Expression of Translocator Protein and [18F]-GE180 Ligand Uptake in Multiple Sclerosis Animal Models


 ,
,  ,
, 
Abstract
:1. Introduction
2. Material and Methods
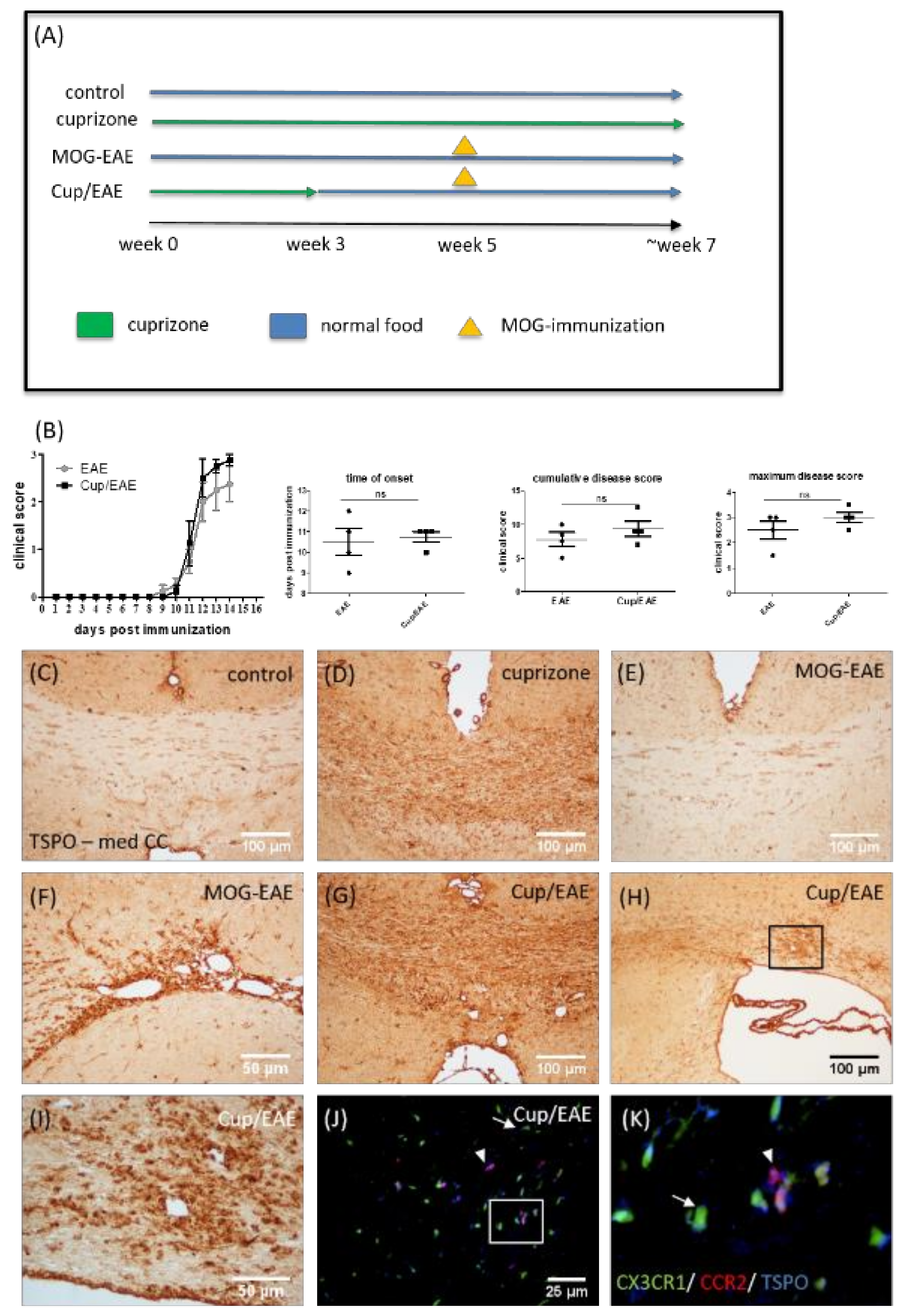
2.1. Animals and Experimental Groups
2.2. EAE and Disease Scoring
2.3. Positron Emission Tomography (PET)—Imaging
2.4. Gene Expression Studies
2.5. Tissue Preparation
2.6. Immunohistochemistry
2.7. Immunofluorescence Labeling
2.8. Ultrastructural Analysis via Serial Block-Face Scanning Electron Microscopy
2.9. Statistical Analysis
3. Results
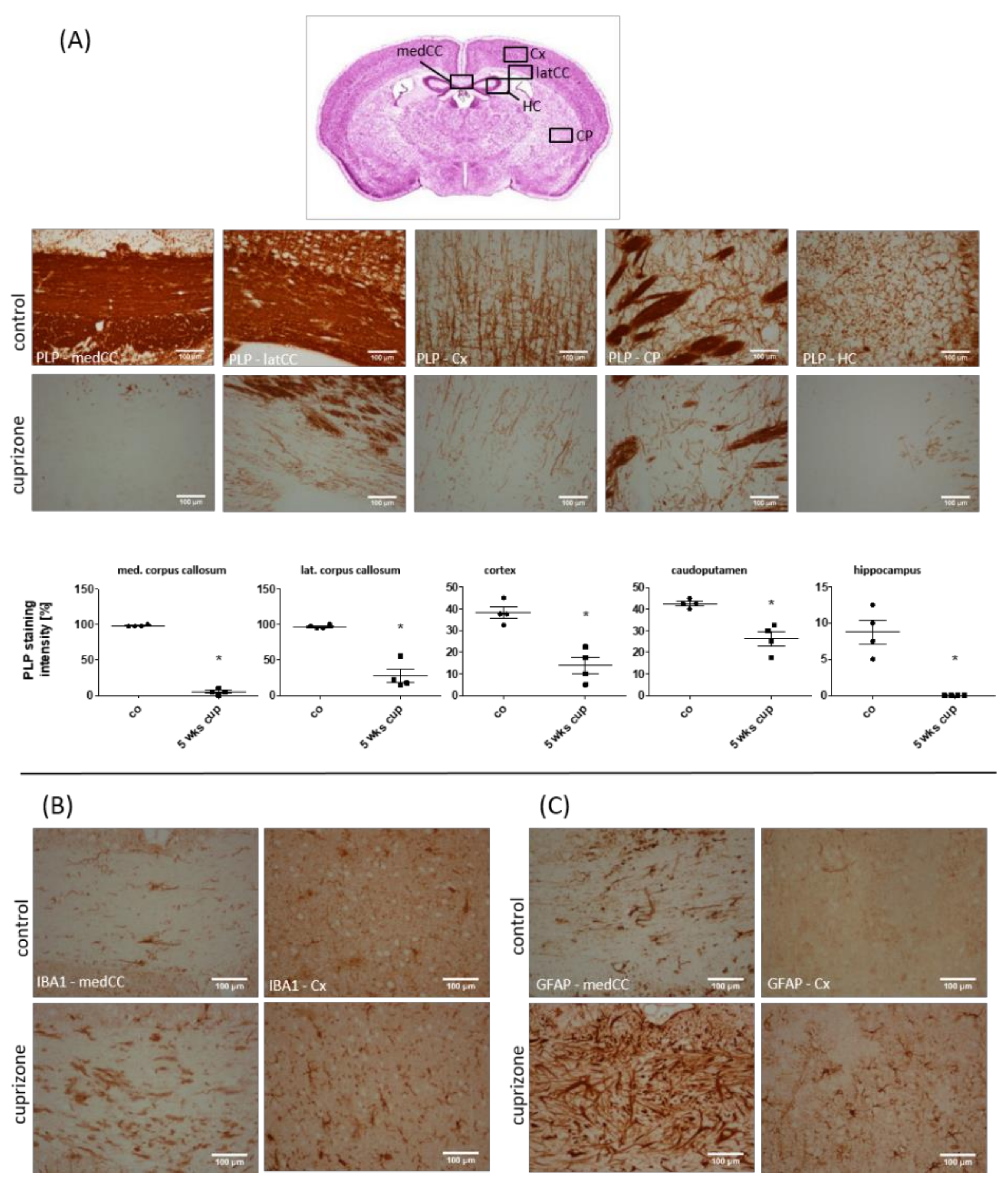
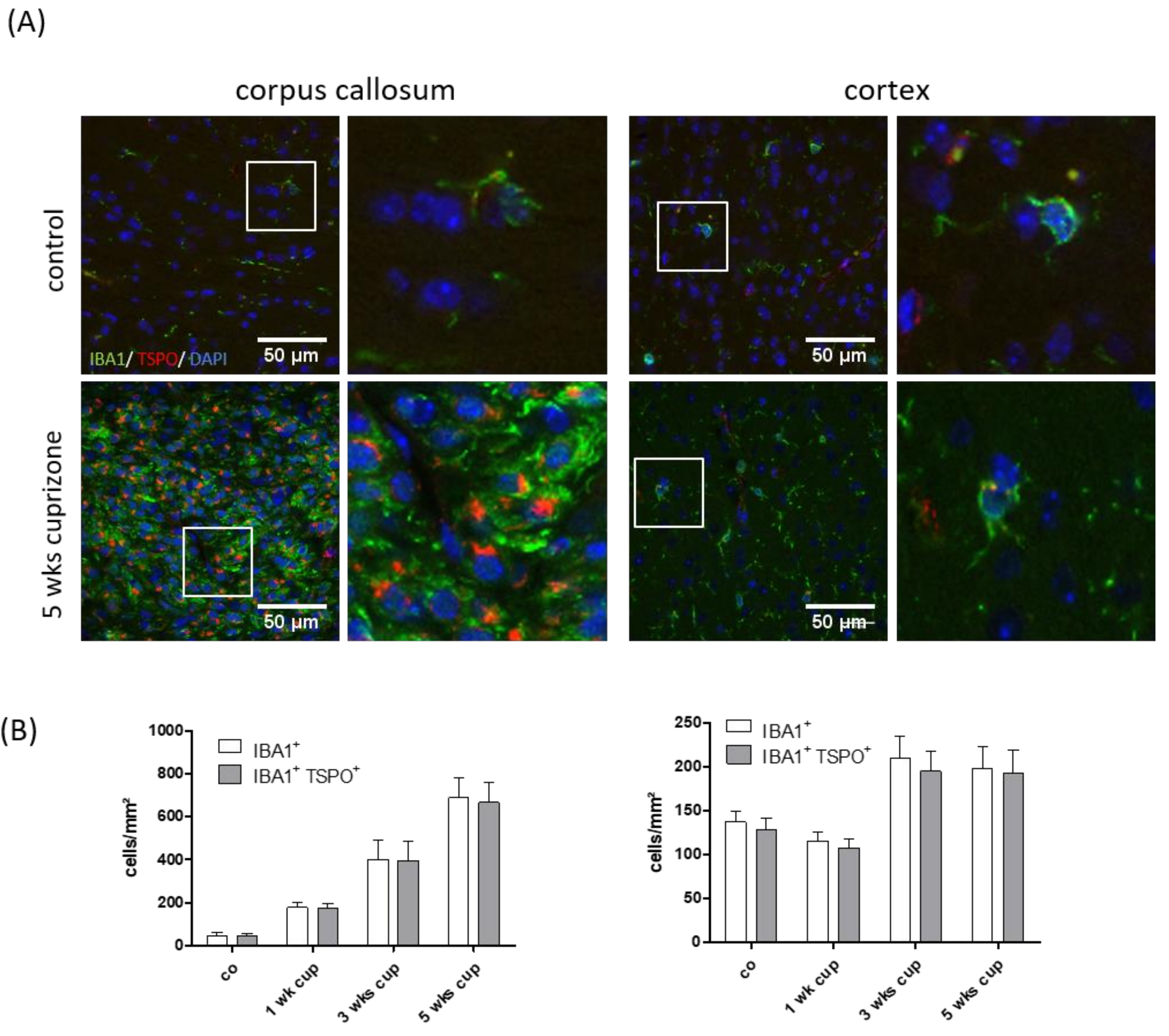
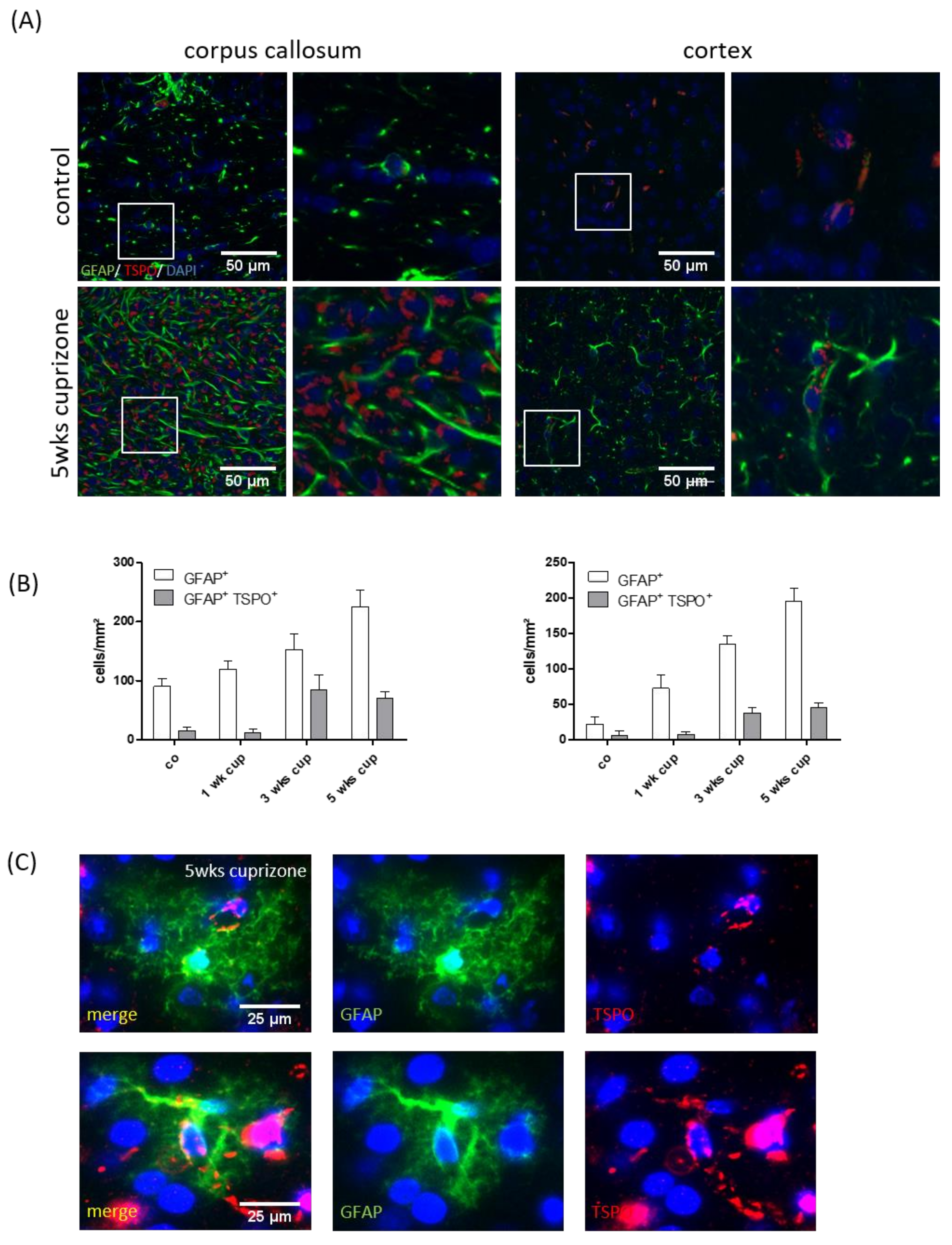
3.1. Cuprizone Intoxication Induces Reproducible Demyelination and Glia Activation
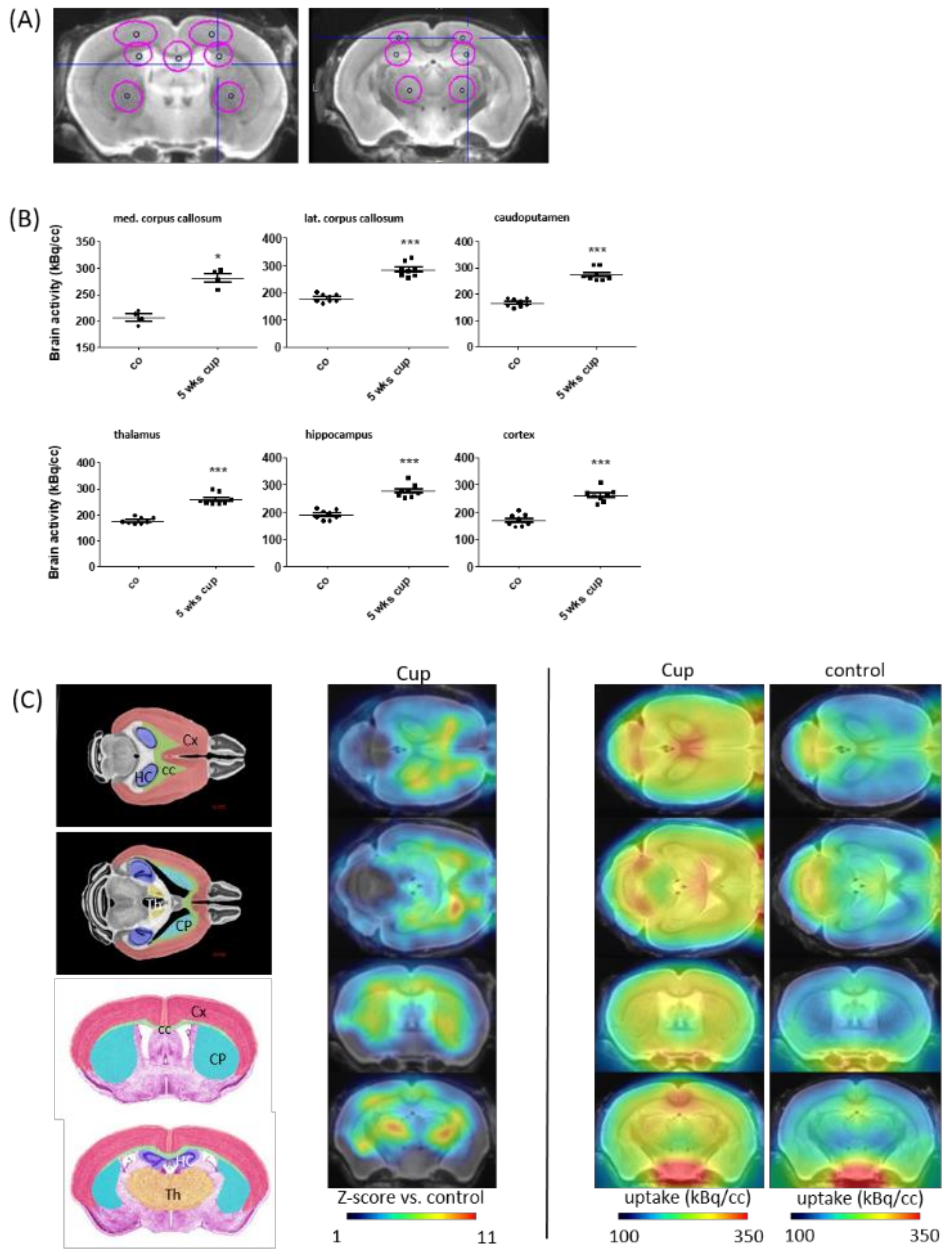
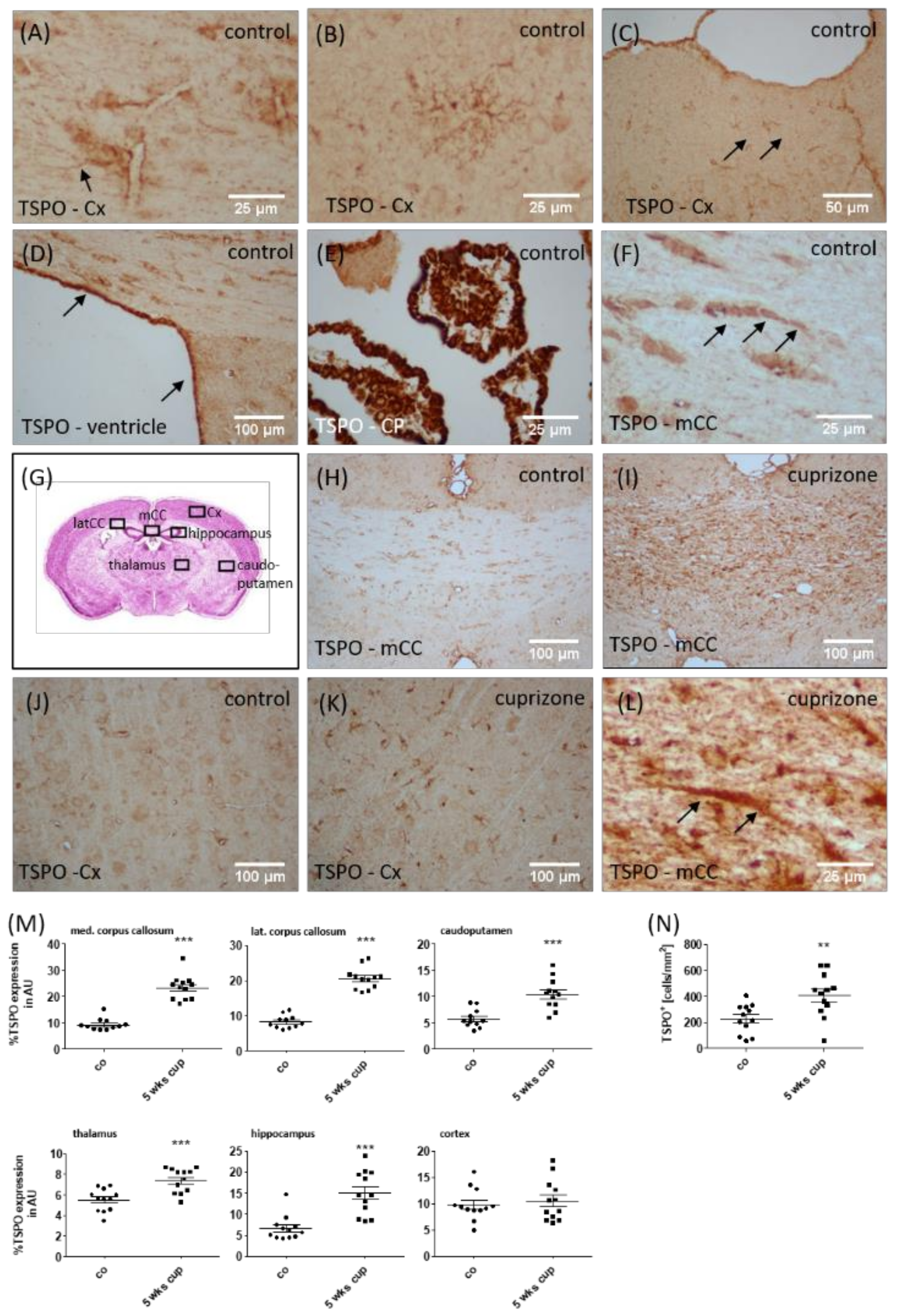
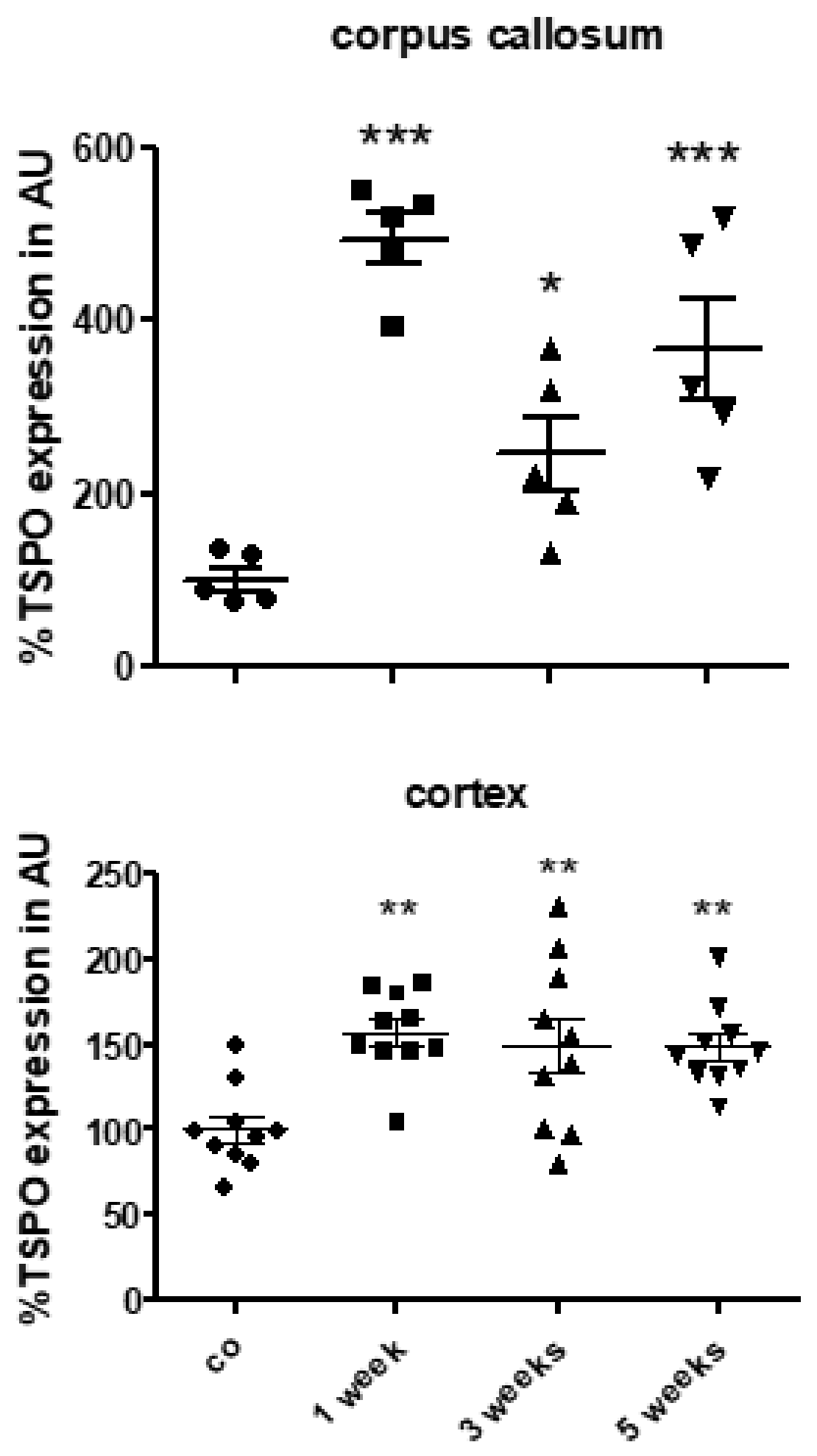
3.2. TSPO Expression and (18F)-GE180 Uptake Correlate with the Degree of Demyelination and Glial Cell Activation
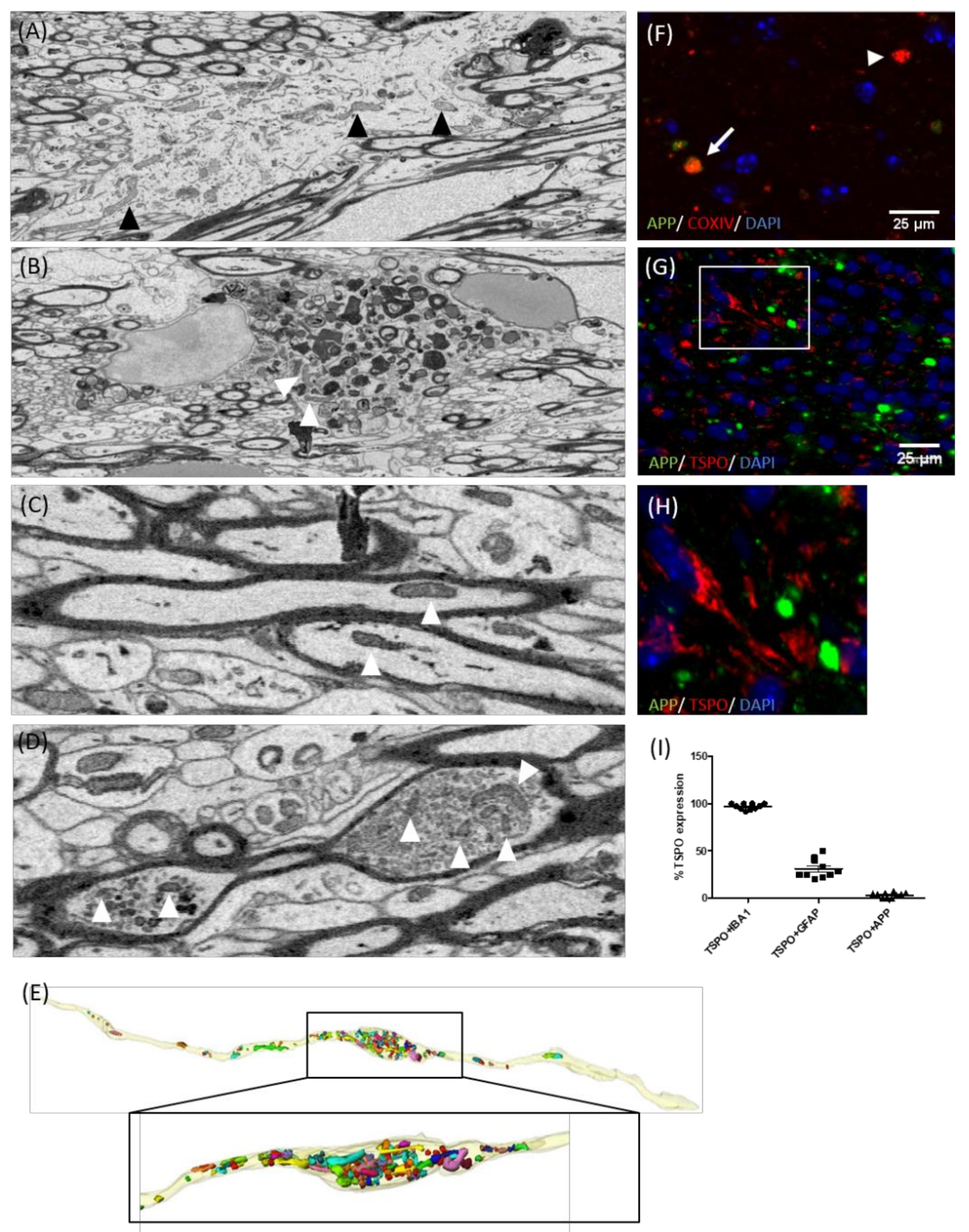
3.3. TSPO Mainly Co-Localizes with Microglia
3.4. TSPO Is Not Expressed in the Mitochondria of Injured Axons
3.5. TSPO Is Expressed by Both Microglia and Monocytes in Inflammatory Lesions
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kipp, M.; Nyamoya, S.; Hochstrasser, T.; Amor, S. Multiple sclerosis animal models: A clinical and histopathological perspective. Brain Pathol. 2017, 27, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Kipp, M.; van der Valk, P.; Amor, S. Pathology of multiple sclerosis. CNS Neurol. Disord. Drug Targets 2012, 11, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Lucchinetti, C.F.; Popescu, B.F.; Bunyan, R.F.; Moll, N.M.; Roemer, S.F.; Lassmann, H.; Bruck, W.; Parisi, J.E.; Scheithauer, B.W.; Giannini, C.; et al. Inflammatory cortical demyelination in early multiple sclerosis. N. Engl. J. Med. 2011, 365, 2188–2197. [Google Scholar] [CrossRef] [PubMed]
- Pirko, I.; Lucchinetti, C.F.; Sriram, S.; Bakshi, R. Gray matter involvement in multiple sclerosis. Neurology 2007, 68, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Vercellino, M.; Plano, F.; Votta, B.; Mutani, R.; Giordana, M.T.; Cavalla, P. Grey matter pathology in multiple sclerosis. J. Neuropathol. Exp. Neurol. 2005, 64, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Kooi, E.J.; Prins, M.; Bajic, N.; Belien, J.A.; Gerritsen, W.H.; van Horssen, J.; Aronica, E.; van Dam, A.M.; Hoozemans, J.J.; Francis, P.T.; et al. Cholinergic imbalance in the multiple sclerosis hippocampus. Acta Neuropathol. 2011, 122, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Kutzelnigg, A.; Lucchinetti, C.F.; Stadelmann, C.; Bruck, W.; Rauschka, H.; Bergmann, M.; Schmidbauer, M.; Parisi, J.E.; Lassmann, H. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 2005, 128, 2705–2712. [Google Scholar] [CrossRef]
- Hasan, K.M.; Halphen, C.; Kamali, A.; Nelson, F.M.; Wolinsky, J.S.; Narayana, P.A. Caudate nuclei volume, diffusion tensor metrics, and T(2) relaxation in healthy adults and relapsing-remitting multiple sclerosis patients: Implications for understanding gray matter degeneration. J. Magn. Reson. Imaging 2009, 29, 70–77. [Google Scholar] [CrossRef]
- Ontaneda, D.; Thompson, A.J.; Fox, R.J.; Cohen, J.A. Progressive multiple sclerosis: Prospects for disease therapy, repair, and restoration of function. Lancet 2017, 389, 1357–1366. [Google Scholar] [CrossRef]
- Liu, J.; Tian, D.; Murugan, M.; Eyo, U.B.; Dreyfus, C.F.; Wang, W.; Wu, L.J. Microglial Hv1 proton channel promotes cuprizone-induced demyelination through oxidative damage. J. Neurochem. 2015, 135, 347–356. [Google Scholar] [CrossRef]
- Xiao, Y.; Jin, J.; Chang, M.; Chang, J.H.; Hu, H.; Zhou, X.; Brittain, G.C.; Stansberg, C.; Torkildsen, O.; Wang, X.; et al. Peli1 promotes microglia-mediated CNS inflammation by regulating Traf3 degradation. Nat. Med. 2013, 19, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.; Sminia, T.; Wouterlood, F.G.; Dijkstra, C.D. Phagocytic activity of macrophages and microglial cells during the course of acute and chronic relapsing experimental autoimmune encephalomyelitis. J. Neurosci. Res. 1994, 38, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Wlodarczyk, A.; Lobner, M.; Cedile, O.; Owens, T. Comparison of microglia and infiltrating CD11c(+) cells as antigen presenting cells for T cell proliferation and cytokine response. J. Neuroinflamm. 2014, 11, 57. [Google Scholar] [CrossRef] [PubMed]
- Aloisi, F.; Ria, F.; Adorini, L. Regulation of T-cell responses by CNS antigen-presenting cells: Different roles for microglia and astrocytes. Immunol. Today 2000, 21, 141–147. [Google Scholar] [CrossRef]
- Frischer, J.M.; Bramow, S.; Dal-Bianco, A.; Lucchinetti, C.F.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Lassmann, H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain J. Neurol. 2009, 132, 1175–1189. [Google Scholar] [CrossRef] [PubMed]
- Hoflich, K.M.; Beyer, C.; Clarner, T.; Schmitz, C.; Nyamoya, S.; Kipp, M.; Hochstrasser, T. Acute axonal damage in three different murine models of multiple sclerosis: A comparative approach. Brain Res. 2016, 1650, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Minghetti, L.; Levi, G. Microglia as effector cells in brain damage and repair: Focus on prostanoids and nitric oxide. Prog. Neurobiol. 1998, 54, 99–125. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Perry, V.H. Microglial physiology: Unique stimuli, specialized responses. Annu. Rev. Immunol. 2009, 27, 119–145. [Google Scholar] [CrossRef]
- Venneti, S.; Lopresti, B.J.; Wiley, C.A. The peripheral benzodiazepine receptor (Translocator protein 18kDa) in microglia: From pathology to imaging. Prog. Neurobiol. 2006, 80, 308–322. [Google Scholar] [CrossRef]
- McEnery, M.W.; Snowman, A.M.; Trifiletti, R.R.; Snyder, S.H. Isolation of the mitochondrial benzodiazepine receptor: Association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc. Natl. Acad. Sci. USA 1992, 89, 3170–3174. [Google Scholar] [CrossRef]
- Yeliseev, A.A.; Kaplan, S. A sensory transducer homologous to the mammalian peripheral-type benzodiazepine receptor regulates photosynthetic membrane complex formation in Rhodobacter sphaeroides 2.4.1. J. Biol. Chem. 1995, 270, 21167–21175. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, V.; Berkovich, A.; Krueger, K.E.; Costa, E.; Guidotti, A. Diazepam binding inhibitor and its processing products stimulate mitochondrial steroid biosynthesis via an interaction with mitochondrial benzodiazepine receptors. Endocrinology 1991, 129, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Denora, N.; Natile, G. An Updated View of Translocator Protein (TSPO). Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- McNeela, A.M.; Bernick, C.; Hines, R.M.; Hines, D.J. TSPO regulation in reactive gliotic diseases. J. Neurosci. Res. 2018, 96, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Cosenza-Nashat, M.; Zhao, M.L.; Suh, H.S.; Morgan, J.; Natividad, R.; Morgello, S.; Lee, S.C. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol. Appl. Neurobiol. 2009, 35, 306–328. [Google Scholar] [CrossRef] [PubMed]
- Maeda, J.; Higuchi, M.; Inaji, M.; Ji, B.; Haneda, E.; Okauchi, T.; Zhang, M.R.; Suzuki, K.; Suhara, T. Phase-dependent roles of reactive microglia and astrocytes in nervous system injury as delineated by imaging of peripheral benzodiazepine receptor. Brain Res. 2007, 1157, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Takaya, S.; Hashikawa, K.; Turkheimer, F.E.; Mottram, N.; Deprez, M.; Ishizu, K.; Kawashima, H.; Akiyama, H.; Fukuyama, H.; Banati, R.B.; et al. The lack of expression of the peripheral benzodiazepine receptor characterises microglial response in anaplastic astrocytomas. J. Neuro-Oncol. 2007, 85, 95–103. [Google Scholar] [CrossRef]
- Kuhlmann, A.C.; Guilarte, T.R. Cellular and subcellular localization of peripheral benzodiazepine receptors after trimethyltin neurotoxicity. J. Neurochem. 2000, 74, 1694–1704. [Google Scholar] [CrossRef]
- Banati, R.B. Visualising microglial activation in vivo. Glia 2002, 40, 206–217. [Google Scholar] [CrossRef]
- Zivadinov, R.; Stosic, M.; Cox, J.L.; Ramasamy, D.P.; Dwyer, M.G. The place of conventional MRI and newly emerging MRI techniques in monitoring different aspects of treatment outcome. J. Neurol. 2008, 255, 61–74. [Google Scholar] [CrossRef]
- Hagens, M.; van Berckel, B.; Barkhof, F. Novel MRI and PET markers of neuroinflammation in multiple sclerosis. Curr. Opin. Neurol. 2016, 29, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Vowinckel, E.; Reutens, D.; Becher, B.; Verge, G.; Evans, A.; Owens, T.; Antel, J.P. PK11195 binding to the peripheral benzodiazepine receptor as a marker of microglia activation in multiple sclerosis and experimental autoimmune encephalomyelitis. J. Neurosci. Res. 1997, 50, 345–353. [Google Scholar] [CrossRef]
- Wickstrom, T.; Clarke, A.; Gausemel, I.; Horn, E.; Jorgensen, K.; Khan, I.; Mantzilas, D.; Rajanayagam, T.; In’t Veld, D.J.; Trigg, W. The development of an automated and GMP compliant FASTlab Synthesis of [(18) F]GE-180; a radiotracer for imaging translocator protein (TSPO). J. Label. Compd. Radiopharm. 2014, 57, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Unterrainer, M.; Mahler, C.; Vomacka, L.; Lindner, S.; Havla, J.; Brendel, M.; Boning, G.; Ertl-Wagner, B.; Kumpfel, T.; Milenkovic, V.M.; et al. TSPO PET with [(18)F]GE-180 sensitively detects focal neuroinflammation in patients with relapsing-remitting multiple sclerosis. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Vomacka, L.; Albert, N.L.; Lindner, S.; Unterrainer, M.; Mahler, C.; Brendel, M.; Ermoschkin, L.; Gosewisch, A.; Brunegraf, A.; Buckley, C.; et al. TSPO imaging using the novel PET ligand [(18)F]GE-180: Quantification approaches in patients with multiple sclerosis. EJNMMI Res. 2017, 7, 89. [Google Scholar] [CrossRef] [PubMed]
- Blume, T.; Focke, C.; Peters, F.; Deussing, M.; Albert, N.L.; Lindner, S.; Gildehaus, F.J.; von Ungern-Sternberg, B.; Ozmen, L.; Baumann, K.; et al. Microglial response to increasing amyloid load saturates with aging: A longitudinal dual tracer in vivo muPET-study. J. Neuroinflamm. 2018, 15, 307. [Google Scholar] [CrossRef] [PubMed]
- Klein, B.; Mrowetz, H.; Barker, C.M.; Lange, S.; Rivera, F.J.; Aigner, L. Age Influences Microglial Activation After Cuprizone-Induced Demyelination. Front. Aging Neurosci. 2018, 10, 278. [Google Scholar] [CrossRef] [PubMed]
- Airas, L.; Dickens, A.M.; Elo, P.; Marjamaki, P.; Johansson, J.; Eskola, O.; Jones, P.A.; Trigg, W.; Solin, O.; Haaparanta-Solin, M.; et al. In vivo PET imaging demonstrates diminished microglial activation after fingolimod treatment in an animal model of multiple sclerosis. J. Nucl. Med. 2015, 56, 305–310. [Google Scholar] [CrossRef]
- Mattner, F.; Staykova, M.; Berghofer, P.; Wong, H.J.; Fordham, S.; Callaghan, P.; Jackson, T.; Pham, T.; Gregoire, M.C.; Zahra, D.; et al. Central nervous system expression and PET imaging of the translocator protein in relapsing-remitting experimental autoimmune encephalomyelitis. J. Nucl. Med. 2013, 54, 291–298. [Google Scholar] [CrossRef]
- Abourbeh, G.; Theze, B.; Maroy, R.; Dubois, A.; Brulon, V.; Fontyn, Y.; Dolle, F.; Tavitian, B.; Boisgard, R. Imaging microglial/macrophage activation in spinal cords of experimental autoimmune encephalomyelitis rats by positron emission tomography using the mitochondrial 18 kDa translocator protein radioligand [(1)(8)F]DPA-714. J. Neurosci. 2012, 32, 5728–5736. [Google Scholar] [CrossRef]
- Mattner, F.; Bandin, D.L.; Staykova, M.; Berghofer, P.; Gregoire, M.C.; Ballantyne, P.; Quinlivan, M.; Fordham, S.; Pham, T.; Willenborg, D.O.; et al. Evaluation of [(1)(2)(3)I]-CLINDE as a potent SPECT radiotracer to assess the degree of astroglia activation in cuprizone-induced neuroinflammation. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1516–1528. [Google Scholar] [CrossRef] [PubMed]
- Notter, T.; Coughlin, J.M.; Gschwind, T.; Weber-Stadlbauer, U.; Wang, Y.; Kassiou, M.; Vernon, A.C.; Benke, D.; Pomper, M.G.; Sawa, A.; et al. Translational evaluation of translocator protein as a marker of neuroinflammation in schizophrenia. Mol. Psychiatry 2018, 23, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Ruther, B.J.; Scheld, M.; Dreymueller, D.; Clarner, T.; Kress, E.; Brandenburg, L.O.; Swartenbroekx, T.; Hoornaert, C.; Ponsaerts, P.; Fallier-Becker, P.; et al. Combination of cuprizone and experimental autoimmune encephalomyelitis to study inflammatory brain lesion formation and progression. Glia 2017, 65, 1900–1913. [Google Scholar] [CrossRef]
- Scheld, M.; Ruther, B.J.; Grosse-Veldmann, R.; Ohl, K.; Tenbrock, K.; Dreymuller, D.; Fallier-Becker, P.; Zendedel, A.; Beyer, C.; Clarner, T.; et al. Neurodegeneration Triggers Peripheral Immune Cell Recruitment into the Forebrain. J. Neurosci. 2016, 36, 1410–1415. [Google Scholar] [CrossRef] [PubMed]
- Nyamoya, S.; Leopold, P.; Becker, B.; Beyer, C.; Hustadt, F.; Schmitz, C.; Michel, A.; Kipp, M. G-Protein-Coupled Receptor Gpr17 Expression in Two Multiple Sclerosis Remyelination Models. Mol. Neurobiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Nolte, C.; Matyash, M.; Pivneva, T.; Schipke, C.G.; Ohlemeyer, C.; Hanisch, U.K.; Kirchhoff, F.; Kettenmann, H. GFAP promoter-controlled EGFP-expressing transgenic mice: A tool to visualize astrocytes and astrogliosis in living brain tissue. Glia 2001, 33, 72–86. [Google Scholar] [CrossRef]
- Saederup, N.; Cardona, A.E.; Croft, K.; Mizutani, M.; Cotleur, A.C.; Tsou, C.L.; Ransohoff, R.M.; Charo, I.F. Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS ONE 2010, 5, e13693. [Google Scholar] [CrossRef]
- Brendel, M.; Probst, F.; Jaworska, A.; Overhoff, F.; Korzhova, V.; Albert, N.L.; Beck, R.; Lindner, S.; Gildehaus, F.J.; Baumann, K.; et al. Glial Activation and Glucose Metabolism in a Transgenic Amyloid Mouse Model: A Triple-Tracer PET Study. J. Nucl. Med. 2016, 57, 954–960. [Google Scholar] [CrossRef]
- Overhoff, F.; Brendel, M.; Jaworska, A.; Korzhova, V.; Delker, A.; Probst, F.; Focke, C.; Gildehaus, F.J.; Carlsen, J.; Baumann, K.; et al. Automated Spatial Brain Normalization and Hindbrain White Matter Reference Tissue Give Improved [(18)F]-Florbetaben PET Quantitation in Alzheimer’s Model Mice. Front. Neurosci. 2016, 10, 45. [Google Scholar] [CrossRef]
- Deussing, M.; Blume, T.; Vomacka, L.; Mahler, C.; Focke, C.; Todica, A.; Unterrainer, M.; Albert, N.L.; Lindner, S.; von Ungern-Sternberg, B.; et al. Coupling between physiological TSPO expression in brain and myocardium allows stabilization of late-phase cerebral [(18)F]GE180 PET quantification. NeuroImage 2017, 165, 83–91. [Google Scholar] [CrossRef]
- Slowik, A.; Schmidt, T.; Beyer, C.; Amor, S.; Clarner, T.; Kipp, M. The sphingosine 1-phosphate receptor agonist FTY720 is neuroprotective after cuprizone-induced CNS demyelination. Br. J. Pharmacol. 2015, 172, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Baertling, F.; Kokozidou, M.; Pufe, T.; Clarner, T.; Windoffer, R.; Wruck, C.J.; Brandenburg, L.O.; Beyer, C.; Kipp, M. ADAM12 is expressed by astrocytes during experimental demyelination. Brain Res. 2010, 1326, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ohno, N.; Chiang, H.; Mahad, D.J.; Kidd, G.J.; Liu, L.; Ransohoff, R.M.; Sheng, Z.H.; Komuro, H.; Trapp, B.D. Mitochondrial immobilization mediated by syntaphilin facilitates survival of demyelinated axons. Proc. Natl. Acad. Sci. USA 2014, 111, 9953–9958. [Google Scholar] [CrossRef] [PubMed]
- Kipp, M.; Clarner, T.; Dang, J.; Copray, S.; Beyer, C. The cuprizone animal model: New insights into an old story. Acta Neuropathol. 2009, 118, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Calsolaro, V.; Atkinson, R.A.; Femminella, G.D.; Waldman, A.; Buckley, C.; Trigg, W.; Brooks, D.J.; Hinz, R.; Edison, P. Flutriciclamide (18F-GE180) PET: First-in-Human PET Study of Novel Third-Generation In Vivo Marker of Human Translocator Protein. J. Nucl. Med. 2016, 57, 1753–1759. [Google Scholar] [CrossRef] [PubMed]
- Fukudome, D.; Hayes, L.N.; Faust, T.E.; Foss, C.A.; Kondo, M.A.; Lee, B.J.; Saito, A.; Kano, S.I.; Coughlin, J.M.; Kamiya, A.; et al. Translocator protein (TSPO) and stress cascades in mouse models of psychosis with inflammatory disturbances. Schizophr. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Vlodavsky, E.; Soustiel, J.F. Immunohistochemical expression of peripheral benzodiazepine receptors in human astrocytomas and its correlation with grade of malignancy, proliferation, apoptosis and survival. J. Neurooncol. 2007, 81, 1–7. [Google Scholar] [CrossRef]
- Chen, M.K.; Baidoo, K.; Verina, T.; Guilarte, T.R. Peripheral benzodiazepine receptor imaging in CNS demyelination: Functional implications of anatomical and cellular localization. Brain 2004, 127, 1379–1392. [Google Scholar] [CrossRef]
- Clarner, T.; Diederichs, F.; Berger, K.; Denecke, B.; Gan, L.; van der Valk, P.; Beyer, C.; Amor, S.; Kipp, M. Myelin debris regulates inflammatory responses in an experimental demyelination animal model and multiple sclerosis lesions. Glia 2012, 60, 1468–1480. [Google Scholar] [CrossRef]
- Mahad, D.J.; Ziabreva, I.; Campbell, G.; Lax, N.; White, K.; Hanson, P.S.; Lassmann, H.; Turnbull, D.M. Mitochondrial changes within axons in multiple sclerosis. Brain 2009, 132, 1161–1174. [Google Scholar] [CrossRef]
- Zambonin, J.L.; Zhao, C.; Ohno, N.; Campbell, G.R.; Engeham, S.; Ziabreva, I.; Schwarz, N.; Lee, S.E.; Frischer, J.M.; Turnbull, D.M.; et al. Increased mitochondrial content in remyelinated axons: Implications for multiple sclerosis. Brain 2011, 134, 1901–1913. [Google Scholar] [CrossRef] [PubMed]
- Hiremath, M.M.; Chen, V.S.; Suzuki, K.; Ting, J.P.; Matsushima, G.K. MHC class II exacerbates demyelination in vivo independently of T cells. J. Neuroimmunol. 2008, 203, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Banati, R.B.; Goerres, G.W.; Myers, R.; Gunn, R.N.; Turkheimer, F.E.; Kreutzberg, G.W.; Brooks, D.J.; Jones, T.; Duncan, J.S. [11C](R)-PK11195 positron emission tomography imaging of activated microglia in vivo in Rasmussen’s encephalitis. Neurology 1999, 53, 2199–2203. [Google Scholar] [CrossRef]
- Junck, L.; Olson, J.M.; Ciliax, B.J.; Koeppe, R.A.; Watkins, G.L.; Jewett, D.M.; McKeever, P.E.; Wieland, D.M.; Kilbourn, M.R.; Starosta-Rubinstein, S.; et al. PET imaging of human gliomas with ligands for the peripheral benzodiazepine binding site. Ann. Neurol. 1989, 26, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Banati, R.B. Neuropathological imaging: In vivo detection of glial activation as a measure of disease and adaptive change in the brain. Br. Med. Bull. 2003, 65, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Banati, R.B.; Newcombe, J.; Gunn, R.N.; Cagnin, A.; Turkheimer, F.; Heppner, F.; Price, G.; Wegner, F.; Giovannoni, G.; Miller, D.H.; et al. The peripheral benzodiazepine binding site in the brain in multiple sclerosis: Quantitative in vivo imaging of microglia as a measure of disease activity. Brain J. Neurol. 2000, 123, 2321–2337. [Google Scholar] [CrossRef]
- Kreisl, W.C.; Fujita, M.; Fujimura, Y.; Kimura, N.; Jenko, K.J.; Kannan, P.; Hong, J.; Morse, C.L.; Zoghbi, S.S.; Gladding, R.L.; et al. Comparison of [(11)C]-(R)-PK 11195 and [(11)C]PBR28, two radioligands for translocator protein (18 kDa) in human and monkey: Implications for positron emission tomographic imaging of this inflammation biomarker. NeuroImage 2010, 49, 2924–2932. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.K.; Guilarte, T.R. Imaging the peripheral benzodiazepine receptor response in central nervous system demyelination and remyelination. Toxicol. Sci. 2006, 91, 532–539. [Google Scholar] [CrossRef]
- Narayan, N.; Owen, D.; Mandhair, H.; Smyth, E.; Carlucci, F.; Saleem, A.; Gunn, R.; Rabiner, E.I.A.; Wells, L.; Dakin, S.; et al. Translocator Protein as an Imaging Marker of Macrophage and Stromal Activation in RA Pannus. J. Nucl. Med. 2018, 59, 1125–1132. [Google Scholar] [CrossRef]
- Thackeray, J.T.; Hupe, H.C.; Wang, Y.; Bankstahl, J.P.; Berding, G.; Ross, T.L.; Bauersachs, J.; Wollert, K.C.; Bengel, F.M. Myocardial Inflammation Predicts Remodeling and Neuroinflammation After Myocardial Infarction. J. Am. Coll. Cardiol. 2018, 71, 263–275. [Google Scholar] [CrossRef]
- Hellberg, S.; Silvola, J.M.U.; Kiugel, M.; Liljenback, H.; Savisto, N.; Li, X.G.; Thiele, A.; Lehmann, L.; Heinrich, T.; Vollmer, S.; et al. 18-kDa translocator protein ligand (18)F-FEMPA: Biodistribution and uptake into atherosclerotic plaques in mice. J. Nucl. Cardiol. 2017, 24, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Fernandez Hurst, N.; Zanetti, S.R.; Baez, N.S.; Bibolini, M.J.; Bouzat, C.; Roth, G.A. Diazepam treatment reduces inflammatory cells and mediators in the central nervous system of rats with experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2017, 313, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Berkovich, A.; Ferrarese, C.; Cavaletti, G.; Alho, H.; Marzorati, C.; Bianchi, G.; Guidotti, A.; Costa, E. Topology of two DBI receptors in human lymphocytes. Life Sci. 1993, 52, 1265–1277. [Google Scholar] [CrossRef]
- Matsushima, G.K.; Morell, P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol. 2001, 11, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Koo, E.H.; Sisodia, S.S.; Archer, D.R.; Martin, L.J.; Weidemann, A.; Beyreuther, K.; Fischer, P.; Masters, C.L.; Price, D.L. Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc. Natl. Acad. Sci. USA 1990, 87, 1561–1565. [Google Scholar] [CrossRef] [PubMed]
- Sherriff, F.E.; Bridges, L.R.; Gentleman, S.M.; Sivaloganathan, S.; Wilson, S. Markers of axonal injury in post mortem human brain. Acta Neuropathol. 1994, 88, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.R.; Singleton, R.H.; Povlishock, J.T. Antibodies to the C-terminus of the beta-amyloid precursor protein (APP): A site specific marker for the detection of traumatic axonal injury. Brain Res. 2000, 871, 288–302. [Google Scholar] [CrossRef]
- Bitsch, A.; Schuchardt, J.; Bunkowski, S.; Kuhlmann, T.; Bruck, W. Acute axonal injury in multiple sclerosis. Correlation with demyelination and inflammation. Brain 2000, 123, 1174–1183. [Google Scholar] [CrossRef]
- Khademi, M.; Dring, A.M.; Gilthorpe, J.D.; Wuolikainen, A.; Al Nimer, F.; Harris, R.A.; Andersson, M.; Brundin, L.; Piehl, F.; Olsson, T.; et al. Intense inflammation and nerve damage in early multiple sclerosis subsides at older age: A reflection by cerebrospinal fluid biomarkers. PLoS ONE 2013, 8, e63172. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sense | Antisense | Bp | AT | |
|---|---|---|---|---|
| TSPO1 | GCCTACTTTGTACGTGGCGAG | CCTCCCAGCTCTTTCCAGAC | 152 | 60 |
| β-Actin 1 | GTACCACCATGTACCCAGGC | AACGCAGCTCAGTAACAGTCC | 247 | 60 |
| Antigen | Species | Dilution | HIER Method | Purchase Number | RRID | Company |
|---|---|---|---|---|---|---|
| Single stains | ||||||
| PLP | Mouse | 1:5,000 | None | MCA839G | AB_2237198 | Bio-Rad Laboratories, Inc., Germany |
| TSPO/PBR | Goat | 1:100 | Tris/EDTA | ab118913 | AB_10898989 | Abcam, UK |
| Iba1 | Rabbit | 1:5,000 | Tris/EDTA | 019–19741 | AB_839504 | Wako, USA |
| GFAP | Chicken | 1:5,000 | Citrate | ab4674 | AB_304558 | Abcam, UK |
| Double stains | ||||||
| TSPO/PBR | Goat | 1:100 | Tris/EDTA | ab118913 | AB_10898989 | Abcam, UK |
| GFAP | Mouse | 1:400 | Tris/EDTA | G3893 | AB_477010 | Sigma-Aldrich, USA |
| Iba1 | Rabbit | 1:2,500 | Tris/EDTA | 019-19741 | AB_839504 | Wako, USA |
| APP | Mouse | 1:1,000 | Tris/EDTA | MAB348 | AB_94882 | Millipore, Germany |
| COXIV | Rabbit | 1:500 | Tris/EDTA | ab16056 | AB_443304 | Abcam, UK |
| Order Number | RRID | Supplier | |
|---|---|---|---|
| Rabbit anti-goat IgG | BA-5000 | AB_2336126 | Vector Laboratories, Burlingame, USA |
| Goat anti-rabbit IgG | BA-1000 | AB_2313606 | Vector Laboratories, Burlingame, USA |
| Goat anti-mouse IgG | BA-9200 | AB_2336171 | Vector Laboratories, Burlingame, USA |
| Goat anti-chicken IgG | BA-9010 | AB_2336114 | Vector Laboratories, Burlingame, USA |
| Cy3 donkey anti-goat IgG | 705-165-147 | AB_2307351 | Jackson ImmunoResearch Labs |
| Alexa Fluor 488 donkey anti-rabbit IgG | A21206 | AB_2535792 | Invitrogen, USA |
| Alexa Fluor 488 donkey anti-mouse IgG | A21202 | AB_141607 | Invitrogen, USA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nack, A.; Brendel, M.; Nedelcu, J.; Daerr, M.; Nyamoya, S.; Beyer, C.; Focke, C.; Deussing, M.; Hoornaert, C.; Ponsaerts, P.; et al. Expression of Translocator Protein and [18F]-GE180 Ligand Uptake in Multiple Sclerosis Animal Models. Cells 2019, 8, 94. https://doi.org/10.3390/cells8020094
Nack A, Brendel M, Nedelcu J, Daerr M, Nyamoya S, Beyer C, Focke C, Deussing M, Hoornaert C, Ponsaerts P, et al. Expression of Translocator Protein and [18F]-GE180 Ligand Uptake in Multiple Sclerosis Animal Models. Cells. 2019; 8(2):94. https://doi.org/10.3390/cells8020094
Chicago/Turabian StyleNack, Anne, Matthias Brendel, Julia Nedelcu, Markus Daerr, Stella Nyamoya, Cordian Beyer, Carola Focke, Maximilian Deussing, Chloé Hoornaert, Peter Ponsaerts, and et al. 2019. "Expression of Translocator Protein and [18F]-GE180 Ligand Uptake in Multiple Sclerosis Animal Models" Cells 8, no. 2: 94. https://doi.org/10.3390/cells8020094
APA StyleNack, A., Brendel, M., Nedelcu, J., Daerr, M., Nyamoya, S., Beyer, C., Focke, C., Deussing, M., Hoornaert, C., Ponsaerts, P., Schmitz, C., Bartenstein, P., Rominger, A., & Kipp, M. (2019). Expression of Translocator Protein and [18F]-GE180 Ligand Uptake in Multiple Sclerosis Animal Models. Cells, 8(2), 94. https://doi.org/10.3390/cells8020094