crazzolara, R.; Bradstock, K.F.; Bendall, L.J. RAD001 (Everolimus) induces autophagy in acute lymphoblastic leukemia. Autophagy 2009, 5, 727–728. [Google Scholar] [CrossRef]
- Jia, L.; Dourmashkin, R.R.; Allen, P.D.; Gray, A.B.; Newland, A.C.; Kelsey, S.M. Inhibition of autophagy abrogates tumour necrosis factor alpha induced apoptosis in human T-lymphoblastic leukaemic cells. Br. J. Haematol. 1997, 98, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Li, F.; Shi, K.; Wu, P.; An, J.; Yang, Y.; Xu, C. ATF4 activation by the p38MAPK-eIF4E axis mediates apoptosis and autophagy induced by selenite in Jurkat cells. FEBS Lett. 2013, 587, 2420–2429. [Google Scholar] [CrossRef]
- Bhadri, V.A.; Trahair, T.N.; Lock, R.B. Glucocorticoid resistance in paediatric acute lymphoblastic leukaemia. J. Paediatr. Child Health 2012, 48, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Heidari, N.; Hicks, M.A.; Harada, H. GX15-070 (obatoclax) overcomes glucocorticoid resistance in acute lymphoblastic leukemia through induction of apoptosis and autophagy. Cell Death Dis. 2010, 1, e76. [Google Scholar] [CrossRef] [PubMed]
- Urtishak, K.A.; Edwards, A.Y.Z.; Wang, L.-S.; Hudome, A.; Robinson, B.W.; Barrett, J.S.; Cao, K.; Cory, L.; Moore, J.S.; Bantly, A.D.; et al. Potent obatoclax cytotoxicity and activation of triple death mode killing across infant acute lymphoblastic leukemia. Blood 2013, 121, 2689–2703. [Google Scholar] [CrossRef] [PubMed]
- Bonapace, L.; Bornhauser, B.C.; Schmitz, M.; Cario, G.; Ziegler, U.; Niggli, F.K.; Schafer, B.W.; Schrappe, M.; Stanulla, M.; Bourquin, J.-P. Induction of autophagy-dependent necroptosis is required for childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance. J. Clin. Investig. 2010, 120, 1310–1323. [Google Scholar] [CrossRef] [PubMed]
- Sarang, Z.; Gyurina, K.; Scholtz, B.; Kiss, C.; Szegedi, I. Altered expression of autophagy-related genes might contribute to glucocorticoid resistance in precursor B-cell-type acute lymphoblastic leukemia. Eur. J. Haematol. 2016, 97, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Wallington-Beddoe, C.T.; Hewson, J.; Bradstock, K.F.; Bendall, L.J. FTY720 produces caspase-independent cell death of acute lymphoblastic leukemia cells. Autophagy 2011, 7, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, C.; Ricci, F.; Tazzari, P.; Chiarini, F.; Battistelli, M.; Falcieri, E.; Ognibene, A.; Pagliaro, P.; Cocco, L.; McCubrey, J.A.; et al. Preclinical testing of the Akt inhibitor triciribine in T-cell acute lymphoblastic leukemia. J. Cell. Physiol. 2011, 226, 822–831. [Google Scholar] [CrossRef]
- Herranz, D.; Ambesi-Impiombato, A.; Sudderth, J.; Sanchez-Martin, M.; Belver, L.; Tosello, V.; Xu, L.; Wendorff, A.A.; Castillo, M.; Haydu, J.E.; et al. Metabolic reprogramming induces resistance to anti-NOTCH1 therapies in T cell acute lymphoblastic leukemia. Nat. Med. 2015, 21, 1182–1189. [Google Scholar] [CrossRef]
- Hongo, T.; Yajima, S.; Sakurai, M.; Horikoshi, Y.; Hanada, R. In vitro drug sensitivity testing can predict induction failure and early relapse of childhood acute lymphoblastic leukemia. Blood 1997, 89, 2959–2965. [Google Scholar]
- Kaspers, G.J.; Veerman, A.J.; Pieters, R.; Van Zantwijk, C.H.; Smets, L.A.; Van Wering, E.R.; Van Der Does-Van Den Berg, A. In vitro cellular drug resistance and prognosis in newly diagnosed childhood acute lymphoblastic leukemia. Blood 1997, 90, 2723–2729. [Google Scholar]
- Takahashi, H.; Inoue, J.; Sakaguchi, K.; Takagi, M.; Mizutani, S.; Inazawa, J. Autophagy is required for cell survival under L-asparaginase-induced metabolic stress in acute lymphoblastic leukemia cells. Oncogene 2017, 36, 4267–4276. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.M.; Catovsky, D.; Daniel, M.T.; Flandrin, G.; Galton, D.A.G.; Gralnick, H.R.; Sultan, C. Proposals for the classification of the myelodysplastic syndromes. Br. J. Haematol. 1982, 51, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Li, J. Myelodysplastic syndrome hematopoietic stem cell. Int. J. Cancer 2013, 133, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Woll, P.S.; Kjällquist, U.; Chowdhury, O.; Doolittle, H.; Wedge, D.C.; Thongjuea, S.; Erlandsson, R.; Ngara, M.; Anderson, K.; Deng, Q.; et al. Myelodysplastic syndromes are propagated by rare and distinct human cancer stem cells in vivo. Cancer Cell 2014, 25, 794–808. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627; quiz 3699. [Google Scholar] [CrossRef] [PubMed]
- Parmar, S.; de Lima, M.; Deeg, H.J.; Champlin, R. Hematopoietic stem cell transplantation for myelodysplastic syndrome: A review. Semin. Oncol. 2011, 38, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Odenike, O.; Onida, F.; Padron, E. Myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms: An update on risk stratification, molecular genetics, and therapeutic approaches including allogeneic hematopoietic stem cell transplantation. Am. Soc. Clin. Oncol. Educ. Book 2015, e398–e412. [Google Scholar] [CrossRef]
- Saliba, J.; Saint-Martin, C.; Di Stefano, A.; Lenglet, G.; Marty, C.; Keren, B.; Pasquier, F.; Valle, V.D.; Secardin, L.; Leroy, G.; et al. Germline duplication of ATG2B and GSKIP predisposes to familial myeloid malignancies. Nat. Genet. 2015, 47, 1131–1140. [Google Scholar] [CrossRef]
- Park, S.M.; Ou, J.; Chamberlain, L.; Simone, T.M.; Yang, H.; Virbasius, C.-M.; Ali, A.M.; Zhu, L.J.; Mukherjee, S.; Raza, A.; et al. U2AF35(S34F) Promotes Transformation by Directing Aberrant ATG7 Pre-mRNA 3′ End Formation. Mol. Cell 2016, 62, 479–490. [Google Scholar] [CrossRef]
- Jiang, H.; Yang, L.; Guo, L.; Cui, N.; Zhang, G.; Liu, C.; Xing, L.; Shao, Z.; Wang, H. Impaired Mitophagy of Nucleated Erythroid Cells Leads to Anemia in Patients with Myelodysplastic Syndromes. Oxid. Med. Cell. Longev. 2018, 2018, 6328051. [Google Scholar] [CrossRef]
- Zhuang, L.; Ma, Y.; Wang, Q.; Zhang, J.; Zhu, C.; Zhang, L.; Xu, X. Atg3 Overexpression Enhances Bortezomib-Induced Cell Death in SKM-1 Cell. PLoS ONE 2016, 11, e0158761. [Google Scholar] [CrossRef] [PubMed]
- Ossenkoppele, G.; Löwenberg, B. How I treat the older patient with acute myeloid leukemia. Blood 2015, 125, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Beau, M.M.L.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Schnegg-Kaufmann, A.; Feller, A.; Baldomero, H.; Rovo, A.; Manz, M.G.; Gregor, M.; Efthymiou, A.; Bargetzi, M.; Hess, U.; Spertini, O.; et al. Improvement of relative survival in elderly patients with acute myeloid leukaemia emerging from population-based cancer registries in Switzerland between 2001 and 2013. Cancer Epidemiol. 2018, 52, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Juliusson, G.; Lazarevic, V.; Hörstedt, A.-S.; Hagberg, O.; Höglund, M.; Swedish Acute Leukemia Registry Group. Acute myeloid leukemia in the real world: Why population-based registries are needed. Blood 2012, 119, 3890–3899. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Britschgi, A.; Schläfli, A.M.; Humbert, M.; Shan-Krauer, D.; Batliner, J.; Federzoni, E.A.; Ernst, M.; Torbett, B.E.; Yousefi, S.; et al. Low Autophagy (ATG) Gene Expression Is Associated with an Immature AML Blast Cell Phenotype and Can Be Restored during AML Differentiation Therapy. Oxid. Med. Cell. Longev. 2018, 2018, 1482795. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.S.; Riffelmacher, T.; Stranks, A.; Williams, O.; De Boer, J.; Cain, K.; MacFarlane, M.; McGouran, J.; Kessler, B.; Khandwala, S.; et al. Autophagy limits proliferation and glycolytic metabolism in acute myeloid leukemia. Cell Death Discov. 2015, 1, 15008. [Google Scholar] [CrossRef]
- Meenhuis, A.; van Veelen, P.A.; de Looper, H.; van Boxtel, N.; van den Berge, I.J.; Sun, S.M.; Taskesen, E.; Stern, P.; de Ru, A.H.; van Adrichem, A.J.; et al. MiR-17/20/93/106 promote hematopoietic cell expansion by targeting sequestosome 1-regulated pathways in mice. Blood 2011, 118, 916–925. [Google Scholar] [CrossRef]
- Trocoli, A.; Bensadoun, P.; Richard, E.; Labrunie, G.; Merhi, F.; Schläfli, A.M.; Brigger, D.; Souquere, S.; Pierron, G.; Pasquet, J.-M.; et al. p62/SQSTM1 upregulation constitutes a survival mechanism that occurs during granulocytic differentiation of acute myeloid leukemia cells. Cell Death Differ. 2014, 21, 1852. [Google Scholar] [CrossRef]
- Ségal-Bendirdjian, E.; Tschan, M.P.; Reiffers, J.; Djavaheri-Mergny, M. Pro-survival role of p62 during granulocytic differentiation of acute myeloid leukemia cells. Mol. Cell. Oncol. 2014, 1, e970066. [Google Scholar] [CrossRef]
- Nguyen, T.D.; Shaid, S.; Vakhrusheva, O.; Koschade, S.E.; Klann, K.; Thölken, M.; Baker, F.; Zhang, J.; Oellerich, T.; Sürün, D.; et al. Loss of the selective autophagy receptor p62 impairs murine myeloid leukemia progression and mitophagy. Blood 2019, 133, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Rudat, S.; Pfaus, A.; Cheng, Y.Y.; Holtmann, J.; Ellegast, J.M.; Bühler, C.; Marcantonio, D.D.; Martinez, E.; Göllner, S.; Wickenhauser, C.; et al. RET-mediated autophagy suppression as targetable co-dependence in acute myeloid leukemia. Leukemia 2018, 32, 2189–2202. [Google Scholar] [CrossRef] [PubMed]
- Larrue, C.; Saland, E.; Boutzen, H.; Vergez, F.; David, M.; Joffre, C.; Hospital, M.-A.; Tamburini, J.; Delabesse, E.; Manenti, S.; et al. Proteasome inhibitors induce FLT3-ITD degradation through autophagy in AML cells. Blood 2016, 127, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Heydt, Q.; Larrue, C.; Saland, E.; Bertoli, S.; Sarry, J.-E.; Besson, A.; Manenti, S.; Joffre, C.; Mansat-De Mas, V. Oncogenic FLT3-ITD supports autophagy via ATF4 in acute myeloid leukemia. Oncogene 2018, 37, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Tan, S.; Yang, Z.; Zhan, Q.; Jin, H.; Xian, J.; Zhang, S.; Yang, L.; Wang, L.; Zhang, L. NPM1 Mutant Mediated PML Delocalization and Stabilization Enhances Autophagy and Cell Survival in Leukemic Cells. Theranostics 2017, 7, 2289–2304. [Google Scholar] [CrossRef] [PubMed]
- Sumitomo, Y.; Koya, J.; Nakazaki, K.; Kataoka, K.; Tsuruta-Kishino, T.; Morita, K.; Sato, T.; Kurokawa, M. Cytoprotective autophagy maintains leukemia-initiating cells in murine myeloid leukemia. Blood 2016, 128, 1614–1624. [Google Scholar] [CrossRef] [PubMed]
- Man, N.; Tan, Y.; Sun, X.-J.; Liu, F.; Cheng, G.; Greenblatt, S.M.; Martinez, C.; Karl, D.L.; Ando, K.; Sun, M.; et al. Caspase-3 controls AML1-ETO-driven leukemogenesis via autophagy modulation in a ULK1-dependent manner. Blood 2017, 129, 2782–2792. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Clark, J.; Wunderlich, M.; Fan, C.; Davis, A.; Chen, S.; Guan, J.-L.; Mulloy, J.C.; Kumar, A.; Zheng, Y. Autophagy is dispensable for Kmt2a/Mll-Mllt3/Af9 AML maintenance and anti-leukemic effect of chloroquine. Autophagy 2017, 13, 955–966. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Chen, L.; Atkinson, J.M.; Claxton, D.F.; Wang, H.-G. Atg5-dependent autophagy contributes to the development of acute myeloid leukemia in an MLL-AF9-driven mouse model. Cell Death Dis. 2016, 7, e2361. [Google Scholar] [CrossRef] [PubMed]
- Piragyte, I.; Clapes, T.; Polyzou, A.; Klein Geltink, R.I.; Lefkopoulos, S.; Yin, N.; Cauchy, P.; Curtis, J.D.; Klaeylé, L.; Langa, X.; et al. A metabolic interplay coordinated by HLX regulates myeloid differentiation and AML through partly overlapping pathways. Nat. Commun. 2018, 9, 3090. [Google Scholar] [CrossRef] [PubMed]
- Helgason, G.V.; Mukhopadhyay, A.; Karvela, M.; Salomoni, P.; Calabretta, B.; Holyoake, T.L. Autophagy in chronic myeloid leukaemia: Stem cell survival and implication in therapy. Curr. Cancer Drug Targets 2013, 13, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Goussetis, D.J.; Gounaris, E.; Wu, E.J.; Vakana, E.; Sharma, B.; Bogyo, M.; Altman, J.K.; Platanias, L.C. Autophagic degradation of the BCR-ABL oncoprotein and generation of antileukemic responses by arsenic trioxide. Blood 2012, 120, 3555–3562. [Google Scholar] [CrossRef] [PubMed]
- Colecchia, D.; Rossi, M.; Sasdelli, F.; Sanzone, S.; Strambi, A.; Chiariello, M. MAPK15 mediates BCR-ABL1-induced autophagy and regulates oncogene-dependent cell proliferation and tumor formation. Autophagy 2015, 11, 1790–1802. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.; Ma, L.; Sun, J.E.; Zhu, L.J.; Green, M.R. BCR-ABL suppresses autophagy through ATF5-mediated regulation of mTOR transcription. Blood 2011, 118, 2840–2848. [Google Scholar] [CrossRef] [PubMed]
- Baquero, P.; Dawson, A.; Mukhopadhyay, A.; Kuntz, E.M.; Mitchell, R.; Olivares, O.; Ianniciello, A.; Scott, M.T.; Dunn, K.; Nicastri, M.C.; et al. Targeting quiescent leukemic stem cells using second generation autophagy inhibitors. Leukemia 2018. [Google Scholar] [CrossRef] [PubMed]
- Ianniciello, A.; Dumas, P.-Y.; Drullion, C.; Guitart, A.; Villacreces, A.; Peytour, Y.; Chevaleyre, J.; Brunet de la Grange, P.; Vigon, I.; Desplat, V.; et al. Chronic myeloid leukemia progenitor cells require autophagy when leaving hypoxia-induced quiescence. Oncotarget 2017, 8, 96984–96992. [Google Scholar] [CrossRef]
- Mourgues, L.; Imbert, V.; Nebout, M.; Colosetti, P.; Neffati, Z.; Lagadec, P.; Verhoeyen, E.; Peng, C.; Duprez, E.; Legros, L.; et al. The BMI1 polycomb protein represses cyclin G2-induced autophagy to support proliferation in chronic myeloid leukemia cells. Leukemia 2015, 29, 1993–2002. [Google Scholar] [CrossRef]
- Cluzeau, T.; Robert, G.; Jacquel, A.; Auberger, P. How recent advances in high-risk myelodysplastic syndrome physiopathology may impact future treatments. Curr. Pharm. Des. 2013, 19, 5362–5373. [Google Scholar] [CrossRef]
- Zeng, W.; Dai, H.; Yan, M.; Cai, X.; Luo, H.; Ke, M.; Liu, Z. Decitabine-Induced Changes in Human Myelodysplastic Syndrome Cell Line SKM-1 Are Mediated by FOXO3A Activation. J. Immunol. Res. 2017, 2017, 4302320. [Google Scholar] [CrossRef]
- Dubois, A.; Furstoss, N.; Calleja, A.; Zerhouni, M.; Cluzeau, T.; Savy, C.; Marchetti, S.; Hamouda, M.A.; Boulakirba, S.; Orange, F.; et al. LAMP2 expression dictates azacytidine response and prognosis in MDS/AML. Leukemia 2019. [Google Scholar] [CrossRef]
- Fabre, C.; Carvalho, G.; Tasdemir, E.; Braun, T.; Adès, L.; Grosjean, J.; Boehrer, S.; Métivier, D.; Souquère, S.; Pierron, G.; et al. NF-κB inhibition sensitizes to starvation-induced cell death in high-risk myelodysplastic syndrome and acute myeloid leukemia. Oncogene 2007, 26, 4071–4083. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Rhyasen, G.; Bolanos, L.; Rasch, C.; Varney, M.; Wunderlich, M.; Goyama, S.; Jansen, G.; Cloos, J.; Rigolino, C.; et al. Cytotoxic effects of bortezomib in myelodysplastic syndrome/acute myeloid leukemia depend on autophagy-mediated lysosomal degradation of TRAF6 and repression of PSMA1. Blood 2012, 120, 858–867. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, C.; Evangelisti, C.; Chiarini, F.; Lonetti, A.; Buontempo, F.; Neri, L.M.; McCubrey, J.A.; Martelli, A.M. Autophagy in acute leukemias: A double-edged sword with important therapeutic implications. Biochim. Biophys. Acta 2015, 1853, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Willems, L.; Chapuis, N.; Puissant, A.; Maciel, T.T.; Green, A.S.; Jacque, N.; Vignon, C.; Park, S.; Guichard, S.; Herault, O.; et al. The dual mTORC1 and mTORC2 inhibitor AZD8055 has anti-tumor activity in acute myeloid leukemia. Leukemia 2012, 26, 1195–1202. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yang, M.; Kang, R.; Wang, Z.; Zhao, Y.; Yu, Y.; Xie, M.; Yin, X.; Livesey, K.M.; Lotze, M.T.; et al. HMGB1-induced autophagy promotes chemotherapy resistance in leukemia cells. Leukemia 2011, 25, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Cheong, J.-W.; Kim, Y.; Eom, J.I.; Jeung, H.-K.; Min, Y.H. Enhanced autophagy in cytarabine arabinoside-resistant U937 leukemia cells and its potential as a target for overcoming resistance. Mol. Med. Rep. 2016, 13, 3433–3440. [Google Scholar] [CrossRef] [PubMed]
- Piya, S.; Kornblau, S.M.; Ruvolo, V.R.; Mu, H.; Ruvolo, P.P.; McQueen, T.; Davis, R.E.; Hail, N.J.; Kantarjian, H.; Andreeff, M.; et al. Atg7 suppression enhances chemotherapeutic agent sensitivity and overcomes stroma-mediated chemoresistance in acute myeloid leukemia. Blood 2016, 128, 1260–1269. [Google Scholar] [CrossRef]
- Chen, L.; Guo, P.; Jia, P.; Tong, J.; Hu, J.; Li, J. Autophagy Is an Important Event for Low Dose Cytarabine Treatment in Acute Myeloid Leukemia U937 Cell Line. Blood 2014, 124, 5209. [Google Scholar]
- Altman, J.K.; Szilard, A.; Goussetis, D.J.; Sassano, A.; Colamonici, M.; Gounaris, E.; Frankfurt, O.; Giles, F.J.; Eklund, E.A.; Beauchamp, E.M.; et al. Autophagy is a survival mechanism of acute myelogenous leukemia precursors during dual mTORC2/mTORC1 targeting. Clin. Cancer Res. 2014, 20, 2400–2409. [Google Scholar] [CrossRef]
- Willems, L.; Jacque, N.; Jacquel, A.; Neveux, N.; Maciel, T.T.; Lambert, M.; Schmitt, A.; Poulain, L.; Green, A.S.; Uzunov, M.; et al. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood 2013, 122, 3521–3532. [Google Scholar] [CrossRef]
- Torgersen, M.L.; Engedal, N.; Bøe, S.-O.; Hokland, P.; Simonsen, A. Targeting autophagy potentiates the apoptotic effect of histone deacetylase inhibitors in t(8;21) AML cells. Blood 2013, 122, 2467–2476. [Google Scholar] [CrossRef] [PubMed]
- Stankov, M.V.; El Khatib, M.; Kumar Thakur, B.; Heitmann, K.; Panayotova-Dimitrova, D.; Schoening, J.; Bourquin, J.P.; Schweitzer, N.; Leverkus, M.; Welte, K.; et al. Histone deacetylase inhibitors induce apoptosis in myeloid leukemia by suppressing autophagy. Leukemia 2014, 28, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, M.; Aust, M.M.; Attkisson, E.; Williams, D.C.; Ferreira-Gonzalez, A.; Grant, S. Inhibition of Bcl-2 antiapoptotic members by obatoclax potently enhances sorafenib-induced apoptosis in human myeloid leukemia cells through a Bim-dependent process. Blood 2012, 119, 6089–6098. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, R.; Hoshijima, H.; Nagasaka, H.; Chowdhury, S.A.; Kikuchi, H.; Kanda, Y.; Kunii, S.; Kawase, M.; Sakagami, H. Induction of non-apoptotic cell death by morphinone in human promyelocytic leukemia HL-60 cells. Anti-Cancer Res. 2006, 26, 3343–3348. [Google Scholar]
- Sujobert, P.; Poulain, L.; Paubelle, E.; Zylbersztejn, F.; Grenier, A.; Lambert, M.; Townsend, E.C.; Brusq, J.M.; Nicodeme, E.; Decrooqc, J.; et al. Co-activation of AMPK and mTORC1 Induces Cytotoxicity in Acute Myeloid Leukemia. Cell Rep. 2015, 11, 1446–1457. [Google Scholar] [CrossRef] [PubMed]
- Goussetis, D.J.; Altman, J.K.; Glaser, H.; McNeer, J.L.; Tallman, M.S.; Platanias, L.C. Autophagy is a critical mechanism for the induction of the antileukemic effects of arsenic trioxide. J. Biol. Chem. 2010, 285, 29989–29997. [Google Scholar] [CrossRef]
- Isakson, P.; Bjørås, M.; Bøe, S.O.; Simonsen, A. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood 2010, 116, 2324–2331. [Google Scholar] [CrossRef]
- Sanz, M.A.; Grimwade, D.; Tallman, M.S.; Lowenberg, B.; Fenaux, P.; Estey, E.H.; Naoe, T.; Lengfelder, E.; Buchner, T.; Dohner, H.; et al. Management of acute promyelocytic leukemia: Recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 2009, 113, 1875–1891. [Google Scholar] [CrossRef]
- Wang, Z.; Cao, L.; Kang, R.; Yang, M.; Liu, L.; Zhao, Y.; Yu, Y.; Xie, M.; Yin, X.; Livesey, K.M.; et al. Autophagy regulates myeloid cell differentiation by p62/SQSTM1-mediated degradation of PML-RARalpha oncoprotein. Autophagy 2011, 7, 401–411. [Google Scholar] [CrossRef]
- Trocoli, A.; Mathieu, J.; Priault, M.; Reiffers, J.; Souquere, S.; Pierron, G.; Besançon, F.; Djavaheri-Mergny, M. ATRA-induced upregulation of Beclin 1 prolongs the life span of differentiated acute promyelocytic leukemia cells. Autophagy 2011, 7, 1108–1114. [Google Scholar] [CrossRef]
- Huang, Y.; Hou, J.-K.; Chen, T.-T.; Zhao, X.-Y.; Yan, Z.-W.; Zhang, J.; Yang, J.; Kogan, S.C.; Chen, G.-Q. PML-RARalpha enhances constitutive autophagic activity through inhibiting the Akt/mTOR pathway. Autophagy 2011, 7, 1132–1144. [Google Scholar] [CrossRef] [PubMed]
- Brigger, D.; Proikas-Cezanne, T.; Tschan, M.P. WIPI-dependent autophagy during neutrophil differentiation of NB4 acute promyelocytic leukemia cells. Cell Death Dis. 2014, 5, e1315. [Google Scholar] [CrossRef]
- Brigger, D.; Torbett, B.E.; Chen, J.; Fey, M.F.; Tschan, M.P. Inhibition of GATE-16 attenuates ATRA-induced neutrophil differentiation of APL cells and interferes with autophagosome formation. Biochem. Biophys. Res. Commun. 2013, 438, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Mueller, C.; Fey, M.F.; Tschan, M.P. Inhibition of damage-regulated autophagy modulator-1 (DRAM-1) impairs neutrophil differentiation of NB4 APL cells. Leuk. Res. 2012, 36, 1552–1556. [Google Scholar] [CrossRef] [PubMed]
- Haimovici, A.; Brigger, D.; Torbett, B.E.; Fey, M.F.; Tschan, M.P. Induction of the autophagy-associated gene MAP1S via PU.1 supports APL differentiation. Leuk. Res. 2014, 38, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ren, W.; Chen, K. MiR-34a Promotes Apoptosis and Inhibits Autophagy by Targeting HMGB1 in Acute Myeloid Leukemia Cells. Cell. Physiol. Biochem. 2017, 41, 1981–1992. [Google Scholar] [CrossRef]
- Zeng, C.W.; Chen, Z.H.; Zhang, X.J.; Han, B.W.; Lin, K.Y.; Li, X.J.; Wei, P.P.; Zhang, H.; Li, Y.; Chen, Y.Q. MIR125B1 represses the degradation of the PML-RARA oncoprotein by an autophagylysosomal pathway in acute promyelocytic leukemia. Autophagy 2014, 10, 1726–1737. [Google Scholar] [CrossRef]
- Chen, Z.-H.; Wang, W.-T.; Huang, W.; Fang, K.; Sun, Y.-M.; Liu, S.-R.; Luo, X.-Q.; Chen, Y.-Q. The lncRNA HOTAIRM1 regulates the degradation of PML-RARA oncoprotein and myeloid cell differentiation by enhancing the autophagy pathway. Cell Death Differ. 2017, 24, 212–224. [Google Scholar] [CrossRef]
- Ganesan, S.; Alex, A.A.; Chendamarai, E.; Balasundaram, N.; Palani, H.K.; David, S.; Kulkarni, U.; Aiyaz, M.; Mugasimangalam, R.; Korula, A.; et al. Rationale and efficacy of proteasome inhibitor combined with arsenic trioxide in the treatment of acute promyelocytic leukemia. Leukemia 2016, 30, 2169–2178. [Google Scholar] [CrossRef]
- Xie, N.; Zhong, L.; Liu, L.; Fang, Y.; Qi, X.; Cao, J.; Yang, B.; He, Q.; Ying, M. Autophagy contributes to dasatinib-induced myeloid differentiation of human acute myeloid leukemia cells. Biochem. Pharmacol. 2014, 89, 74–85. [Google Scholar] [CrossRef]
- Lau, A.; Zheng, Y.; Tao, S.; Wang, H.; Whitman, S.A.; White, E.; Zhang, D.D. Arsenic inhibits autophagic flux, activating the Nrf2-Keap1 pathway in a p62-dependent manner. Mol. Cell. Biol. 2013, 33, 2436–2446. [Google Scholar] [CrossRef] [PubMed]
- Elzinga, B.M.; Nyhan, M.J.; Crowley, L.C.; O’Donovan, T.R.; Cahill, M.R.; McKenna, S.L. Induction of autophagy by Imatinib sequesters Bcr-Abl in autophagosomes and down-regulates Bcr-Abl protein. Am. J. Hematol. 2013, 88, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Carew, J.S.; Nawrocki, S.T.; Kahue, C.N.; Zhang, H.; Yang, C.; Chung, L.; Houghton, J.A.; Huang, P.; Giles, F.J.; Cleveland, J.L. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood 2007, 110, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Bellodi, C.; Lidonnici, M.R.; Hamilton, A.; Helgason, G.V.; Soliera, A.R.; Ronchetti, M.; Galavotti, S.; Young, K.W.; Selmi, T.; Yacobi, R.; et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J. Clin. Investig. 2009, 119, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Helgason, G.V.; Karvela, M.; Holyoake, T.L. Kill one bird with two stones: Potential efficacy of BCR-ABL and autophagy inhibition in CML. Blood 2011, 118, 2035–2043. [Google Scholar] [CrossRef] [PubMed]
- Calabretta, B.; Salomoni, P. Inhibition of autophagy: A new strategy to enhance sensitivity of chronic myeloid leukemia stem cells to tyrosine kinase inhibitors. Leuk. Lymphoma 2011, 52 (Suppl. 1), 54–59. [Google Scholar] [CrossRef]
- Crowley, L.C.; O’Donovan, T.R.; Nyhan, M.J.; McKenna, S.L. Pharmacological agents with inherent anti-autophagic activity improve the cytotoxicity of imatinib. Oncol. Rep. 2013, 29, 2261–2268. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Zhao, H.; Li, Y.; Fan, J.; Sun, Y.; Wang, S.; Wang, Z.; Song, P.; Ju, D. Targeting Hedgehog signaling pathway and autophagy overcomes drug resistance of BCR-ABLpositive chronic myeloid leukemia. Autophagy 2015, 11, 355–372. [Google Scholar] [CrossRef]
- Puissant, A.; Auberger, P. AMPK- and p62/SQSTM1-dependent autophagy mediate resveratrol-induced cell death in chronic myelogenous leukemia. Autophagy 2010, 6, 655–657. [Google Scholar] [CrossRef]
- Puissant, A.; Robert, G.; Fenouille, N.; Luciano, F.; Cassuto, J.P.; Raynaud, S.; Auberger, P. Resveratrol promotes autophagic cell death in chronic myelogenous leukemia cells via JNK-mediated p62/SQSTM1 expression and AMPK activation. Cancer Res. 2010, 70, 1042–1052. [Google Scholar] [CrossRef]
- Robert, G.; Ben Sahra, I.; Puissant, A.; Colosetti, P.; Belhacene, N.; Gounon, P.; Hofman, P.; Bost, F.; Cassuto, J.P.; Auberger, P. Acadesine kills Chronic Myelogenous Leukemia (CML) cells through PKC-dependent induction of autophagic cell death. PLoS ONE 2009, 4, e7889. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; You, L.; Liu, H.; Li, L.; Meng, H.; Qian, Q.; Qian, W. Potent antitumor activity of oncolytic adenovirus expressing Beclin-1 via induction of autophagic cell death in leukemia. Oncotarget 2013, 4, 860–874. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, A. Autophagy and Disease. J. Biol. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.-J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [PubMed]
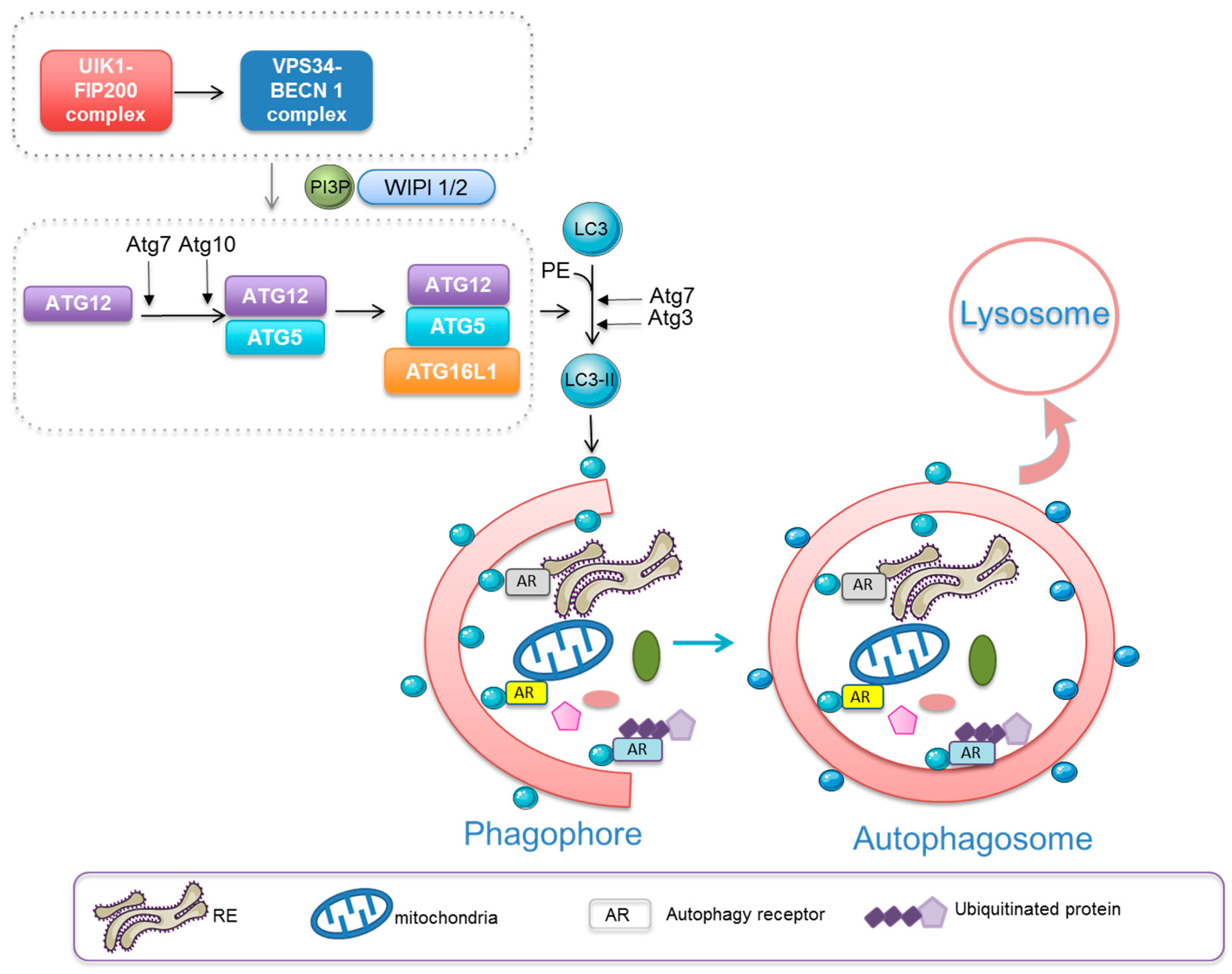


{kind=link}
{kind=link}
{kind=link}
| Haematological Malignancies | Therapeutic Modulation of Autophagy Single Drug or Combination | Clinical Trials | ||
|---|---|---|---|---|
| Number | Phase | |||
| Chronic lymphocytic leukaemia | Autophagy Inhibition | Hydroxychloroquine (HCQ) | NCT00771056 | II |
| Multiple myeloma | HCQ + Bortezomib | NCT00568880 | I/II | |
| HCQ + Cyclophosphamide + Dexamethasone + Rapamycin | NCT01689987 | I | ||
| Lymphoma | Vinblastine | NCT00059839 | III | |
| Chronic myeloid leukaemia | HCQ + Imatinib | NCT01227135 | II | |
| Acute myelogenous leukaemia | HCQ + Mitoxantone + Etoposide | NCT02631252 | I | |
| Chronic myeloid leukaemia | Autophagy Induction | Everolimus (RAD001, mTORC1 inhibitor) | NCT01188889 | I/II |
| Chronic lymphocytic leukaemia | Everolimus + Alemtuzumab | NCT00935792 | I/II | |
| CAL-101 (PI3Kd inhibitor) | NCT01539512 | III | ||
| Perfosine | NCT00873457 | II | ||
| Relapsed follicular or mantle cell lymphoma | Temsirolimus (mTORC1 inhibitor) | NCT01078142 | I | |
| Multiple myeloma | Everolimus + Panobinostat (HDAC inhibitor) | NCT00918333 | I/II | |
| Multiple myeloma, Lymphoma | Everolimus + Sorafenib (multikinase inhibitor) | NCT00474929 | I/II | |
| Acute monoblastic leukaemia | Lithium | NCT01820624 | I | |
| Advanced haematological malignancies | Triciribine | NCT00642031 | I | |
| Myelodysplasic syndrome and | Autophagy inducer + Azacitidine | NCT01210274 | Recruiting | |
| Acute myelogenous leukaemia | ||||
| Relapsed/Refractory NHL or HL | CAL-101 | NCT01306643/NCT01393106 | I/II | |
| NCT01282424 | ||||
| Relapsed/Refractory NHL | CAL-101 + Rituximab +/− Bendamustine | NCT01088048/NCT01732913 | I | |
| NCT01732926 | ||||
| CAL-101 + GS-9973 (Syk inhibitor) | NCT01796470 | II | ||
| Lymphoma malignancies | IPI-145 (PI3Kd and PI3Kg inhibitor) | NCT01476657 | I | |
| CC-223 (dual mTORC1 and mTORC2 inhibitor) | NCT01177397 | I/II | ||
| Relapsed/Refractory/Newly diagnosed lymphoma | Everolimus in combination therapies (+/− Antibodies +/− TKIs +/− Chemotherapy) | NCT00869999/NCT01334502 | I/II | |
| NCT01198665/NCT01665768 | ||||
| NCT01854606/NCT01341834 | ||||
| NCT01075321/NCT01567475 | ||||
| NCT00352443/NCT01453504 | ||||
| Relapsed/Refractory lymphoma | Everolimus | NCT00436618 | II | |
| Everolimus + Panobinostat | NCT00962507/NCT00978432 | I/II | ||
| Ridaforolimus (mTORC1 inhibitor) | NCT00060632/NCT00060645 NCT00086125 | I/II | ||
| Ridaforolimus + Vorinostat (HDAC inhibitor) | NCT 01169532 | I/II | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Djavaheri-Mergny, M.; Giuriato, S.; Tschan, M.P.; Humbert, M. Therapeutic Modulation of Autophagy in Leukaemia and Lymphoma. Cells 2019, 8, 103. https://doi.org/10.3390/cells8020103
Djavaheri-Mergny M, Giuriato S, Tschan MP, Humbert M. Therapeutic Modulation of Autophagy in Leukaemia and Lymphoma. Cells. 2019; 8(2):103. https://doi.org/10.3390/cells8020103
Chicago/Turabian StyleDjavaheri-Mergny, Mojgan, Sylvie Giuriato, Mario P. Tschan, and Magali Humbert. 2019. "Therapeutic Modulation of Autophagy in Leukaemia and Lymphoma" Cells 8, no. 2: 103. https://doi.org/10.3390/cells8020103
APA StyleDjavaheri-Mergny, M., Giuriato, S., Tschan, M. P., & Humbert, M. (2019). Therapeutic Modulation of Autophagy in Leukaemia and Lymphoma. Cells, 8(2), 103. https://doi.org/10.3390/cells8020103