Targeting the Hedgehog Pathway in Cancer: Current Evidence and Future Perspectives


Abstract
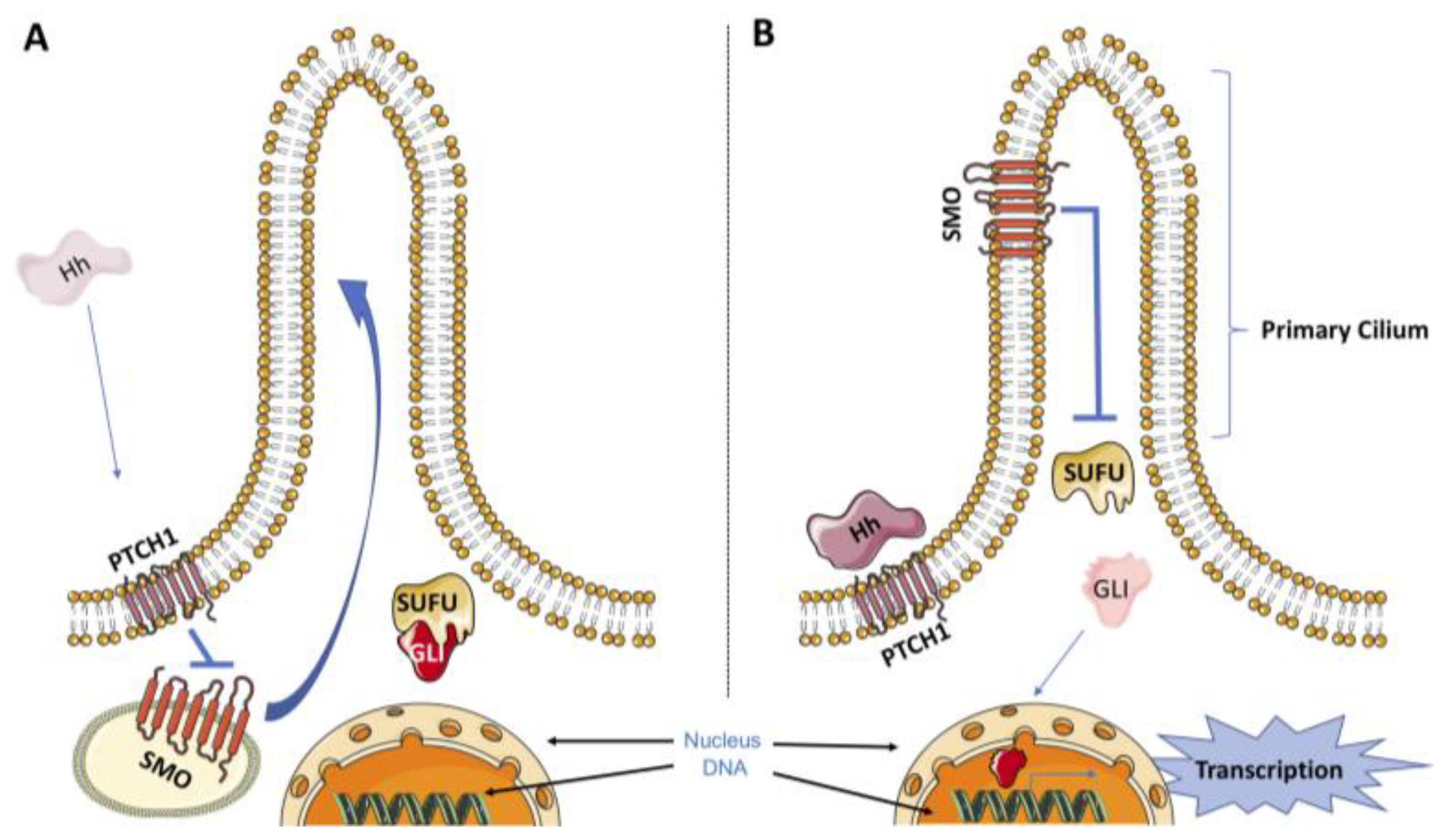
:1. Introduction
2. Characterization of the HhP and Its Relation with Carcinogenesis
3. Other Implications of the HhP and Cancer
4. SMO Inhibitors
4.1. Cyclopamine
4.2. Vismodegib (GDC-0449)
4.3. Sonidegib (LDE-225)
4.4. Saridegib (IPI-926)
4.5. Taladegib (LY2940680)
4.6. Glasdegib (PF-04449913)
4.7. Itraconazole
5. GLI Inhibitors
6. Combination Therapy
7. Conclusion and Future Perspectives
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| AKT1 | V-akt murine thymoma viral oncogene homolog 1 |
| ATO | Arsenic trioxide |
| BCC | Basal cell carcinoma |
| BET | Bromodomain and extra terminal |
| BRD4 | Bromodomain-containing protein 4 |
| CK1 | Casein kinase 1 |
| DHH | Desert Hedgehog |
| EFS | Event-free survival |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial-to-mesenchymal transition |
| GLI | Glioma-associated oncogene |
| GSK3β | Glycogen synthase kinase 3β |
| HhP | Hedgehog pathway |
| HIF2α | Hypoxia-inducible factor 2α |
| HNSCC | Head and neck squamous cell carcinoma |
| HPI | Hedgehog pathway inhibitors |
| HR | Hazard ratio |
| IHH | Indian Hedgehog |
| IL-1β | Interleukin-1β |
| KLF4 | Kruppel-like factor 4 |
| LSC | Leukemic stem cell |
| mTOR | Mechanistic target of rapamycin |
| NF-κB | Nuclear factor kappa light chain enhancer of activated B cells |
| OCT4 | Octamer binding transcription factor 4 |
| ORR | Overall response rate |
| OS | Overall survival |
| PFS | Progression-free survival |
| PKA | Protein kinase A |
| PTCH1 | Patched1 |
| RFS | Relapse-free survival |
| S6K1 | Ribosomal protein S6 kinase 1 |
| SHH | Sonic Hedgehog |
| SMO | Smoothened |
| SUFU | Suppressor of fused |
| TGF-β | Transforming growth factor-beta |
| TLR | Toll-like receptor |
| TNF-α | Tumor necrosis factor-α |
References
- Gan, G.N.; Jimeno, A. Emerging from their burrow: Hedgehog pathway inhibitors for cancer. Expert. Opin. Investig. Drugs 2016, 25, 1153–1166. [Google Scholar] [CrossRef]
- Ramalho-Santos, M.; Melton, D.A.; McMahon, A.P. Hedgehog signals regulate multiple aspects of gastrointestinal development. Development 2000, 127, 2763–2772. [Google Scholar] [PubMed]
- Jiang, J.; Hui, C.C. Hedgehog signaling in development and cancer. Dev. Cell 2008, 15, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Raffel, C.; Jenkins, R.B.; Frederick, L.; Hebrink, D.; Alderete, B.; Fults, D.W.; James, C.D. Sporadic medulloblastomas contain PTCH mutations. Cancer Res. 1997, 57, 842–845. [Google Scholar] [PubMed]
- Reifenberger, J.; Wolter, M.; Weber, R.G.; Megahed, M.; Ruzicka, T.; Lichter, P.; Reifenberger, G. Missense mutations in SMOH in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res. 1998, 58, 1798–1803. [Google Scholar] [PubMed]
- Tostar, U.; Malm, C.J.; Meis-Kindblom, J.M.; Kindblom, L.G.; Toftgard, R.; Unden, A.B. Deregulation of the hedgehog signalling pathway: A possible role for the PTCH and SUFU genes in human rhabdomyoma and rhabdomyosarcoma development. J. Pathol. 2006, 208, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Murone, M.; Luoh, S.M.; Ryan, A.; Gu, Q.; Zhang, C.; Bonifas, J.M.; Lam, C.W.; Hynes, M.; Goddard, A.; et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 1998, 391, 90–92. [Google Scholar] [CrossRef]
- Basset-Seguin, N.; Hauschild, A.; Grob, J.J.; Kunstfeld, R.; Dreno, B.; Mortier, L.; Ascierto, P.A.; Licitra, L.; Dutriaux, C.; Thomas, L.; et al. Vismodegib in patients with advanced basal cell carcinoma (STEVIE): A pre-planned interim analysis of an international, open-label trial. Lancet Oncol. 2015, 16, 729–736. [Google Scholar] [CrossRef]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef]
- Echelard, Y.; Epstein, D.J.; St-Jacques, B.; Shen, L.; Mohler, J.; McMahon, J.A.; McMahon, A.P. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 1993, 75, 1417–1430. [Google Scholar] [CrossRef]
- Chen, M.H.; Wilson, C.W.; Li, Y.J.; Law, K.K.; Lu, C.S.; Gacayan, R.; Zhang, X.; Hui, C.C.; Chuang, P.T. Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev. 2009, 23, 1910–1928. [Google Scholar] [CrossRef] [PubMed]
- Robbins, D.J.; Fei, D.L.; Riobo, N.A. The Hedgehog signal transduction network. Sci. Signal. 2012, 5, re6. [Google Scholar] [CrossRef] [PubMed]
- Aza-Blanc, P.; Lin, H.Y.; Ruiz i Altaba, A.; Kornberg, T.B. Expression of the vertebrate Gli proteins in Drosophila reveals a distribution of activator and repressor activities. Development 2000, 127, 4293–4301. [Google Scholar]
- Kogerman, P.; Grimm, T.; Kogerman, L.; Krause, D.; Undén, A.B.; Sandstedt, B.; Toftgård, R.; Zaphiropoulos, P.G. Mammalian Suppressor-of-Fused modulates nuclear–cytoplasmic shuttling of GLI-1. Nat. Cell Biol. 1999, 1, 312. [Google Scholar] [CrossRef] [PubMed]
- Gonnissen, A.; Isebaert, S.; Haustermans, K. Targeting the Hedgehog signaling pathway in cancer: Beyond Smoothened. Oncotarget 2015, 6, 13899–13913. [Google Scholar] [CrossRef] [PubMed]
- Teperino, R.; Aberger, F.; Esterbauer, H.; Riobo, N.; Pospisilik, J.A. Canonical and non-canonical Hedgehog signalling and the control of metabolism. Semin. Cell Dev. Biol. 2014, 33, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ding, Q.; Yen, C.J.; Xia, W.; Izzo, J.G.; Lang, J.Y.; Li, C.W.; Hsu, J.L.; Miller, S.A.; Wang, X.; et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell 2012, 21, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, N.K.; Qu, C.; Kunkalla, K.; Liu, Y.; Vega, F. Transcriptional regulation of serine/threonine protein kinase (AKT) genes by glioma-associated oncogene homolog 1. J. Biol. Chem. 2013, 288, 15390–15401. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jin, G.; Li, Q.; Wang, Z.; Hu, W.; Li, P.; Li, S.; Wu, H.; Kong, X.; Gao, J.; et al. Hedgehog Signaling Non-Canonical Activated by Pro-Inflammatory Cytokines in Pancreatic Ductal Adenocarcinoma. J. Cancer 2016, 7, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
- Colavito, S.A.; Zou, M.R.; Yan, Q.; Nguyen, D.X.; Stern, D.F. Significance of glioma-associated oncogene homolog 1 (GLI1) expression in claudin-low breast cancer and crosstalk with the nuclear factor kappa-light-chain-enhancer of activated B cells (NFkappaB) pathway. Breast Cancer Res. 2014, 16, 444. [Google Scholar] [CrossRef] [PubMed]
- Dennler, S.; Andre, J.; Alexaki, I.; Li, A.; Magnaldo, T.; ten Dijke, P.; Wang, X.J.; Verrecchia, F.; Mauviel, A. Induction of sonic hedgehog mediators by transforming growth factor-beta: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 2007, 67, 6981–6986. [Google Scholar] [CrossRef] [PubMed]
- Yoo, Y.A.; Kang, M.H.; Kim, J.S.; Oh, S.C. Sonic hedgehog signaling promotes motility and invasiveness of gastric cancer cells through TGF-beta-mediated activation of the ALK5-Smad 3 pathway. Carcinogenesis 2008, 29, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; He, M.; Sheng, T.; Zhang, X.; Sinha, M.; Luxon, B.; Zhao, X.; Xie, J. Requirement of TGFbeta signaling for SMO-mediated carcinogenesis. J. Biol. Chem. 2010, 285, 36570–36576. [Google Scholar] [CrossRef]
- Ji, Z.; Mei, F.C.; Xie, J.; Cheng, X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J. Biol. Chem. 2007, 282, 14048–14055. [Google Scholar] [CrossRef] [PubMed]
- Schnidar, H.; Eberl, M.; Klingler, S.; Mangelberger, D.; Kasper, M.; Hauser-Kronberger, C.; Regl, G.; Kroismayr, R.; Moriggl, R.; Sibilia, M.; et al. Epidermal growth factor receptor signaling synergizes with Hedgehog/GLI in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer Res. 2009, 69, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.W.; Gallant, M.; Lamm, M.L.; Iannaccone, S.; Vieux, K.F.; Proytcheva, M.; Hyjek, E.; Iannaccone, P.; Walterhouse, D. Noncanonical regulation of the Hedgehog mediator GLI1 by c-MYC in Burkitt lymphoma. Mol. Cancer Res. 2013, 11, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.H.; Dignam, J.J.; Elizabeth Hammond, M.; Klimowicz, A.C.; Petrillo, S.K.; Magliocco, A.; Jordan, R.; Trotti, A.; Spencer, S.; Cooper, J.S.; et al. Glioma-Associated Oncogene Family Zinc Finger 1 Expression and Metastasis in Patients With Head and Neck Squamous Cell Carcinoma Treated With Radiation Therapy (RTOG 9003). J. Clin. Oncol. 2011, 29, 1326–1334. [Google Scholar] [CrossRef]
- Mori, Y.; Okumura, T.; Tsunoda, S.; Sakai, Y.; Shimada, Y. Gli-1 expression is associated with lymph node metastasis and tumor progression in esophageal squamous cell carcinoma. Oncology 2006, 70, 378–389. [Google Scholar] [CrossRef]
- Ding, Y.-l.; Zhou, Y.; Xiang, L.; Ji, Z.-p.; Luo, Z.-h. Expression of Glioma-associated Oncogene Homolog 1 is Associated with Invasion and Postoperative Liver Metastasis in Colon Cancer. Int. J. Med. Sci. 2012, 9, 334–338. [Google Scholar] [CrossRef]
- Wellbrock, J.; Latuske, E.; Kohler, J.; Wagner, K.; Stamm, H.; Vettorazzi, E.; Vohwinkel, G.; Klokow, M.; Uibeleisen, R.; Ehm, P.; et al. Expression of Hedgehog Pathway Mediator GLI Represents a Negative Prognostic Marker in Human Acute Myeloid Leukemia and Its Inhibition Exerts Antileukemic Effects. Clin. Cancer Res. 2015, 21, 2388–2398. [Google Scholar] [CrossRef]
- Steg, A.D.; Katre, A.A.; Bevis, K.S.; Ziebarth, A.; Dobbin, Z.C.; Shah, M.M.; Alvarez, R.D.; Landen, C.N. Smoothened antagonists reverse taxane resistance in ovarian cancer. Mol. Cancer Ther. 2012, 11, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Katoh, M. Hedgehog signaling, epithelial-to-mesenchymal transition and miRNA. Int. J. Mol. Med. 2008, 22, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Ginnebaugh, K.R.; Ahmad, A.; Sarkar, F.H. The therapeutic potential of targeting the epithelial-mesenchymal transition in cancer. Expert Opin. Ther. Targets 2014, 18, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Kurrey, N.K.; Jalgaonkar, S.P.; Joglekar, A.V.; Ghanate, A.D.; Chaskar, P.D.; Doiphode, R.Y.; Bapat, S.A. Snail and slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells 2009, 27, 2059–2068. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhou, B.P. Snail: More than EMT. Cell Adh. Migr. 2010, 4, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Nanta, R.; Sharma, J.; Gunewardena, S.; Singh, K.P.; Shankar, S.; Srivastava, R.K. PI3K/AKT/mTOR and sonic hedgehog pathways cooperate together to inhibit human pancreatic cancer stem cell characteristics and tumor growth. Oncotarget 2015, 6, 32039–32060. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.W.; Mizumoto, K.; Urashima, T.; Nagai, E.; Maehara, N.; Sato, N.; Nakajima, M.; Tanaka, M. Radiation-induced increase in invasive potential of human pancreatic cancer cells and its blockade by a matrix metalloproteinase inhibitor, CGS27023. Clin. Cancer Res. 2002, 8, 1223–1227. [Google Scholar]
- Wild-Bode, C.; Weller, M.; Rimner, A.; Dichgans, J.; Wick, W. Sublethal irradiation promotes migration and invasiveness of glioma cells: Implications for radiotherapy of human glioblastoma. Cancer Res. 2001, 61, 2744–2750. [Google Scholar]
- Steg, A.D.; Bevis, K.S.; Katre, A.A.; Ziebarth, A.; Dobbin, Z.C.; Alvarez, R.D.; Zhang, K.; Conner, M.; Landen, C.N. Stem cell pathways contribute to clinical chemoresistance in ovarian cancer. Clin. Cancer Res. 2012, 18, 869–881. [Google Scholar] [CrossRef]
- De Boeck, A.; Narine, K.; De Neve, W.; Mareel, M.; Bracke, M.; De Wever, O. Resident and bone marrow-derived mesenchymal stem cells in head and neck squamous cell carcinoma. Oral Oncol. 2010, 46, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.E.; Ailles, L.E. Cancer stem cells in head and neck squamous cell cancer. J. Clin. Oncol. 2008, 26, 2871–2875. [Google Scholar] [CrossRef] [PubMed]
- Peacock, C.D.; Wang, Q.; Gesell, G.S.; Corcoran-Schwartz, I.M.; Jones, E.; Kim, J.; Devereux, W.L.; Rhodes, J.T.; Huff, C.A.; Beachy, P.A.; et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc. Natl. Acad. Sci. USA 2007, 104, 4048–4053. [Google Scholar] [CrossRef] [PubMed]
- Varnat, F.; Siegl-Cachedenier, I.; Malerba, M.; Gervaz, P.; Ruiz i Altaba, A. Loss of WNT-TCF addiction and enhancement of HH-GLI1 signalling define the metastatic transition of human colon carcinomas. EMBO Mol. Med. 2010, 2, 440–457. [Google Scholar] [CrossRef] [PubMed]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Altaba, A.R.i. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Gan, G.N.; Eagles, J.; Keysar, S.B.; Wang, G.; Glogowska, M.J.; Altunbas, C.; Anderson, R.T.; Le, P.N.; Morton, J.J.; Frederick, B.; et al. Hedgehog signaling drives radioresistance and stroma-driven tumor repopulation in head and neck squamous cancers. Cancer Res. 2014, 74, 7024–7036. [Google Scholar] [CrossRef]
- Keysar, S.B.; Le, P.N.; Anderson, R.T.; Morton, J.J.; Bowles, D.W.; Paylor, J.J.; Vogler, B.W.; Thorburn, J.; Fernandez, P.; Glogowska, M.J.; et al. Hedgehog signaling alters reliance on EGF receptor signaling and mediates anti-EGFR therapeutic resistance in head and neck cancer. Cancer Res. 2013, 73, 3381–3392. [Google Scholar] [CrossRef]
- Bora-Singhal, N.; Perumal, D.; Nguyen, J.; Chellappan, S. Gli1-Mediated Regulation of Sox2 Facilitates Self-Renewal of Stem-Like Cells and Confers Resistance to EGFR Inhibitors in Non–Small Cell Lung Cancer. Neoplasia 2015, 17, 538–551. [Google Scholar] [CrossRef]
- Ohta, H.; Aoyagi, K.; Fukaya, M.; Danjoh, I.; Ohta, A.; Isohata, N.; Saeki, N.; Taniguchi, H.; Sakamoto, H.; Shimoda, T.; et al. Cross talk between hedgehog and epithelial–mesenchymal transition pathways in gastric pit cells and in diffuse-type gastric cancers. Br. J. Cancer 2009, 100, 389–398. [Google Scholar] [CrossRef]
- Isohata, N.; Aoyagi, K.; Mabuchi, T.; Daiko, H.; Fukaya, M.; Ohta, H.; Ogawa, K.; Yoshida, T.; Sasaki, H. Hedgehog and epithelial-mesenchymal transition signaling in normal and malignant epithelial cells of the esophagus. Int. J. Cancer 2009, 125, 1212–1221. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, A.; Yokoe, T.; Tanaka, K.; Saigusa, S.; Toiyama, Y.; Yasuda, H.; Inoue, Y.; Miki, C.; Kusunoki, M. Radiation induces epithelial-mesenchymal transition in colorectal cancer cells. Oncol. Rep. 2012, 27, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Amakye, D.; Jagani, Z.; Dorsch, M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat. Med. 2013, 19, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, J.M.; Segal, R.J.; Zeitouni, N.C. Combination vismodegib and photodynamic therapy for multiple basal cell carcinomas. Photodiagnosis Photodyn. Ther. 2018, 21, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Basset-Seguin, N.; Hauschild, A.; Kunstfeld, R.; Grob, J.; Dreno, B.; Mortier, L.; Ascierto, P.A.; Licitra, L.; Dutriaux, C.; Thomas, L.; et al. Vismodegib in patients with advanced basal cell carcinoma: Primary analysis of STEVIE, an international, open-label trial. Eur. J. Cancer 2017, 86, 334–348. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Lewis, L.D.; LoRusso, P.; Maitland, M.; Chandra, P.; Cheeti, S.; Colburn, D.; Williams, S.; Simmons, B.; Graham, R.A. Pharmacokinetics and safety of vismodegib in patients with advanced solid malignancies and hepatic impairment. Cancer Chemother. Pharmacol. 2017, 80, 29–36. [Google Scholar] [CrossRef]
- Sekulic, A.; Migden, M.R.; Basset-Seguin, N.; Garbe, C.; Gesierich, A.; Lao, C.D.; Miller, C.; Mortier, L.; Murrell, D.F.; Hamid, O.; et al. Long-term safety and efficacy of vismodegib in patients with advanced basal cell carcinoma: Final update of the pivotal ERIVANCE BCC study. BMC Cancer 2017, 17, 332. [Google Scholar] [CrossRef]
- Dreno, B.; Kunstfeld, R.; Hauschild, A.; Fosko, S.; Zloty, D.; Labeille, B.; Grob, J.J.; Puig, S.; Gilberg, F.; Bergstrom, D.; et al. Two intermittent vismodegib dosing regimens in patients with multiple basal-cell carcinomas (MIKIE): A randomised, regimen-controlled, double-blind, phase 2 trial. Lancet Oncol. 2017, 18, 404–412. [Google Scholar] [CrossRef]
- Tang, J.Y.; Ally, M.S.; Chanana, A.M.; Mackay-Wiggan, J.M.; Aszterbaum, M.; Lindgren, J.A.; Ulerio, G.; Rezaee, M.R.; Gildengorin, G.; Marji, J.; et al. Inhibition of the hedgehog pathway in patients with basal-cell nevus syndrome: Final results from the multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2016, 17, 1720–1731. [Google Scholar] [CrossRef]
- Maughan, B.L.; Suzman, D.L.; Luber, B.; Wang, H.; Glavaris, S.; Hughes, R.; Sullivan, R.; Harb, R.; Boudadi, K.; Paller, C.; et al. Pharmacodynamic study of the oral hedgehog pathway inhibitor, vismodegib, in patients with metastatic castration-resistant prostate cancer. Cancer Chemother. Pharmacol. 2016, 78, 1297–1304. [Google Scholar] [CrossRef]
- Kwon, G.P.; Ally, M.S.; Bailey-Healy, I.; Oro, A.E.; Kim, J.; Chang, A.L.; Aasi, S.; Tang, J.Y. Update to an open-label clinical trial of vismodegib as neoadjuvant before surgery for high-risk basal cell carcinoma (BCC). J. Am. Acad. Dermatol. 2016, 75, 213–215. [Google Scholar] [CrossRef]
- Belani, C.P.; Dahlberg, S.E.; Rudin, C.M.; Fleisher, M.; Chen, H.X.; Takebe, N.; Velasco, M.R., Jr.; Tester, W.J.; Sturtz, K.; Hann, C.L.; et al. Vismodegib or cixutumumab in combination with standard chemotherapy for patients with extensive-stage small cell lung cancer: A trial of the ECOG-ACRIN Cancer Research Group (E1508). Cancer 2016, 122, 2371–2378. [Google Scholar] [CrossRef]
- Houot, R.; Soussain, C.; Tilly, H.; Haioun, C.; Thieblemont, C.; Casasnovas, O.; Bouabdallah, K.; Morschhauser, F.; Le Gouill, S.; Salles, G.A.; et al. Inhibition of Hedgehog signaling for the treatment of lymphoma and CLL: A phase II study from the LYSA. Ann. Oncol. 2016, 27, 1349–1350. [Google Scholar] [CrossRef] [PubMed]
- Catenacci, D.V.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized phase Ib/II study of gemcitabine plus placebo or vismodegib, a hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer. J. Clin. Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef] [PubMed]
- Sofen, H.; Gross, K.G.; Goldberg, L.H.; Sharata, H.; Hamilton, T.K.; Egbert, B.; Lyons, B.; Hou, J.; Caro, I. A phase II, multicenter, open-label, 3-cohort trial evaluating the efficacy and safety of vismodegib in operable basal cell carcinoma. J. Am. Acad. Dermatol. 2015, 73, 99–105. [Google Scholar] [CrossRef]
- Kim, E.J.; Sahai, V.; Abel, E.V.; Griffith, K.A.; Greenson, J.K.; Takebe, N.; Khan, G.N.; Blau, J.L.; Craig, R.; Balis, U.G.; et al. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin. Cancer Res. 2014, 20, 5937–5945. [Google Scholar] [CrossRef]
- Italiano, A.; Le Cesne, A.; Bellera, C.; Piperno-Neumann, S.; Duffaud, F.; Penel, N.; Cassier, P.; Domont, J.; Takebe, N.; Kind, M.; et al. GDC-0449 in patients with advanced chondrosarcomas: A French sarcoma group/US and French national cancer institute single-arm phase II collaborative study. Ann. Oncol. 2013, 24, 2922–2926. [Google Scholar] [CrossRef]
- Gajjar, A.; Stewart, C.F.; Ellison, D.W.; Kaste, S.; Kun, L.E.; Packer, R.J.; Goldman, S.; Chintagumpala, M.; Wallace, D.; Takebe, N.; et al. Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: A pediatric brain tumor consortium study. Clin. Cancer Res. 2013, 19, 6305–6312. [Google Scholar] [CrossRef]
- Thacker, C.A.; Weiss, G.J.; Tibes, R.; Blaydorn, L.; Downhour, M.; White, E.; Baldwin, J.; Hoff, D.D.; Korn, R.L. 18-FDG PET/CT assessment of basal cell carcinoma with vismodegib. Cancer Med. 2012, 1, 230–236. [Google Scholar] [CrossRef]
- Berlin, J.; Bendell, J.C.; Hart, L.L.; Firdaus, I.; Gore, I.; Hermann, R.C.; Mulcahy, M.F.; Zalupski, M.M.; Mackey, H.M.; Yauch, R.L.; et al. A randomized phase II trial of vismodegib versus placebo with FOLFOX or FOLFIRI and bevacizumab in patients with previously untreated metastatic colorectal cancer. Clin. Cancer Res. 2013, 19, 258–267. [Google Scholar] [CrossRef]
- LoRusso, P.M.; Piha-Paul, S.A.; Mita, M.; Colevas, A.D.; Malhi, V.; Colburn, D.; Yin, M.; Low, J.A.; Graham, R.A. Co-administration of vismodegib with rosiglitazone or combined oral contraceptive in patients with locally advanced or metastatic solid tumors: A pharmacokinetic assessment of drug-drug interaction potential. Cancer Chemother. Pharmacol. 2013, 71, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Kaye, S.B.; Fehrenbacher, L.; Holloway, R.; Amit, A.; Karlan, B.; Slomovitz, B.; Sabbatini, P.; Fu, L.; Yauch, R.L.; Chang, I.; et al. A phase II, randomized, placebo-controlled study of vismodegib as maintenance therapy in patients with ovarian cancer in second or third complete remission. Clin. Cancer Res. 2012, 18, 6509–6518. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Yauch, R.L.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N. Engl. J. Med. 2009, 361, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Lear, J.T.; Migden, M.R.; Lewis, K.D.; Chang, A.L.S.; Guminski, A.; Gutzmer, R.; Dirix, L.; Combemale, P.; Stratigos, A.; Plummer, R.; et al. Long-term efficacy and safety of sonidegib in patients with locally advanced and metastatic basal cell carcinoma: 30-month analysis of the randomized phase 2 BOLT study. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Kieran, M.W.; Chisholm, J.; Casanova, M.; Brandes, A.A.; Aerts, I.; Bouffet, E.; Bailey, S.; Leary, S.; MacDonald, T.J.; Mechinaud, F.; et al. Phase I study of oral sonidegib (LDE225) in pediatric brain and solid tumors and a phase II study in children and adults with relapsed medulloblastoma. Neuro Oncol. 2017, 19, 1542–1552. [Google Scholar] [CrossRef] [PubMed]
- Stathis, A.; Hess, D.; von Moos, R.; Homicsko, K.; Griguolo, G.; Joerger, M.; Mark, M.; Ackermann, C.J.; Allegrini, S.; Catapano, C.V.; et al. Phase I trial of the oral smoothened inhibitor sonidegib in combination with paclitaxel in patients with advanced solid tumors. Invest. New Drugs 2017, 35, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Minami, H.; Ando, Y.; Ma, B.B.; Hsiang Lee, J.; Momota, H.; Fujiwara, Y.; Li, L.; Fukino, K.; Ito, K.; Tajima, T.; et al. Phase I, multicenter, open-label, dose-escalation study of sonidegib in Asian patients with advanced solid tumors. Cancer Sci. 2016, 107, 1477–1483. [Google Scholar] [CrossRef] [PubMed]
- Pietanza, M.C.; Litvak, A.M.; Varghese, A.M.; Krug, L.M.; Fleisher, M.; Teitcher, J.B.; Holodny, A.I.; Sima, C.S.; Woo, K.M.; Ng, K.K.; et al. A phase I trial of the Hedgehog inhibitor, sonidegib (LDE225), in combination with etoposide and cisplatin for the initial treatment of extensive stage small cell lung cancer. Lung Cancer 2016, 99, 23–30. [Google Scholar] [CrossRef]
- Ko, A.H.; LoConte, N.; Tempero, M.A.; Walker, E.J.; Kate Kelley, R.; Lewis, S.; Chang, W.C.; Kantoff, E.; Vannier, M.W.; Catenacci, D.V.; et al. A Phase I Study of FOLFIRINOX Plus IPI-926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas 2016, 45, 370–375. [Google Scholar] [CrossRef]
- Sasaki, K.; Gotlib, J.R.; Mesa, R.A.; Newberry, K.J.; Ravandi, F.; Cortes, J.E.; Kelly, P.; Kutok, J.L.; Kantarjian, H.M.; Verstovsek, S. Phase II evaluation of IPI-926, an oral Hedgehog inhibitor, in patients with myelofibrosis. Leuk. Lymphoma 2015, 56, 2092–2097. [Google Scholar] [CrossRef]
- Bowles, D.W.; Keysar, S.B.; Eagles, J.R.; Wang, G.; Glogowska, M.J.; McDermott, J.D.; Le, P.N.; Gao, D.; Ray, C.E.; Rochon, P.J.; et al. A pilot study of cetuximab and the hedgehog inhibitor IPI-926 in recurrent/metastatic head and neck squamous cell carcinoma. Oral Oncol. 2016, 53, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, A.; Weiss, G.J.; Miller, W.H., Jr.; Gettinger, S.; Eigl, B.J.; Chang, A.L.; Dunbar, J.; Devens, S.; Faia, K.; Skliris, G.; et al. Phase I study of the Hedgehog pathway inhibitor IPI-926 in adult patients with solid tumors. Clin. Cancer Res. 2013, 19, 2766–2774. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Hong, H.; Kim, W.; Zhang, L.; Friedlander, T.W.; Fong, L.; Lin, A.M.; Small, E.J.; Wei, X.X.; Rodvelt, T.J.; et al. Itraconazole as a Noncastrating Treatment for Biochemically Recurrent Prostate Cancer: A Phase 2 Study. Clin. Genitourin. Cancer 2018, 7, e92–e96. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Kim, J.; Spaunhurst, K.; Montoya, J.; Khodosh, R.; Chandra, K.; Fu, T.; Gilliam, A.; Molgo, M.; Beachy, P.A.; et al. Open-label, exploratory phase II trial of oral itraconazole for the treatment of basal cell carcinoma. J. Clin. Oncol. 2014, 32, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Brahmer, J.R.; Juergens, R.A.; Hann, C.L.; Ettinger, D.S.; Sebree, R.; Smith, R.; Aftab, B.T.; Huang, P.; Liu, J.O. Phase 2 study of pemetrexed and itraconazole as second-line therapy for metastatic nonsquamous non-small-cell lung cancer. J. Thorac. Oncol. 2013, 8, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Heath, E.I.; Smith, D.C.; Rathkopf, D.; Blackford, A.L.; Danila, D.C.; King, S.; Frost, A.; Ajiboye, A.S.; Zhao, M.; et al. Repurposing itraconazole as a treatment for advanced prostate cancer: A noncomparative randomized phase II trial in men with metastatic castration-resistant prostate cancer. Oncologist 2013, 18, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.; Andre, V.; Ho, A.; Kudchadkar, R.; Migden, M.; Infante, J.; Tiu, R.V.; Pitou, C.; Tucker, T.; Brail, L.; et al. Phase I Study of LY2940680, a Smo Antagonist, in Patients with Advanced Cancer Including Treatment-Naive and Previously Treated Basal Cell Carcinoma. Clin. Cancer Res. 2018, 24, 2082–2091. [Google Scholar] [CrossRef]
- Savona, M.R.; Pollyea, D.A.; Stock, W.; Oehler, V.G.; Schroeder, M.A.; Lancet, J.; McCloskey, J.; Kantarjian, H.M.; Ma, W.W.; Shaik, M.N.; et al. Phase Ib Study of Glasdegib, a Hedgehog Pathway Inhibitor, in Combination with Standard Chemotherapy in Patients with AML or High-Risk MDS. Clin. Cancer Res. 2018, 24, 2294–2303. [Google Scholar] [CrossRef]
- Cortes, J.E.; Douglas Smith, B.; Wang, E.S.; Merchant, A.; Oehler, V.G.; Arellano, M.; DeAngelo, D.J.; Pollyea, D.A.; Sekeres, M.A.; Robak, T.; et al. Glasdegib in combination with cytarabine and daunorubicin in patients with AML or high-risk MDS: Phase 2 study results. Am. J. Hematol. 2018, 93, 1301–1310. [Google Scholar] [CrossRef]
- Wagner, A.J.; Messersmith, W.A.; Shaik, M.N.; Li, S.; Zheng, X.; McLachlan, K.R.; Cesari, R.; Courtney, R.; Levin, W.J.; El-Khoueiry, A.B. A phase I study of PF-04449913, an oral hedgehog inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2015, 21, 1044–1051. [Google Scholar] [CrossRef]
- Incardona, J.P.; Gaffield, W.; Kapur, R.P.; Roelink, H. The teratogenic Veratrum alkaloid cyclopamine inhibits sonic hedgehog signal transduction. Development 1998, 125, 3553–3562. [Google Scholar] [PubMed]
- Cooper, M.K.; Porter, J.A.; Young, K.E.; Beachy, P.A. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science 1998, 280, 1603–1607. [Google Scholar] [CrossRef] [PubMed]
- Taipale, J.; Chen, J.K.; Cooper, M.K.; Wang, B.; Mann, R.K.; Milenkovic, L.; Scott, M.P.; Beachy, P.A. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 2000, 406, 1005–1009. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, P.; Ruiz i Altaba, A. In vivo inhibition of endogenous brain tumors through systemic interference of Hedgehog signaling in mice. Mech. Dev. 2005, 122, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Karhadkar, S.S.; Bova, G.S.; Abdallah, N.; Dhara, S.; Gardner, D.; Maitra, A.; Isaacs, J.T.; Berman, D.M.; Beachy, P.A. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature 2004, 431, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Berman, D.M.; Karhadkar, S.S.; Maitra, A.; Montes De Oca, R.; Gerstenblith, M.R.; Briggs, K.; Parker, A.R.; Shimada, Y.; Eshleman, J.R.; Watkins, D.N.; et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003, 425, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Watkins, D.N.; Berman, D.M.; Burkholder, S.G.; Wang, B.; Beachy, P.A.; Baylin, S.B. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 2003, 422, 313–317. [Google Scholar] [CrossRef]
- Fan, Q.; Gu, D.; He, M.; Liu, H.; Sheng, T.; Xie, G.; Li, C.X.; Zhang, X.; Wainwright, B.; Garrossian, A.; et al. Tumor shrinkage by cyclopamine tartrate through inhibiting hedgehog signaling. Chin. J. Cancer 2011, 30, 472–481. [Google Scholar] [CrossRef]
- Alam, M.M.; Sohoni, S.; Kalainayakan, S.P.; Garrossian, M.; Zhang, L. Cyclopamine tartrate, an inhibitor of Hedgehog signaling, strongly interferes with mitochondrial function and suppresses aerobic respiration in lung cancer cells. BMC Cancer 2016, 16, 150. [Google Scholar] [CrossRef]
- Di Magno, L.; Coni, S.; Di Marcotullio, L.; Canettieri, G. Digging a hole under Hedgehog: Downstream inhibition as an emerging anticancer strategy. Biochim. Biophys. Acta 2015, 1856, 62–72. [Google Scholar] [CrossRef]
- Mohan, S.V.; Chang, J.; Li, S.; Henry, A.S.; Wood, D.J.; Chang, A.L. Increased Risk of Cutaneous Squamous Cell Carcinoma After Vismodegib Therapy for Basal Cell Carcinoma. JAMA Dermatol. 2016, 152, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, T.; Abrouk, M.; Sima, C.S.; Sadetsky, N.; Hou, J.; Caro, I.; Chren, M.M.; Arron, S.T. Risk of cutaneous squamous cell carcinoma after treatment of basal cell carcinoma with vismodegib. J. Am. Acad. Dermatol. 2017, 77, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.L.; Oro, A.E. Initial assessment of tumor regrowth after vismodegib in advanced Basal cell carcinoma. Arch. Dermatol. 2012, 148, 1324–1325. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; Tawbi, H.A.; Thomas, A.L.; Stoller, R.G.; Turtschi, C.P.; Baselga, J.; Sarantopoulos, J.; Mahalingam, D.; Shou, Y.; Moles, M.A.; et al. A phase I, multicenter, open-label, first-in-human, dose-escalation study of the oral smoothened inhibitor Sonidegib (LDE225) in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 1900–1909. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Lefrancois, P. Efficacy, safety, and comparison of sonic hedgehog inhibitors in basal cell carcinomas: A systematic review and meta-analysis. J. Am. Acad. Dermatol. 2018, 79, 1089–1100. [Google Scholar] [CrossRef]
- Danial, C.; Sarin, K.Y.; Oro, A.E.; Chang, A.L. An Investigator-Initiated Open-Label Trial of Sonidegib in Advanced Basal Cell Carcinoma Patients Resistant to Vismodegib. Clin. Cancer Res. 2016, 22, 1325–1329. [Google Scholar] [CrossRef] [PubMed]
- Della Corte, C.M.; Bellevicine, C.; Vicidomini, G.; Vitagliano, D.; Malapelle, U.; Accardo, M.; Fabozzi, A.; Fiorelli, A.; Fasano, M.; Papaccio, F.; et al. SMO gene amplification and activation of the hedgehog pathway as novel mechanisms of resistance to anti-epidermal growth factor receptor drugs in human lung cancer. Clin. Cancer Res. 2015, 21, 4686–4697. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Rodova, M.; Nanta, R.; Meeker, D.; Van Veldhuizen, P.J.; Srivastava, R.K.; Shankar, S. NPV-LDE-225 (Erismodegib) inhibits epithelial mesenchymal transition and self-renewal of glioblastoma initiating cells by regulating miR-21, miR-128, and miR-200. Neuro Oncol. 2013, 15, 691–706. [Google Scholar] [CrossRef]
- D’Amato, C.; Rosa, R.; Marciano, R.; D’Amato, V.; Formisano, L.; Nappi, L.; Raimondo, L.; Di Mauro, C.; Servetto, A.; Fulciniti, F.; et al. Inhibition of Hedgehog signalling by NVP-LDE225 (Erismodegib) interferes with growth and invasion of human renal cell carcinoma cells. Br. J. Cancer 2014, 111, 1168–1179. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef]
- Wang, C.; Zhu, M.; Lu, X.; Wang, H.; Zhao, W.; Zhang, X.; Dong, X. Synthesis and evaluation of novel dimethylpyridazine derivatives as hedgehog signaling pathway inhibitors. Bioorg. Med. Chem. 2018, 26, 3308–3320. [Google Scholar] [CrossRef] [PubMed]
- Munchhof, M.J.; Li, Q.; Shavnya, A.; Borzillo, G.V.; Boyden, T.L.; Jones, C.S.; LaGreca, S.D.; Martinez-Alsina, L.; Patel, N.; Pelletier, K.; et al. Discovery of PF-04449913, a Potent and Orally Bioavailable Inhibitor of Smoothened. ACS Med. Chem. Lett. 2012, 3, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Chaudhry, P.; Merchant, A.A. Primary cilia are present on human blood and bone marrow cells and mediate Hedgehog signaling. Exp. Hematol. 2016, 44, 1181–1187 e1182. [Google Scholar] [CrossRef] [PubMed]
- Sadarangani, A.; Pineda, G.; Lennon, K.M.; Chun, H.J.; Shih, A.; Schairer, A.E.; Court, A.C.; Goff, D.J.; Prashad, S.L.; Geron, I.; et al. GLI2 inhibition abrogates human leukemia stem cell dormancy. J. Transl. Med. 2015, 13, 98. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, N.; Minami, Y.; Kakiuchi, S.; Kuwatsuka, Y.; Hayakawa, F.; Jamieson, C.; Kiyoi, H.; Naoe, T. Small-molecule Hedgehog inhibitor attenuates the leukemia-initiation potential of acute myeloid leukemia cells. Cancer Sci. 2016, 107, 1422–1429. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Tang, J.Y.; Gong, R.; Kim, J.; Lee, J.J.; Clemons, K.V.; Chong, C.R.; Chang, K.S.; Fereshteh, M.; Gardner, D.; et al. Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell 2010, 17, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Tsubamoto, H.; Sonoda, T.; Yamasaki, M.; Inoue, K. Impact of combination chemotherapy with itraconazole on survival for patients with recurrent or persistent ovarian clear cell carcinoma. Anticancer Res. 2014, 34, 2007–2014. [Google Scholar] [PubMed]
- Tsubamoto, H.; Sonoda, T.; Inoue, K. Impact of itraconazole on the survival of heavily pre-treated patients with triple-negative breast cancer. Anticancer Res. 2014, 34, 3839–3844. [Google Scholar]
- Tsubamoto, H.; Sonoda, T.; Ikuta, S.; Tani, S.; Inoue, K.; Yamanaka, N. Combination chemotherapy with itraconazole for treating metastatic pancreatic cancer in the second-line or additional setting. Anticancer Res. 2015, 35, 4191–4196. [Google Scholar]
- Tsubamoto, H.; Sonoda, T.; Ikuta, S.; Tani, S.; Inoue, K.; Yamanaka, N. impact of itraconazole after first-line chemotherapy on survival of patients with metastatic biliary tract cancer. Anticancer Res. 2015, 35, 4923–4927. [Google Scholar]
- Yauch, R.L.; Dijkgraaf, G.J.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 2009, 326, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Dijkgraaf, G.J.; Alicke, B.; Weinmann, L.; Januario, T.; West, K.; Modrusan, Z.; Burdick, D.; Goldsmith, R.; Robarge, K.; Sutherlin, D.; et al. Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 2011, 71, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Buonamici, S.; Williams, J.; Morrissey, M.; Wang, A.; Guo, R.; Vattay, A.; Hsiao, K.; Yuan, J.; Green, J.; Ospina, B.; et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci. Transl. Med. 2010, 2, 51ra70. [Google Scholar] [CrossRef] [PubMed]
- Lauth, M.; Bergstrom, A.; Shimokawa, T.; Toftgard, R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Walter, V.; Hayes, D.N.; Onaitis, M. Hedgehog-GLI signaling inhibition suppresses tumor growth in squamous lung cancer. Clin. Cancer Res. 2014, 20, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Latuske, E.M.; Stamm, H.; Klokow, M.; Vohwinkel, G.; Muschhammer, J.; Bokemeyer, C.; Jucker, M.; Kebenko, M.; Fiedler, W.; Wellbrock, J. Combined inhibition of GLI and FLT3 signaling leads to effective anti-leukemic effects in human acute myeloid leukemia. Oncotarget 2017, 8, 29187–29201. [Google Scholar] [CrossRef]
- Srivastava, R.K.; Kaylani, S.Z.; Edrees, N.; Li, C.; Talwelkar, S.S.; Xu, J.; Palle, K.; Pressey, J.G.; Athar, M. GLI inhibitor GANT-61 diminishes embryonal and alveolar rhabdomyosarcoma growth by inhibiting Shh/AKT-mTOR axis. Oncotarget 2014, 5, 12151–12165. [Google Scholar] [CrossRef]
- Wickstrom, M.; Dyberg, C.; Shimokawa, T.; Milosevic, J.; Baryawno, N.; Fuskevag, O.M.; Larsson, R.; Kogner, P.; Zaphiropoulos, P.G.; Johnsen, J.I. Targeting the hedgehog signal transduction pathway at the level of GLI inhibits neuroblastoma cell growth in vitro and in vivo. Int. J. Cancer 2013, 132, 1516–1524. [Google Scholar] [CrossRef]
- Benvenuto, M.; Masuelli, L.; De Smaele, E.; Fantini, M.; Mattera, R.; Cucchi, D.; Bonanno, E.; Di Stefano, E.; Frajese, G.V.; Orlandi, A.; et al. In vitro and in vivo inhibition of breast cancer cell growth by targeting the Hedgehog/GLI pathway with SMO (GDC-0449) or GLI (GANT-61) inhibitors. Oncotarget 2016, 7, 9250–9270. [Google Scholar] [CrossRef]
- Mazumdar, T.; Sandhu, R.; Qadan, M.; DeVecchio, J.; Magloire, V.; Agyeman, A.; Li, B.; Houghton, J.A. Hedgehog signaling regulates telomerase reverse transcriptase in human cancer cells. PLoS ONE 2013, 8, e75253. [Google Scholar] [CrossRef]
- Yan, M.; Wang, L.; Zuo, H.; Zhang, Z.; Chen, W.; Mao, L.; Zhang, P. HH/GLI signalling as a new therapeutic target for patients with oral squamous cell carcinoma. Oral Oncol. 2011, 47, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Peng, X.; Yuan, X.; Huang, D.; Chen, J.; Lu, Q.; Lv, N.; Luo, S. Suppression of growth and migration by blocking the Hedgehog signaling pathway in gastric cancer cells. Cell. Oncol. 2013, 36, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, T.; DeVecchio, J.; Shi, T.; Jones, J.; Agyeman, A.; Houghton, J.A. Hedgehog signaling drives cellular survival in human colon carcinoma cells. Cancer Res. 2011, 71, 1092–1102. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Rodova, M.; Roy, S.K.; Sharma, J.; Singh, K.P.; Srivastava, R.K.; Shankar, S. GANT-61 inhibits pancreatic cancer stem cell growth in vitro and in NOD/SCID/IL2R gamma null mice xenograft. Cancer Lett. 2013, 330, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Samarzija, I.; Beard, P. Hedgehog pathway regulators influence cervical cancer cell proliferation, survival and migration. Biochem. Biophys. Res. Commun. 2012, 425, 64–69. [Google Scholar] [CrossRef]
- Santini, R.; Pietrobono, S.; Pandolfi, S.; Montagnani, V.; D’Amico, M.; Penachioni, J.Y.; Vinci, M.C.; Borgognoni, L.; Stecca, B. SOX2 regulates self-renewal and tumorigenicity of human melanoma-initiating cells. Oncogene 2014, 33, 4697–4708. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, A.N.; Bernardazzi, C.; Carneiro, A.J.; Elia, C.C.; Martinusso, C.A.; Ventura, G.M.; Castelo-Branco, M.T.; de Souza, H.S. Hedgehog pathway signaling regulates human colon carcinoma HT-29 epithelial cell line apoptosis and cytokine secretion. PLoS ONE 2012, 7, e45332. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Chen, Z. Differentiation and apoptosis induction therapy in acute promyelocytic leukaemia. Lancet Oncol. 2000, 1, 101–106. [Google Scholar] [CrossRef]
- Kim, J.; Lee, J.J.; Kim, J.; Gardner, D.; Beachy, P.A. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc. Natl. Acad. Sci. USA 2010, 107, 13432–13437. [Google Scholar] [CrossRef]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glassberg, M.K.; Kardatzke, D.; King, T.E., Jr.; Lancaster, L.; Sahn, S.A.; Szwarcberg, J.; et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): Two randomised trials. Lancet 2011, 377, 1760–1769. [Google Scholar] [CrossRef]
- Didiasova, M.; Singh, R.; Wilhelm, J.; Kwapiszewska, G.; Wujak, L.; Zakrzewicz, D.; Schaefer, L.; Markart, P.; Seeger, W.; Lauth, M.; et al. Pirfenidone exerts antifibrotic effects through inhibition of GLI transcription factors. FASEB J. 2017, 31, 1916–1928. [Google Scholar] [CrossRef] [PubMed]
- Polydorou, C.; Mpekris, F.; Papageorgis, P.; Voutouri, C.; Stylianopoulos, T. Pirfenidone normalizes the tumor microenvironment to improve chemotherapy. Oncotarget 2017, 8, 24506–24517. [Google Scholar] [CrossRef]
- Iwata, T.; Yoshida, S.; Fujiwara, T.; Wada, H.; Nakajima, T.; Suzuki, H.; Yoshino, I. Effect of Perioperative Pirfenidone Treatment in Lung Cancer Patients With Idiopathic Pulmonary Fibrosis. Ann. Thorac. Surg. 2016, 102, 1905–1910. [Google Scholar] [CrossRef]
- Kozono, S.; Ohuchida, K.; Eguchi, D.; Ikenaga, N.; Fujiwara, K.; Cui, L.; Mizumoto, K.; Tanaka, M. Pirfenidone inhibits pancreatic cancer desmoplasia by regulating stellate cells. Cancer Res. 2013, 73, 2345–2356. [Google Scholar] [CrossRef] [PubMed]
- Mediavilla-Varela, M.; Boateng, K.; Noyes, D.; Antonia, S.J. The anti-fibrotic agent pirfenidone synergizes with cisplatin in killing tumor cells and cancer-associated fibroblasts. BMC Cancer 2016, 16, 176. [Google Scholar] [CrossRef] [PubMed]
- Burghardt, I.; Tritschler, F.; Opitz, C.A.; Frank, B.; Weller, M.; Wick, W. Pirfenidone inhibits TGF-beta expression in malignant glioma cells. Biochem. Biophys. Res. Commun. 2007, 354, 542–547. [Google Scholar] [CrossRef]
- Zou, W.J.; Huang, Z.; Jiang, T.P.; Shen, Y.P.; Zhao, A.S.; Zhou, S.; Zhang, S. Pirfenidone Inhibits Proliferation and Promotes Apoptosis of Hepatocellular Carcinoma Cells by Inhibiting the Wnt/beta-Catenin Signaling Pathway. Med. Sci. Monit. 2017, 23, 6107–6113. [Google Scholar] [CrossRef]
- Hyman, J.M.; Firestone, A.J.; Heine, V.M.; Zhao, Y.; Ocasio, C.A.; Han, K.; Sun, M.; Rack, P.G.; Sinha, S.; Wu, J.J.; et al. Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc. Natl. Acad. Sci. USA 2009, 106, 14132–14137. [Google Scholar] [CrossRef]
- Li, B.; Fei, D.L.; Flaveny, C.A.; Dahmane, N.; Baubet, V.; Wang, Z.; Bai, F.; Pei, X.H.; Rodriguez-Blanco, J.; Hang, B.; et al. Pyrvinium attenuates Hedgehog signaling downstream of smoothened. Cancer Res. 2014, 74, 4811–4821. [Google Scholar] [CrossRef]
- Wolff, F.; Loipetzberger, A.; Gruber, W.; Esterbauer, H.; Aberger, F.; Frischauf, A.M. Imiquimod directly inhibits Hedgehog signalling by stimulating adenosine receptor/protein kinase A-mediated GLI phosphorylation. Oncogene 2013, 32, 5574–5581. [Google Scholar] [CrossRef] [PubMed]
- Nayak, A.; Satapathy, S.R.; Das, D.; Siddharth, S.; Tripathi, N.; Bharatam, P.V.; Kundu, C. Nanoquinacrine induced apoptosis in cervical cancer stem cells through the inhibition of hedgehog-GLI1 cascade: Role of GLI-1. Sci. Rep. 2016, 6, 20600. [Google Scholar] [CrossRef]
- Long, J.; Li, B.; Rodriguez-Blanco, J.; Pastori, C.; Volmar, C.-H.; Wahlestedt, C.; Capobianco, A.; Bai, F.; Pei, X.-H.; Ayad, N.G.; et al. The BET Bromodomain Inhibitor I-BET151 Acts Downstream of Smoothened Protein to Abrogate the Growth of Hedgehog Protein-driven Cancers. J. Biol. Chem. 2014, 289, 35494–35502. [Google Scholar] [CrossRef]
- Tang, Y.; Gholamin, S.; Schubert, S.; Willardson, M.I.; Lee, A.; Bandopadhayay, P.; Bergthold, G.; Masoud, S.; Nguyen, B.; Vue, N.; et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat. Med. 2014, 20, 732–740. [Google Scholar] [CrossRef]
- Pan, D.; Li, Y.; Li, Z.; Wang, Y.; Wang, P.; Liang, Y. Gli inhibitor GANT61 causes apoptosis in myeloid leukemia cells and acts in synergy with rapamycin. Leuk. Res. 2012, 36, 742–748. [Google Scholar] [CrossRef]
- Zuo, M.; Rashid, A.; Churi, C.; Vauthey, J.N.; Chang, P.; Li, Y.; Hung, M.C.; Li, D.; Javle, M. Novel therapeutic strategy targeting the Hedgehog signalling and mTOR pathways in biliary tract cancer. Br. J. Cancer 2015, 112, 1042–1051. [Google Scholar] [CrossRef]
- Zhou, J.; Wu, K.; Gao, D.; Zhu, G.; Wu, D.; Wang, X.; Chen, Y.; Du, Y.; Song, W.; Ma, Z.; et al. Reciprocal regulation of hypoxia-inducible factor 2alpha and GLI1 expression associated with the radioresistance of renal cell carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2014, 90, 942–951. [Google Scholar] [CrossRef]
- Zeng, J.; Aziz, K.; Chettiar, S.T.; Aftab, B.T.; Armour, M.; Gajula, R.; Gandhi, N.; Salih, T.; Herman, J.M.; Wong, J.; et al. Hedgehog pathway inhibition radiosensitizes non-small cell lung cancers. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 143–149. [Google Scholar] [CrossRef]
- Ally, M.S.; Ransohoff, K.; Sarin, K.; Atwood, S.X.; Rezaee, M.; Bailey-Healy, I.; Kim, J.; Beachy, P.A.; Chang, A.L.S.; Oro, A.; et al. Effects of combined treatment with arsenic trioxide and itraconazole in patients with refractory metastatic basal cell carcinoma. JAMA Dermatol. 2016, 152, 452–456. [Google Scholar] [CrossRef]


{kind=link}
| Study | Phase | Type of Cancer | Experimental Arm | Control Arm | Results of Primary EP |
|---|---|---|---|---|---|
| NCT02639117 | Phase 1 | Multiple BCC | Vismodegib + photodynamic therapy sessions + topical application of 20% 5-aminolevulinic acid (ALA) | Combination PDT-vismodegib therapy was overall well tolerated (50% dysgeusia, 50% myalgia, 75% flu-like symptoms) [54]. | |
| STEVIE NCT01367665 | Phase 2 | Locally advanced and metastatic BCC | Vismodegib | Serious side effects (grade ≥ 3) in 289 patients (23.8%) and death in 46 patients (3.8%) [55]. | |
| NCT01546519 | Phase 1b | Advanced solid malignancies and hepatic impairment | Vismodegib | 96.8% in all groups, experienced at least one AE. 67.7% of all AEs reported were grade 3 or 4 [56]. | |
| ERIVANCE BCC NCT00833417 | Phase 2 | Locally advanced and metastatic BCC | Vismodegib | ORR of 60.3% in patients with locally advanced BCC and 48.5% metastatic BCC [57]. | |
| MIKIE NCT01815840 | Phase 2 | Multiple BCC | A. Vismodegib 12 w - placebo 8 w - vismodegib 12 w B. Vismodegib 24 w - placebo 8 w - vismodegib 8 w | The mean number of BCC lesions at week 73 was reduced from baseline by 62.7% in group A and 54% in group B [58]. | |
| NCT00957229 | Phase 2 | Basal cell nevus syndrome (BCNS) | Vismodegib | Placebo | Reduced rate of new surgically eligible BCC (2 vs 34 per patient per year) [59]. |
| NCT02115828 | Phase 2 | Metastatic castration-resistant prostate cancer | Vismodegib | Gli1 mRNA was significantly suppressed by vismodegib in both tumor tissue (57%) and benign skin biopsies (75%) [60]. | |
| NCT01631331 | Phase 1 | BCC | Neoadjuvant vismodegib | Reduction of the final surgical defect size by 34.8% compared with baseline [61]. | |
| E1508 NCT00887159 | Phase 2 | Extensive stage small cell lung carcinoma | A. Cisplatin + etoposide B. Vismodegib C. Cixutumumab | The median PFS times in arms A, B, and C were 4.4, 4.4, and 4.6 months, respectively [62]. | |
| VISMOLY NCT01944943 | Phase 2 | Refractory or relapsed B-cell lymphoma or chronic lymphocytic leukemia | Vismodegib | The best overall response: DLBCL: 0 (0%), iNHL: 1 (16.7%), PCNSL: 0 (0%), CLL: (0%), all: 1 (3.2%) [63]. | |
| NCT01064622 | Phase 1b/2 | Metastatic pancreatic cancer | Gemcitabine + vismodegib | Gemcitabine plus Placebo | Median PFS was 4.0 and 2.5 months for GV and GP arms, respectively [64] |
| NCT01201915 | Phase 2 | BCC | Neoadjuvant vismodegib
| Complete histologic clearance was achieved by 42%, 16%, and 44% of patients in cohorts 1, 2, and 3, respectively [65]. | |
| NCT01195415 | Phase 2 | Metastatic pancreatic adenocarcinoma | Vismodegib plus gemcitabine | GLI1 and PTCH1 decreased in 95.6% and 82.6%, respectively [66]. | |
| NCT01267955 | Phase 2 | Advanced chondrosarcoma | Vismodegib | The 6-month clinical benefit rate was 25.6% [67]. | |
| NCT00822458 | Phase 1 | Medulloblastoma | Vismodegib | 3 dose-limiting toxicities but no drug-related bone toxicity. The median vismodegib penetration in the CSF was 0.53 (ratio of the concentration of vismodegib in the CSF to that of the unbound drug in plasma) [68]. | |
| NCT00607724 | Phase 1 | BCC | Vismodegib | SUVmax decreased (median 33%, SD ± 45%) with metabolic activity normalizing or disappearing in 42% of lesions [69] | |
| NCT00636610 | Phase 2 | Metastatic colorectal cancer | Vismodegib + FOLFOX or FOLFIRI + bevacizumab | Placebo + FOLFOX or FOLFIRI + bevacizumab | Median PFS hazard ratio (HR) was 1.25 [70]. |
| NCT01209143 | Phase 1b | Solid cancers |
| Systemic exposure of rosiglitazone or oral contraceptive is not altered with concomitant vismodegib [71]. | |
| NCT00739661 | Phase 2 | Ovarian cancer | Vismodegib | Placebo | Median PFS in vismodegib and placebo groups were 7.5 months and 5.8 months, respectively [72]. |
| NCT00607724 | Phase 1 | Solid tumor | Vismodegib | 8 grade 3 adverse events in 6 patients. 1 patient withdrew from the study because of adverse events [73] | |
| BOLT NCT01327053 | Phase 2 | BCC | Sonidegib 200 mg and 800 mg | Sonidegib 200 mg (approved dose), objective response rates were 56.1% (central) and 71.2% (investigator) in laBCC and 7.7% (central) and 23.1% (investigator) in mBCC [74]. | |
| NCT01125800 | Phase 1/2 | Medulloblastoma, rhabdomyosarcoma, neuroblastoma, hepatoblastoma, glioma, astrocytoma | Sonidegib 233 mg/m2 daily; 372 mg/m2 daily; 425 mg/m2 daily; 680 mg/m2 daily; 800 mg/m2 daily | The recommended phase II dose in pediatric patients was 680 mg/m2 once daily. The results were 4 complete responders (2 pediatric and 2 adult) and 1 partial response (adult) [75]. | |
| NCT01954355 | Phase 1 | Solid tumorovarian cancer | Sonidegib 400, 600 and 800 mg + paclitaxel | The recommended phase II dose was 800 mg in combination with paclitaxel [76]. | |
| NCT01208831 | Phase 1 | Advanced solid tumor cancers, medulloblastoma, BCC | Sonidegib | The recommended dose in East Asian patients (400 mg) was lower than in patients from Europe and the USA (800 mg and 250 mg, respectively, twice daily) [77]. | |
| NCT01579929 | Phase 1 | Lung cancer | Sonidegib + etoposide + cisplatin | Sonidegib 800 mg daily was the maximum tolerated dose when administered with EP [78]. | |
| NCT01383538 | Phase 1 | Advanced pancreatic adenocarcinoma | Saridegib + FOLFIRINOX | The combination was active and safe [79]. | |
| NCT01371617 | Phase 2 | Primary myelofibrosis | Saridegib | Nine out of fourteen patients (79%) did not respond. Saridegib is not active in myelofibrosis as monotherapy [80]. | |
| NCT01255800 | Phase 1 | Recurrent head and neck cancer | Saridegib + cetuximab | The recommended phase 2 dose was 160 mg, the same as the single-agent saridegib maximum tolerated dose [81]. | |
| NCT00761696 | Phase 1 | Solid tumor | Saridegib | The maximum tolerated dose (MTD) of saridegib was 160 mg QD within 28-day cycles [82]. | |
| NCT01787331 | Phase 2 | Biochemically relapsed prostate cancer | Itraconazole | One patient (5%) had a > 50% PSA decline [83]. | |
| NCT01108094 | Phase 2 | BCC | Itraconazole | Itraconazole reduced cell proliferation by 45%, HH pathway activity by 65% and reduced tumor area by 24% [84]. | |
| NCT00769600 | Phase 2 | Recurrent non-small cell lung cancer | Itraconazole + pemetrexed | Pemetrexed | Median PFS was 5.5 months (itraconazole) versus 2.8 months (control). There were no evident differences in toxicity between the study arms [85]. |
| NCT00887458 | Phase 2 | Metastatic castration-resistant prostate cancer | Itraconazole 200 mg daily and 600 mg daily | The PSA PFS rates at 24 weeks were 11.8% in the low-dose arm and 48.0% in the high-dose arm [86]. | |
| NCT01919398 | Phase 1 | Metastatic solid tumor | Taladegib | No dose-limiting toxicities were observed at doses of 100 mg or 200 mg; 3 of the 9 patients evaluable for DLTs at the 400 mg dose level experienced DLTs [83]. | |
| NCT01226485 | Phase 1 | Advanced cancer | Taladegib | The maximum tolerable dose was 400 mg [87]. | |
| NCT01546038 | Phase 1/2 | Acute myeloid leukemia High-risk myelodysplastic syndromes | A: Glasdegib + low-dose ARA-C B: Glasdegib + decitabine C: Glasdegib + daunorubicin + cytarabine | No dose-limiting toxicities (DLT) were observed in arms A and B; 1 DLT (grade 4 neuropathy) occurred in arm C. 46.4% of patient achieved investigator-reported. Among patients ≥55 years old (n = 60), 40.0% achieved complete remission [88,89]. | |
| NCT01286467 | Phase 1 | Solid tumors | Glasdegib | The first-cycle DLT rate at the 640 mg dose level was 33.3%, and the O maximum tolerable dose was estimated to be 320 mg once daily [90]. |
| Study | Phase | Type of Cancer | Experimental Arm | Control Arm | Status |
|---|---|---|---|---|---|
| NCT02138929 | Phase 1 | Advanced gastroesophageal cancer | Sonidegib + everolimus | Active, not recruiting | |
| NCT01485744 | Phase 1 | Advanced pancreatic adenocarcinoma | Sonidegib + FOLFIRINOX | Active, not recruiting | |
| NCT01431794 | Phase 1/2 | Pancreatic | Neoadjuvant gemcitabine, nab-paclitaxel and sonidegib | Neoadjuvant gemcitabine, nab-paclitaxel | Active, not recruiting |
| NCT02151864 | Phase 1 | Hepatocellular carcinoma | Sonidegib in patients intolerant to sorafenib | Active, not recruiting | |
| NCT02303041 | Phase 2 | Metastatic basal cell cancer | Sonidegib + buparlisib | Completed | |
| NCT01576666 | Phase 1 | Advanced solid tumors | Sonidegib + buparlisib | Completed | |
| NCT03434262 | Phase 1 | Recurrent brain tumors | Sonidegib among others | Recruiting | |
| NCT01787331 | Phase 2 | Biochemically relapsed prostate cancer | Itraconazole | Active, not recruiting | |
| NCT02735356 | Early phase 1 | Basal cell cancer | Topical itraconazole | Active, not recruiting | |
| NCT02357836 | Early phase 1 | Non-small cell lung cancer | Neoadjuvant itraconazole | Recruiting | |
| NCT02749513 | Early phase 1 | Esophagus | Itraconazole | Recruiting | |
| NCT01835626 | Phase 2 | Basal cell cancer | Vismodegib + radiotherapy | Recruiting | |
| NCT03052478 | Phase 2 | Advanced gastric cancer | Vismodegib | Recruiting | |
| NCT01878617 | Phase 2 | Medulloblastoma | Vismodegib among others | Recruiting | |
| NCT03035188 | Phase 2 | Basal cell cancer | Neoadjuvant vismodegib | Recruiting | |
| NCT02694224 | Phase 2 | Triple-negative breast cancer | Neoadjuvant vismodegib + paclitaxel, epirubicin and cyclophosphamide | Neoadjuvant paclitaxel, epirubicin and cyclophosphamide | Recruiting |
| NCT01791894 | Phase 1/2 | Basal cell cancer | Arsenic trioxide | Completed | |
| NCT01470248 | Phase 2 | Advanced small cell lung cancer | Arsenic trioxide | Completed | |
| NCT03503864 | Phase 2 | Advanced neuroblastoma | Arsenic trioxide + conventional induction chemotherapy | Conventional induction chemotherapy | Recruiting |
| NCT03466450 | Phase 1/2 | Glioblastoma | Glasdegib + temozolomide | Recruiting | |
| NCT02530437 | Phase 1/2 | Localized esophageal or gastroesophageal junction cancer | Taladegib + carboplatin, paclitaxel and radiation | Active, not recruiting | |
| NCT01130142 | Phase 1/2 | Metastatic pancreatic adenocarcinoma | Saridegib + gemcitabine | Gemcitabine | Completed |
| NCT01383538 | Phase 1 | Metastatic pancreatic adenocarcinoma | Saridegib + FOLFIRINOX | Completed | |
| NCT01310816 | Phase 2 | Metastatic or locally advanced chondrosarcoma | Saridegib | Placebo | Completed |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Girardi, D.; Barrichello, A.; Fernandes, G.; Pereira, A. Targeting the Hedgehog Pathway in Cancer: Current Evidence and Future Perspectives. Cells 2019, 8, 153. https://doi.org/10.3390/cells8020153
Girardi D, Barrichello A, Fernandes G, Pereira A. Targeting the Hedgehog Pathway in Cancer: Current Evidence and Future Perspectives. Cells. 2019; 8(2):153. https://doi.org/10.3390/cells8020153
Chicago/Turabian StyleGirardi, Daniel, Adriana Barrichello, Gustavo Fernandes, and Allan Pereira. 2019. "Targeting the Hedgehog Pathway in Cancer: Current Evidence and Future Perspectives" Cells 8, no. 2: 153. https://doi.org/10.3390/cells8020153
APA StyleGirardi, D., Barrichello, A., Fernandes, G., & Pereira, A. (2019). Targeting the Hedgehog Pathway in Cancer: Current Evidence and Future Perspectives. Cells, 8(2), 153. https://doi.org/10.3390/cells8020153