Signals from the Niche: Insights into the Role of IGF-1 and IL-6 in Modulating Skeletal Muscle Fibrosis




Abstract
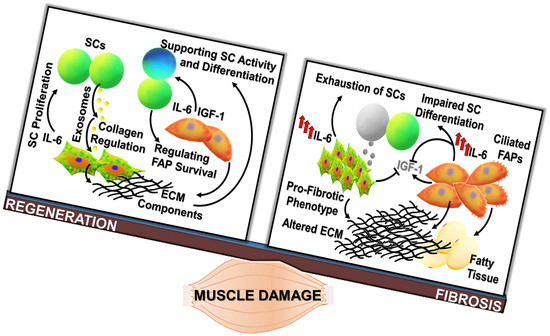
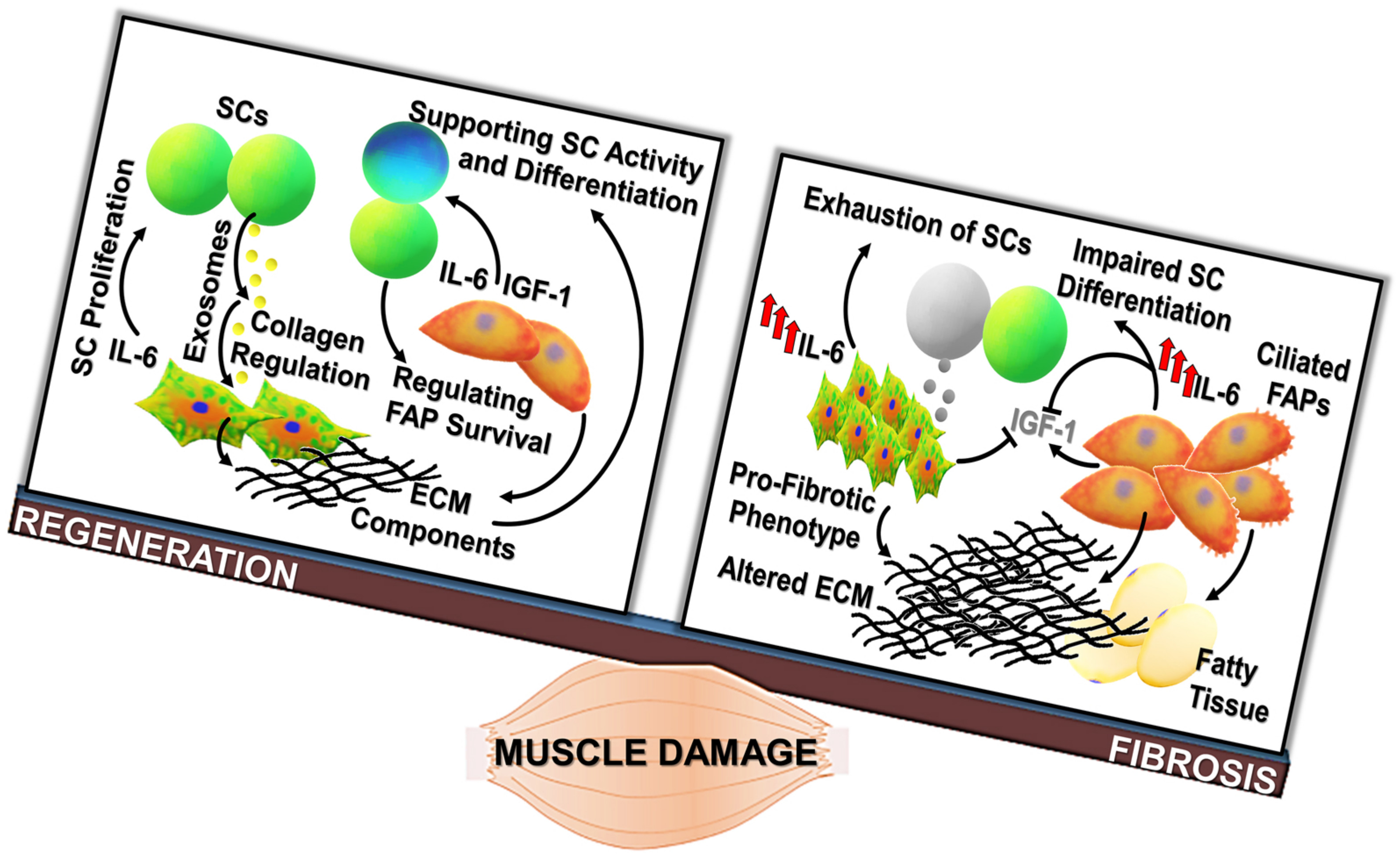
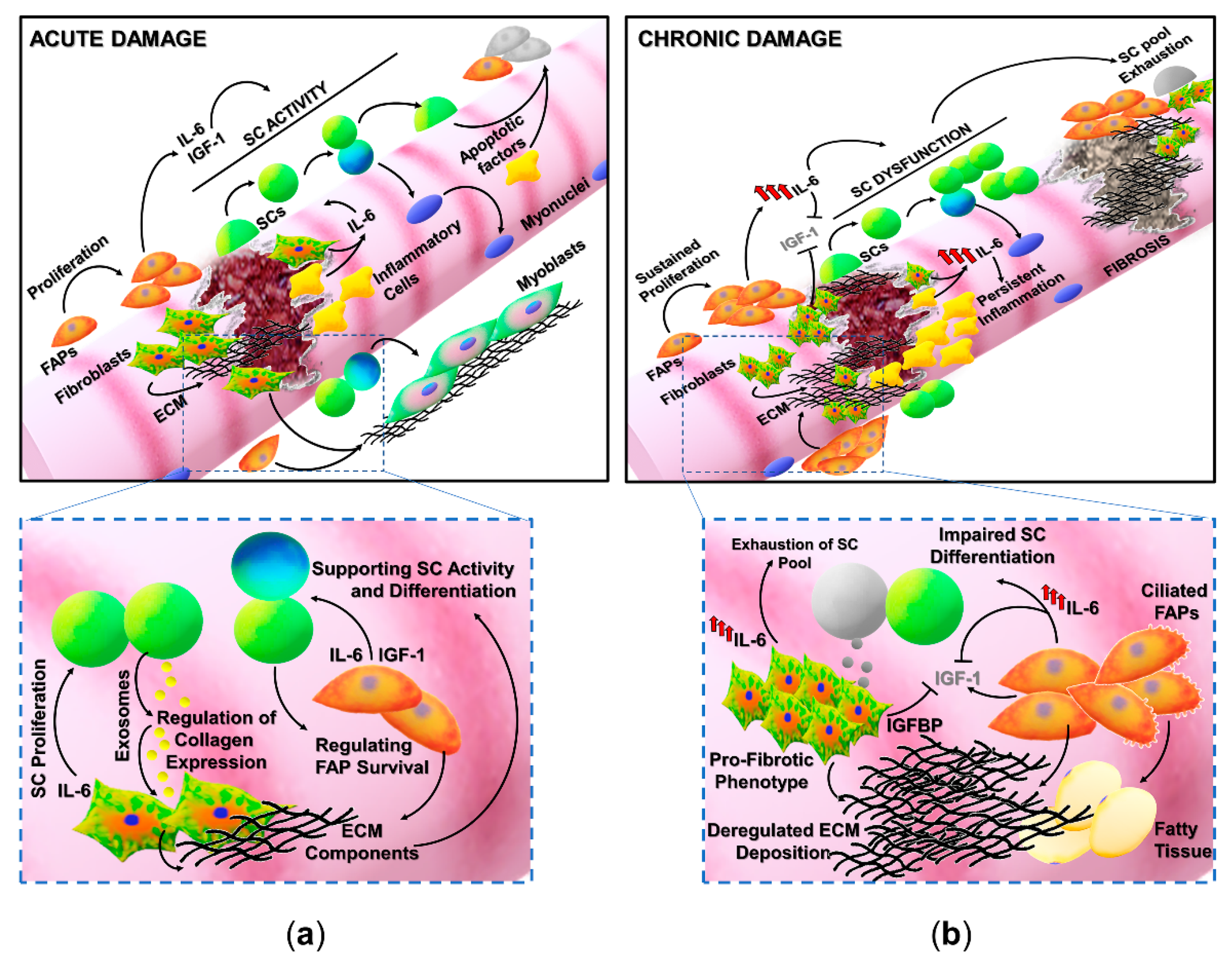
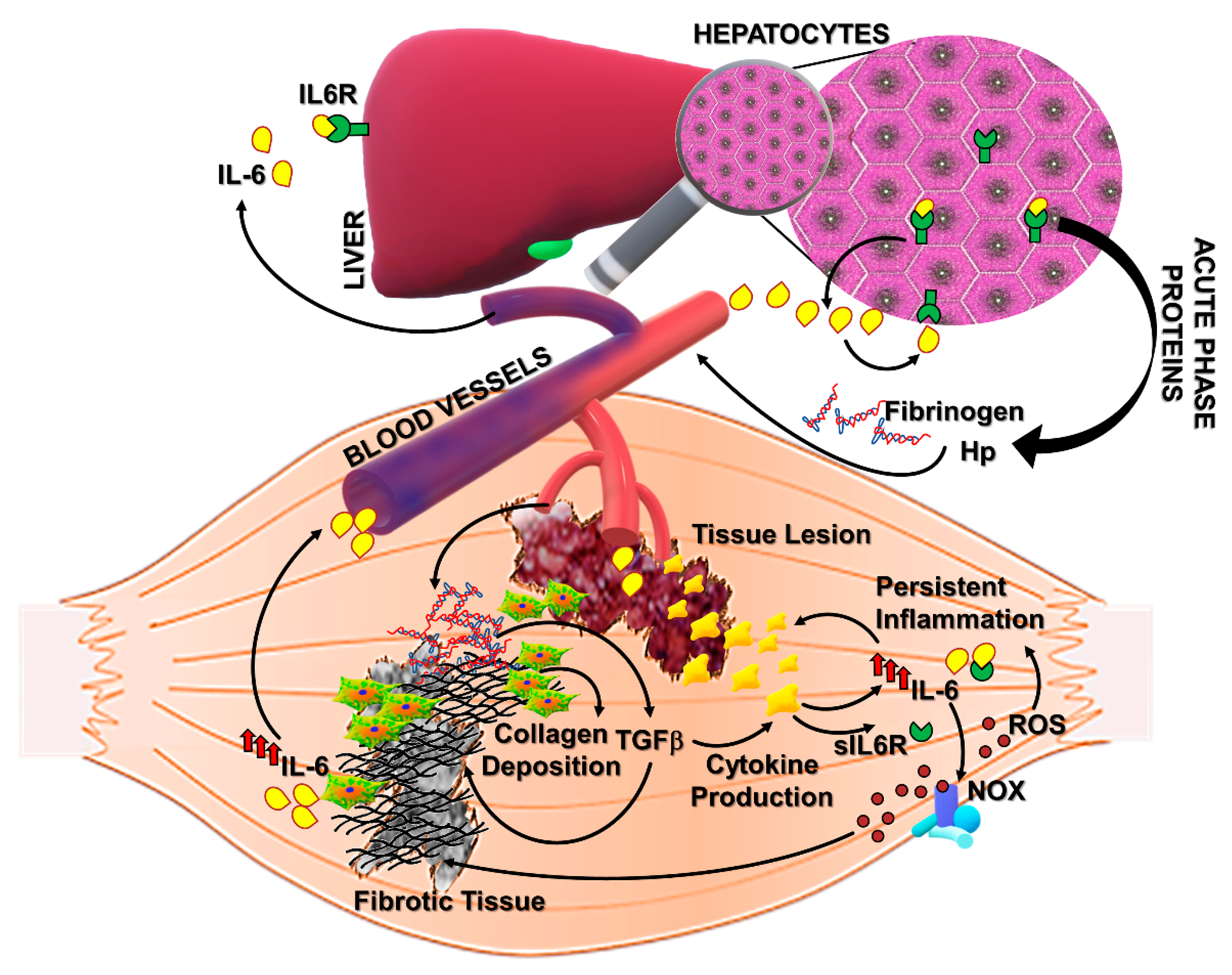
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Cellular Mediators of Regenerative Fibrogenesis and Fibrosis
Reciprocal Regulation of Fibroblasts and Satellite Cells Activity
3. Pro- and Anti-Fibrotic Factors Influencing the Balance between Regeneration and Fibrosis
4. Interleukin-6: Spectrum of Pro-Fibrotic Actions
4.1. Immune Response and Fibrosis: A Fatal Interplay
4.2. Oxidative Stress
5. IGF-1 As a Modulatory Factor in Muscle Niche, Promoting Regenerative Events
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mann, C.J.; Perdiguero, E.; Kharraz, Y.; Aguilar, S.; Pessina, P.; Serrano, A.L.; Muñoz-Cánoves, P. Aberrant repair and fibrosis development in skeletal muscle. Skelet. Muscle 2011, 1, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stearns-Reider, K.M.; D’Amore, A.; Beezhold, K.; Rothrauff, B.; Cavalli, L.; Wagner, W.R.; Vorp, D.A.; Tsamis, A.; Shinde, S.; Zhang, C.; et al. Aging of the skeletal muscle extracellular matrix drives a stem cell fibrogenic conversion. Aging Cell 2017, 16, 518–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahdy, M.A.A. Skeletal muscle fibrosis: An overview. Cell Tissue Res. 2019, 375, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Saul, D.; Böker, K.O.; Ernst, J.; Lehman, W.; Schilling, A.F. Current Methods for Skeletal Muscle Tissue Repair and Regeneration. BioMed Res. Int. 2018, 2018, 1984879. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, R.I.; Christensen, J.L.; Conboy, I.M.; Conboy, M.J.; Rando, T.A.; Weissman, I.L.; Wagers, A.J. Isolation of Adult Mouse Myogenic Progenitors. Cell 2004, 119, 543–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentzinger, C.F.; Wang, Y.X.; Dumont, N.A.; Rudnicki, M.A. Cellular dynamics in the muscle satellite cell niche. EMBO Rep. 2013, 14, 1062–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, H.; Price, F.; Rudnicki, M.A. Satellite Cells and the Muscle Stem Cell Niche. Physiol. Rev. 2013, 93, 23–67. [Google Scholar] [CrossRef] [PubMed]
- Joe, A.W.B.; Yi, L.; Natarajan, A.; Le Grand, F.; So, L.; Wang, J.; Rudnicki, M.A.; Rossi, F.M.V. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 2010, 12, 153–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uezumi, A.; Ikemoto-Uezumi, M.; Tsuchida, K. Roles of nonmyogenic mesenchymal progenitors in pathogenesis and regeneration of skeletal muscle. Front. Physiol. 2014, 5, 68. [Google Scholar] [CrossRef] [PubMed]
- Farup, J.; Madaro, L.; Puri, P.L.; Mikkelsen, U.R. Interactions between muscle stem cells, mesenchymal-derived cells and immune cells in muscle homeostasis, regeneration and disease. Cell Death Dis. 2015, 6, e1830. [Google Scholar] [CrossRef] [PubMed]
- Formicola, L.; Marazzi, G.; Sassoon, D.A. The extraocular muscle stem cell niche is resistant to ageing and disease. Front. Aging Neurosci. 2014, 6, 328. [Google Scholar] [CrossRef] [PubMed]
- Malecova, B.; Gatto, S.; Etxaniz, U.; Passafaro, M.; Cortez, A.; Nicoletti, C.; Giordani, L.; Torcinaro, A.; De Bardi, M.; Bicciato, S.; et al. Dynamics of cellular states of fibro-adipogenic progenitors during myogenesis and muscular dystrophy. Nat. Commun. 2018, 9, 3670. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Steensberg, A.; Schjerling, P. Muscle-derived interleukin-6: Possible biological effects. J. Physiol. 2001, 536, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Haddad, F.; Zaldivar, F.; Cooper, D.M.; Adams, G.R. IL-6-induced skeletal muscle atrophy. J. Appl. Physiol. 2005, 98, 911–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carson, J.A.; Baltgalvis, K.A. Interleukin-6 as a Key Regulator of Muscle Mass during Cachexia. Exerc. Sport Sci. Rev. 2010, 38, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.-H.; Song, J.L.; Delafontaine, P.; Godard, M.P. The therapeutic potential of IGF-I in skeletal muscle repair. Trends Endocrinol. Metab. 2013, 24, 310–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aboalola, D.; Han, V.K.M. Different Effects of Insulin-Like Growth Factor-1 and Insulin-Like Growth Factor-2 on Myogenic Differentiation of Human Mesenchymal Stem Cells. Stem Cells Int. 2017, 2017, 8286248. [Google Scholar] [CrossRef] [PubMed]
- Musarò, A. The Basis of Muscle Regeneration. Adv. Biol. 2014, 2014, 612471. [Google Scholar] [CrossRef]
- Fu, X.; Wang, H.; Hu, P. Stem cell activation in skeletal muscle regeneration. Cell. Mol. Life Sci. 2015, 72, 1663–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scicchitano, B.M.; Sica, G.; Musarò, A. Stem cells and tissue niche in muscle regeneration Stem cells and tissue niche: Two faces of the same coin of muscle regeneration. Eur. J. Transl. Myol. 2016, 26, 6125. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Hu, P. Skeletal muscle regeneration is modulated by inflammation. J. Orthop. Transl. 2018, 13, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Forcina, L.; Miano, C.; Pelosi, L.; Musarò, A. An Overview About the Biology of Skeletal Muscle Satellite Cells. Curr. Genom. 2019, 20, 24–37. [Google Scholar] [CrossRef]
- Mauro, A. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 1961, 9, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Katz, B. The Terminations of the Afferent Nerve Fibre in the Muscle Spindle of the Frog. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1961, 243, 221–240. [Google Scholar]
- Uezumi, A.; Fukada, S.; Yamamoto, N.; Takeda, S.; Tsuchida, K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat. Cell Biol. 2010, 12, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Heredia, J.E.; Mukundan, L.; Chen, F.M.; Mueller, A.A.; Deo, R.C.; Locksley, R.M.; Rando, T.A.; Chawla, A. Type 2 Innate Signals Stimulate Fibro/Adipogenic Progenitors to Facilitate Muscle Regeneration. Cell 2013, 153, 376–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemos, D.R.; Paylor, B.; Chang, C.; Sampaio, A.; Underhill, T.M.; Rossi, F.M.V. Functionally Convergent White Adipogenic Progenitors of Different Lineages Participate in a Diffused System Supporting Tissue Regeneration. Stem Cells 2012, 30, 1152–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wosczyna, M.N.; Rando, T.A. A Muscle Stem Cell Support Group: Coordinated Cellular Responses in Muscle Regeneration. Dev. Cell 2018, 46, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Fiore, D.; Judson, R.N.; Low, M.; Lee, S.; Zhang, E.; Hopkins, C.; Xu, P.; Lenzi, A.; Rossi, F.M.V.; Lemos, D.R. Pharmacological blockage of fibro/adipogenic progenitor expansion and suppression of regenerative fibrogenesis is associated with impaired skeletal muscle regeneration. Stem Cell Res. 2016, 17, 161–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemos, D.R.; Babaeijandaghi, F.; Low, M.; Chang, C.-K.; Lee, S.T.; Fiore, D.; Zhang, R.-H.; Natarajan, A.; Nedospasov, S.A.; Rossi, F.M.V. Nilotinib reduces muscle fibrosis in chronic muscle injury by promoting TNF-mediated apoptosis of fibro/adipogenic progenitors. Nat. Med. 2015, 21, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Saccone, V.; Consalvi, S.; Giordani, L.; Mozzetta, C.; Barozzi, I.; Sandoná, M.; Ryan, T.; Rojas-Muñ, A.; Madaro, L.; Fasanaro, P.; et al. HDAC-regulated myomiRs control BAF60 variant exchange and direct the functional phenotype of fibro-adipogenic progenitors in dystrophic muscles. Genes Dev. 2014, 28, 841–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madaro, L.; Passafaro, M.; Sala, D.; Etxaniz, U.; Lugarini, F.; Proietti, D.; Alfonsi, M.V.; Nicoletti, C.; Gatto, S.; De Bardi, M.; et al. Denervation-activated STAT3–IL-6 signalling in fibro-adipogenic progenitors promotes myofibres atrophy and fibrosis. Nat. Cell Biol. 2018, 20, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, L.; Berardinelli, M.G.; Forcina, L.; Spelta, E.; Rizzuto, E.; Nicoletti, C.; Camilli, C.; Testa, E.; Catizone, A.; De Benedetti, F.; et al. Increased levels of interleukin-6 exacerbate the dystrophic phenotype in mdx mice. Hum. Mol. Genet. 2015, 24, 6041–6053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contreras, O.; Rebolledo, D.L.; Oyarzún, J.E.; Olguín, H.C.; Brandan, E. Connective tissue cells expressing fibro/adipogenic progenitor markers increase under chronic damage: Relevance in fibroblast-myofibroblast differentiation and skeletal muscle fibrosis. Cell Tissue Res. 2016, 364, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Perdiguero, E.; Serrano, A.L.; Muñoz-Cánoves, P. Cilia Control Fat Deposition during Tissue Repair. Dev. Cell 2017, 42, 114–116. [Google Scholar] [CrossRef] [PubMed]
- Kopinke, D.; Roberson, E.C.; Reiter, J.F. Ciliary Hedgehog Signaling Restricts Injury-Induced Adipogenesis. Cell 2017, 170, 340–351.e12. [Google Scholar] [CrossRef] [PubMed]
- Dalbay, M.T.; Thorpe, S.D.; Connelly, J.T.; Chapple, J.P.; Knight, M.M. Adipogenic Differentiation of hMSCs is Mediated by Recruitment of IGF-1r Onto the Primary Cilium Associated With Cilia Elongation. Stem Cells 2015, 33, 1952–1961. [Google Scholar] [CrossRef] [PubMed]
- Malicki, J.J.; Johnson, C.A. The Cilium: Cellular Antenna and Central Processing Unit. Trends Cell Biol. 2017, 27, 126–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fry, C.S.; Kirby, T.J.; Kosmac, K.; McCarthy, J.J.; Peterson, C.A. Myogenic Progenitor Cells Control Extracellular Matrix Production by Fibroblasts during Skeletal Muscle Hypertrophy. Cell Stem Cell 2017, 20, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.M.; Lawson, J.A.; Mathew, S.J.; Hutcheson, D.A.; Kardon, G. Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development 2011, 138, 3625–3637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusner, L.L.; Young, A.; Tjoe, S.; Leahy, P.; Kaminski, H.J. Perimysial Fibroblasts of Extraocular Muscle, as Unique as the Muscle Fibers. Investig. Ophthalmol. Vis. Sci. 2010, 51, 192–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Reilly, S.; Ciechomska, M.; Cant, R.; van Laar, J.M. Interleukin-6 (IL-6) Trans Signaling Drives a STAT3-dependent Pathway That Leads to Hyperactive Transforming Growth Factor-β (TGF-β) Signaling Promoting SMAD3 Activation and Fibrosis via Gremlin Protein. J. Biol. Chem. 2014, 289, 9952–9960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.F.; Tao, Y.; Li, J.; Deng, Z.; Yan, Z.; Xiao, X.; Wang, D.-Z. microRNA-1 and microRNA-206 regulate skeletal muscle satellite cell proliferation and differentiation by repressing Pax7. J. Cell Biol. 2010, 190, 867–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.; Williams, A.H.; Maxeiner, J.M.; Bezprozvannaya, S.; Shelton, J.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. microRNA-206 promotes skeletal muscle regeneration and delays progression of Duchenne muscular dystrophy in mice. J. Clin. Investig. 2012, 122, 2054–2065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingber, D.E. Cellular mechanotransduction: Putting all the pieces together again. FASEB J. 2006, 20, 811–827. [Google Scholar] [CrossRef] [PubMed]
- Zanotti, S.; Gibertini, S.; Bragato, C.; Mantegazza, R.; Morandi, L.; Mora, M. Fibroblasts from the muscles of Duchenne muscular dystrophy patients are resistant to cell detachment apoptosis. Exp. Cell Res. 2011, 317, 2536–2547. [Google Scholar] [CrossRef] [PubMed]
- Zanotti, S.; Gibertini, S.; Mora, M. Altered production of extra-cellular matrix components by muscle-derived Duchenne muscular dystrophy fibroblasts before and after TGF-β1 treatment. Cell Tissue Res. 2010, 339, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Melone, M.A.B.; Peluso, G.; Galderisi, U.; Petillo, O.; Cotrufo, R. Increased expression of IGF-binding protein-5 in Duchenne Muscular Dystrophy (DMD) fibroblasts correlates with the fibroblast-induced downregulation of DMD myoblast growth: An in vitro analysis. J. Cell. Physiol. 2000, 185, 143–153. [Google Scholar] [CrossRef]
- Kim, J.; Lee, J. Role of transforming growth factor-β in muscle damage and regeneration: Focused on eccentric muscle contraction. J. Exerc. Rehabil. 2017, 13, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Gumucio, J.P.; Flood, M.D.; Phan, A.C.; Brooks, S.V.; Mendias, C.L. Targeted inhibition of TGF-β results in an initial improvement but long-term deficit in force production after contraction-induced skeletal muscle injury. J. Appl. Physiol. 2013, 115, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Bernasconi, P.; Torchiana, E.; Confalonieri, P.; Brugnoni, R.; Barresi, R.; Mora, M.; Cornelio, F.; Morandi, L.; Mantegazza, R. Expression of transforming growth factor-beta 1 in dystrophic patient muscles correlates with fibrosis. Pathogenetic role of a fibrogenic cytokine. J. Clin. Investig. 1995, 96, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Epstein, F.H.; Border, W.A.; Noble, N.A. Transforming Growth Factor β in Tissue Fibrosis. N. Engl. J. Med. 1994, 331, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Florini, J.R.; Ewton, D.Z.; Magri, K.A. Hormones, Growth Factors, and Myogenic Differentiation. Annu. Rev. Physiol. 1991, 53, 201–216. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Black, B.L.; Derynck, R. TGF-beta inhibits muscle differentiation through functional repression of myogenic transcription factors by Smad3. Genes Dev. 2001, 15, 2950–2966. [Google Scholar] [CrossRef] [PubMed]
- McCroskery, S.; Thomas, M.; Maxwell, L.; Sharma, M.; Kambadur, R. Myostatin negatively regulates satellite cell activation and self-renewal. J. Cell. Biol. 2003, 162, 1135–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gumucio, J.P.; Sugg, K.B.; Mendias, C.L. TGF-β superfamily signaling in muscle and tendon adaptation to resistance exercise. Exerc. Sport Sci. Rev. 2015, 43, 93–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Foster, W.; Deasy, B.M.; Chan, Y.; Prisk, V.; Tang, Y.; Cummins, J.; Huard, J. Transforming Growth Factor-1 Induces the Differentiation of Myogenic Cells into Fibrotic Cells in Injured Skeletal Muscle A Key Event in Muscle Fibrogenesis. Am. J. Pathol. 2004, 164, 1007–1019. [Google Scholar] [CrossRef]
- Li, Z.B.; Kollias, H.D.; Wagner, K.R. Myostatin Directly Regulates Skeletal Muscle Fibrosis. J. Biol. Chem. 2008, 283, 19371–19378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Li, Y.; Shen, W.; Qiao, C.; Ambrosio, F.; Lavasani, M.; Nozaki, M.; Branca, M.F.; Huard, J. Relationships between TGF-β1, Myostatin, and Decorin: Implications for skeletal muscle fibrosis. J. Biol. Chem. 2007, 282, 25852–25863. [Google Scholar] [CrossRef] [PubMed]
- Bo Li, Z.; Zhang, J.; Wagner, K.R. Inhibition of myostatin reverses muscle fibrosis through apoptosis. J. Cell Sci. 2012, 125, 3957–3965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Malhotra, S.; Kumar, A. Nuclear factor-kappa B signaling in skeletal muscle atrophy. J. Mol. Med. 2008, 86, 1113–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCroskery, S.; Thomas, M.; Platt, L.; Hennebry, A.; Nishimura, T.; McLeay, L.; Sharma, M.; Kambadur, R. Improved muscle healing through enhanced regeneration and reduced fibrosis in myostatin-null mice. J. Cell Sci. 2005, 118, 3531–3541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillies, A.R.; Lieber, R.L. Structure and function of the skeletal muscle extracellular matrix. Muscle Nerve 2011, 44, 318–331. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, K.; Badlani, N.; Usas, A.; Riano, F.; Fu, F.H.; Huard, J. The Use of an Antifibrosis Agent to Improve Muscle Recovery after Laceration. Am. J. Sports Med. 2001, 29, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Li, Y.; Lu, A.; Gharaibeh, B.; Ma, J.; Kobayashi, T.; Quintero, A.J.; Huard, J. Follistatin Improves Skeletal Muscle Healing after Injury and Disease through an Interaction with Muscle Regeneration, Angiogenesis, and Fibrosis. Am. J. Pathol. 2011, 179, 915–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Shanti, N.; Stewart, C.E. Inhibitory effects of IL-6 on IGF-1 activity in skeletal myoblasts could be mediated by the activation of SOCS-3. J. Cell. Biochem. 2012, 113, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, T. Interleukin-6: Discovery of a pleiotropic cytokine. Arthritis Res. Ther. 2006, 8, S2. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Canoves, P.; Scheele, C.; Pedersen, B.K.; Serrano, A.L. Interleukin-6 myokine signaling in skeletal muscle: A double-edged sword? FEBS J. 2013, 280, 4131–4148. [Google Scholar] [CrossRef] [PubMed]
- Forcina, L.; Miano, C.; Musarò, A. The physiopathologic interplay between stem cells and tissue niche in muscle regeneration and the role of IL-6 on muscle homeostasis and diseases. Cytokine Growth Fact. Rev. 2018, 41, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Behrmann, I.; Müller-Newen, G.; Schaper, F.; Graeve, L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 1998, 334, 297–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Müller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.L.; Baeza-Raja, B.; Perdiguero, E.; Jardí, M.; Muñoz-Cánoves, P. Interleukin-6 Is an Essential Regulator of Satellite Cell-Mediated Skeletal Muscle Hypertrophy. Cell Metab. 2008, 7, 33–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose-John, S.; Heinrich, P.C. Soluble receptors for cytokines and growth factors: Generation and biological function. Biochem. J. 1994, 300, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Douglas, M.R.; Morrison, K.E.; Salmon, M.; Buckley, C.D. Why does inflammation persist: A dominant role for the stromal microenvironment? Expert Rev. Mol. Med. 2002, 4, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. IL-6 Trans-Signaling via the Soluble IL-6 Receptor: Importance for the Pro-Inflammatory Activities of IL-6. Int. J. Biol. Sci. 2012, 8, 1237–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbers, C.; Rose-John, S. Dissecting Interleukin-6 Classic- and Trans-Signaling in Inflammation and Cancer. Methods Mol. Biol. 2018, 1725, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, S.; Pelosi, L.; Piemonte, F.; Travaglini, L.; Forcina, L.; Catteruccia, M.; Petrini, S.; Verardo, M.; D’Amico, A.; Musarò, A.; et al. Oxidative stress in Duchenne muscular dystrophy: Focus on the NRF2 redox pathway. Hum. Mol. Genet. 2017, 26, 2781–2790. [Google Scholar] [CrossRef] [PubMed]
- Gabay, C. Interleukin-6 and chronic inflammation. Arthritis Res. Ther. 2006, 8, S3. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, L.; Forcina, L.; Nicoletti, C.; Scicchitano, B.M.; Musarò, A. Increased Circulating Levels of Interleukin-6 Induce Perturbation in Redox-Regulated Signaling Cascades in Muscle of Dystrophic Mice. Oxid. Med. Cell. Longev. 2017, 2017, 1987218. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Naka, T.; Kishimoto, T. IL-6-dependent and -independent pathways in the development of interleukin 17-producing T helper cells. Proc. Natl. Acad. Sci. USA 2007, 104, 12099–12104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGFβ in the Context of an Inflammatory Cytokine Milieu Supports De Novo Differentiation of IL-17-Producing T Cells. Immunity 2006, 24, 179–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fielding, C.A.; Jones, G.W.; McLoughlin, R.M.; McLeod, L.; Hammond, V.J.; Uceda, J.; Williams, A.S.; Lambie, M.; Foster, T.L.; Liao, C.-T.; et al. Interleukin-6 Signaling Drives Fibrosis in Unresolved Inflammation. Immunity 2014, 40, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Huu, D.; Matsushita, T.; Jin, G.; Hamaguchi, Y.; Hasegawa, M.; Takehara, K.; Fujimoto, M. IL-6 Blockade Attenuates the Development of Murine Sclerodermatous Chronic Graft-Versus-Host Disease. J. Investig. Dermatol. 2012, 132, 2752–2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef] [PubMed]
- Schaper, F.; Rose-John, S. Interleukin-6: Biology, signaling and strategies of blockade. Cytokine Growth Fact. Rev. 2015, 26, 475–487. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, S.; Ciechomska, M.; Cant, R.; Hügle, T.; van Laar, J.M. Interleukin-6, its role in fibrosing conditions. Cytokine Growth Fact. Rev. 2012, 23, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Castellt, J.V.; Andust, T. Interleukin-6 and the acute phase response. Biochem. J. 1990, 265, 621–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davalos, D.; Akassoglou, K. Fibrinogen as a key regulator of inflammation in disease. Semin. Immunopathol. 2012, 34, 43–62. [Google Scholar] [CrossRef] [PubMed]
- Weisel, J.W.; Litvinov, R.I. Mechanisms of fibrin polymerization and clinical implications. Blood 2013, 121, 1712–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobotta, S.; Raue, A.; Huang, X.; Vanlier, J.; Jünger, A.; Bohl, S.; Albrecht, U.; Hahnel, M.J.; Wolf, S.; Mueller, N.S.; et al. Model Based Targeting of IL-6-Induced Inflammatory Responses in Cultured Primary Hepatocytes to Improve Application of the JAK Inhibitor Ruxolitinib. Front. Physiol. 2017, 8, 775. [Google Scholar] [CrossRef] [PubMed]
- Vidal, B.; Serrano, A.L.; Tjwa, M.; Suelves, M.; Ardite, E.; De Mori, R.; Baeza-Raja, B.; Martínez De Lagrán, M.; Lafuste, P.; Ruiz-Bonilla, V.; et al. Fibrinogen drives dystrophic muscle fibrosis via a TGF/ alternative macrophage activation pathway. Genes Dev. 2008, 22, 1747–1752. [Google Scholar] [CrossRef] [PubMed]
- Fish, R.J.; Neerman-Arbez, M. Fibrinogen gene regulation. Thromb. Haemost. 2012, 108, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Cronjé, H.T.; Nienaber-Rousseau, C.; Zandberg, L.; de Lange, Z.; Green, F.R.; Pieters, M. Fibrinogen and clot-related phenotypes determined by fibrinogen polymorphisms: Independent and IL-6-interactive associations. PLoS ONE 2017, 12, e0187712. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, D.A.; Roy, C.N.; Fleming, M.D.; Loda, M.F.; Wolfsdorf, J.I.; Andrews, N.C. Inappropriate expression of hepcidin is associated with iron refractory anemia: Implications for the anemia of chronic disease. Blood 2002, 100, 3776–3781. [Google Scholar] [CrossRef] [PubMed]
- Kemna, E.; Pickkers, P.; Nemeth, E.; van der Hoeven, H.; Swinkels, D. Time-course analysis of hepcidin, serum iron, and plasma cytokine levels in humans injected with LPS. Blood 2005, 106, 1864–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, H.; Song, Y.; Colangelo, C.M.; Wu, T.; Bruce, C.; Scabia, G.; Galan, A.; Maffei, M.; Goldstein, D.R. Haptoglobin activates innate immunity to enhance acute transplant rejection in mice. J. Clin. Investig. 2012, 122, 383–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, S.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Proteomic serum biomarkers for neuromuscular diseases. Expert Rev. Proteom. 2018, 15, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Srirangan, S.; Choy, E.H. The role of interleukin 6 in the pathophysiology of rheumatoid arthritis. Ther. Adv. Musculoskelet. Dis. 2010, 2, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, L.; Berardinelli, M.G.; De Pasquale, L.; Nicoletti, C.; D’Amico, A.; Carvello, F.; Moneta, G.M.; Catizone, A.; Bertini, E.; De Benedetti, F.; et al. Functional and Morphological Improvement of Dystrophic Muscle by Interleukin 6 Receptor Blockade. EBioMedicine 2015, 2, 285–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleischmann, R. Interleukin-6 inhibition for rheumatoid arthritis. Lancet 2017, 389, 1168–1170. [Google Scholar] [CrossRef]
- Narazaki, M.; Tanaka, T.; Kishimoto, T. The role and therapeutic targeting of IL-6 in rheumatoid arthritis. Expert Rev. Clin. Immunol. 2017, 13, 535–551. [Google Scholar] [CrossRef] [PubMed]
- Wada, E.; Tanihata, J.; Iwamura, A.; Takeda, S.; Hayashi, Y.K.; Matsuda, R. Treatment with the anti-IL-6 receptor antibody attenuates muscular dystrophy via promoting skeletal muscle regeneration in dystrophin-/utrophin-deficient mice. Skelet. Muscle 2017, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Richter, K.; Kietzmann, T. Reactive oxygen species and fibrosis: Further evidence of a significant liaison. Cell Tissue Res. 2016, 365, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Scicchitano, B.M.; Pelosi, L.; Sica, G.; Musarò, A. The physiopathologic role of oxidative stress in skeletal muscle. Mech. Ageing Dev. 2018, 170, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Yamamoto, M. Nrf2–Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yamamoto, M. Molecular basis of the Keap1–Nrf2 system. Free Radic. Biol. Med. 2015, 88, 93–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forcina, L.; Pelosi, L.; Miano, C.; Musarò, A.; Forcina, L.; Pelosi, L.; Miano, C.; Musarò, A. Insights into the Pathogenic Secondary Symptoms Caused by the Primary Loss of Dystrophin. J. Funct. Morphol. Kinesiol. 2017, 2, 44. [Google Scholar] [CrossRef]
- Hecker, L.; Logsdon, N.J.; Kurundkar, D.; Kurundkar, A.; Bernard, K.; Hock, T.; Meldrum, E.; Sanders, Y.Y.; Thannickal, V.J. Reversal of Persistent Fibrosis in Aging by Targeting Nox4-Nrf2 Redox Imbalance. Sci. Transl. Med. 2014, 6, 231ra47. [Google Scholar] [CrossRef] [PubMed]
- Cabello-Verrugio, C.; Acuña, M.J.; Morales, M.G.; Becerra, A.; Simon, F.; Brandan, E. Fibrotic response induced by angiotensin-II requires NAD(P)H oxidase-induced reactive oxygen species (ROS) in skeletal muscle cells. Biochem. Biophys. Res. Commun. 2011, 410, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Florini, J.R.; Ewton, D.Z.; Coolican, S.A. Growth Hormone and the Insulin-Like Growth Factor System in Myogenesis. Endocr. Rev. 1996, 17, 481–517. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, M.V.; Davis, B.S.; Booth, F.W. IGF-I restores satellite cell proliferative potential in immobilized old skeletal muscle. J. Appl. Physiol. 2000, 89, 1365–1379. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, L.; Giacinti, C.; Nardis, C.; Borsellino, G.; Rizzuto, E.; Nicoletti, C.; Wannenes, F.; Battistini, L.; Rosenthal, N.; Molinaro, M.; et al. Local expression of IGF-1 accelerates muscle regeneration by rapidly modulating inflammatory cytokines and chemokines. FASEB J. 2007, 21, 1393–1402. [Google Scholar] [CrossRef] [PubMed]
- Scicchitano, B.M.; Dobrowolny, G.; Sica, G.; Musaro, A. Molecular Insights into Muscle Homeostasis, Atrophy and Wasting. Curr. Genom. 2018, 19, 356–369. [Google Scholar] [CrossRef] [PubMed]
- Philippou, A.; Barton, E.R. Optimizing IGF-I for skeletal muscle therapeutics. Growth Horm. IGF Res. 2014, 24, 157–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tidball, J.G.; Welc, S.S. Macrophage-Derived IGF-1 Is a Potent Coordinator of Myogenesis and Inflammation in Regenerating Muscle. Mol. Ther. 2015, 23, 1134–1135. [Google Scholar] [CrossRef] [PubMed]
- Tonkin, J.; Temmerman, L.; Sampson, R.D.; Gallego-Colon, E.; Barberi, L.; Bilbao, D.; Schneider, M.D.; Musarò, A.; Rosenthal, N. Monocyte/Macrophage-derived IGF-1 Orchestrates Murine Skeletal Muscle Regeneration and Modulates Autocrine Polarization. Mol. Ther. 2015, 23, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Mavalli, M.D.; DiGirolamo, D.J.; Fan, Y.; Riddle, R.C.; Campbell, K.S.; van Groen, T.; Frank, S.J.; Sperling, M.A.; Esser, K.A.; Bamman, M.M.; et al. Distinct growth hormone receptor signaling modes regulate skeletal muscle development and insulin sensitivity in mice. J. Clin. Investig. 2010, 120, 4007–4020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, B.T.; Lauritzen, H.P.M.M.; Hirshman, M.F.; Smyth, G.; Goodyear, L.J.; Kahn, C.R. Differential Role of Insulin/IGF-1 Receptor Signaling in Muscle Growth and Glucose Homeostasis. Cell Rep. 2015, 11, 1220–1235. [Google Scholar] [CrossRef] [PubMed]
- Arnold, L.; Henry, A.; Poron, F.; Baba-Amer, Y.; van Rooijen, N.; Plonquet, A.; Gherardi, R.K.; Chazaud, B. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med. 2007, 204, 1057–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chazaud, B.; Brigitte, M.; Yacoub-Youssef, H.; Arnold, L.; Gherardi, R.; Sonnet, C.; Lafuste, P.; Chretien, F. Dual and Beneficial Roles of Macrophages During Skeletal Muscle Regeneration. Exerc. Sport Sci. Rev. 2009, 37, 18–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef] [PubMed]
- Ramprasad, M.P.; Fischer, W.; Witztum, J.L.; Sambrano, G.R.; Quehenberger, O.; Steinberg, D. The 94- to 97-kDa mouse macrophage membrane protein that recognizes oxidized low density lipoprotein and phosphatidylserine-rich liposomes is identical to macrosialin, the mouse homologue of human CD68. Proc. Natl. Acad. Sci. USA 1995, 92, 9580–9584. [Google Scholar] [CrossRef] [PubMed]
- Rigamonti, E.; Zordan, P.; Sciorati, C.; Rovere-Querini, P.; Brunelli, S. Macrophage plasticity in skeletal muscle repair. BioMed Res. Int. 2014, 2014, 560629. [Google Scholar] [CrossRef] [PubMed]
- Juban, G.; Chazaud, B. Metabolic regulation of macrophages during tissue repair: Insights from skeletal muscle regeneration. FEBS Lett. 2017, 591, 3007–3021. [Google Scholar] [CrossRef] [PubMed]
- Haddad, F.; Adams, G.R. Aging-sensitive cellular and molecular mechanisms associated with skeletal muscle hypertrophy. J. Appl. Physiol. 2006, 100, 1188–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eliakim, A.; Oh, Y.; Cooper, D.M. Effect of single wrist exercise on fibroblast growth factor-2, insulin-like growth factor, and growth hormone. Am. J. Physiol. Integr. Comp. Physiol. 2000, 279, R548–R553. [Google Scholar] [CrossRef] [PubMed]
- Baumann, A.P.; Ibebunjo, C.; Grasser, W.A.; Paralkar, V.M. Myostatin expression in age and denervation-induced skeletal muscle atrophy. J. Musculoskelet. Neuronal Interact. 2003, 3, 8–16. [Google Scholar] [PubMed]
- De Souza Cordeiro Oliveira, L.; Gomes Côrtes, G.; Gomes de Souza Vale, R.; Henrique Martin Dantas, E. Níveis séricos de IGF-1 em gerontes. Fit. Perform. J. 2003, 2, 289–291. [Google Scholar] [CrossRef]
- Fornelli, G.; Isaia, G.C.; D’amelio, P. Ageing, muscle and bone. J. Gerontol. Geriatr. 2016, 64, 75–80. [Google Scholar]
- Castro, J.B.P.; Vale, R.G.S. Insulin-like growth factor i (igf-1) in older adults: A review. MOJ Gerontol. Geriatr. 2017, 1, 175–176. [Google Scholar] [CrossRef]
- Musarò, A.; McCullagh, K.; Paul, A.; Houghton, L.; Dobrowolny, G.; Molinaro, M.; Barton, E.R.; Sweeney, H.L.; Rosenthal, N. Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat. Genet. 2001, 27, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Burks, T.N.; Andres-Mateos, E.; Marx, R.; Mejias, R.; Van Erp, C.; Simmers, J.L.; Walston, J.D.; Ward, C.W.; Cohn, R.D. Losartan Restores Skeletal Muscle Remodeling and Protects Against Disuse Atrophy in Sarcopenia. Sci. Transl. Med. 2011, 3, 82ra37. [Google Scholar] [CrossRef] [PubMed]
- Accorsi, A.; Kumar, A.; Rhee, Y.; Miller, A.; Girgenrath, M. IGF-1/GH axis enhances losartan treatment in Lama2-related muscular dystrophy. Hum. Mol. Genet. 2016, 25, ddw291. [Google Scholar] [CrossRef] [PubMed]
- Barton, E.R.; Morris, L.; Musaro, A.; Rosenthal, N.; Sweeney, H.L. Muscle-specific expression of insulin-like growth factor I counters muscle decline in mdx mice. J. Cell Biol. 2002, 157, 137–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelosi, L.; Coggi, A.; Forcina, L.; Musarò, A. MicroRNAs modulated by local mIGF-1 expression in mdx dystrophic mice. Front. Aging Neurosci. 2015, 7, 69. [Google Scholar] [CrossRef] [PubMed]
- Huh, J.Y.; Panagiotou, G.; Mougios, V.; Brinkoetter, M.; Vamvini, M.T.; Schneider, B.E.; Mantzoros, C.S. FNDC5 and irisin in humans: I. Predictors of circulating concentrations in serum and plasma and II. mRNA expression and circulating concentrations in response to weight loss and exercise. Metabolism 2012, 61, 1725–1738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huh, J.Y.; Dincer, F.; Mesfum, E.; Mantzoros, C.S. Irisin stimulates muscle growth-related genes and regulates adipocyte differentiation and metabolism in humans. Int. J. Obes. 2014, 38, 1538–1544. [Google Scholar] [CrossRef] [PubMed]
- Jedrychowski, M.P.; Wrann, C.D.; Paulo, J.A.; Gerber, K.K.; Szpyt, J.; Robinson, M.M.; Nair, K.S.; Gygi, S.P.; Spiegelman, B.M. Detection and Quantitation of Circulating Human Irisin by Tandem Mass Spectrometry. Cell Metab. 2015, 22, 734–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reza, M.M.; Sim, C.M.; Subramaniyam, N.; Ge, X.; Sharma, M.; Kambadur, R.; McFarlane, C. Irisin treatment improves healing of dystrophic skeletal muscle. Oncotarget 2017, 8, 98553–98566. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forcina, L.; Miano, C.; Scicchitano, B.M.; Musarò, A. Signals from the Niche: Insights into the Role of IGF-1 and IL-6 in Modulating Skeletal Muscle Fibrosis. Cells 2019, 8, 232. https://doi.org/10.3390/cells8030232
Forcina L, Miano C, Scicchitano BM, Musarò A. Signals from the Niche: Insights into the Role of IGF-1 and IL-6 in Modulating Skeletal Muscle Fibrosis. Cells. 2019; 8(3):232. https://doi.org/10.3390/cells8030232
Chicago/Turabian StyleForcina, Laura, Carmen Miano, Bianca Maria Scicchitano, and Antonio Musarò. 2019. "Signals from the Niche: Insights into the Role of IGF-1 and IL-6 in Modulating Skeletal Muscle Fibrosis" Cells 8, no. 3: 232. https://doi.org/10.3390/cells8030232
APA StyleForcina, L., Miano, C., Scicchitano, B. M., & Musarò, A. (2019). Signals from the Niche: Insights into the Role of IGF-1 and IL-6 in Modulating Skeletal Muscle Fibrosis. Cells, 8(3), 232. https://doi.org/10.3390/cells8030232