Cellular and Animal Models of Striated Muscle Laminopathies



Abstract
:1. Introduction
2. Cellular Models
2.1. Evidence of Abnormal Nuclear Morphology Coupled with Aberrant Lamin A/C Phenotype and Mislocalization of Several Lamin A/C Binding Proteins in LMNA-Related Cardiac and Skeletal Muscle Disease
2.2. Evidence of Disrupted Lamin A/C Interaction with Binding Partners, which Can Result in Multiple Tissue Phenotypes
2.3. Evidence of Impaired Lamin A/C Structural Stability and Dynamics in Striated Muscle Laminopathies
2.4. Evidence of Compromised Nuclear and Cytoskeletal Mechanics in the Presence of LMNA Mutations Causing Muscular Laminopathies
2.5. Evidence of Altered Lamin A/C Post-Translational Modification Status in Striated Muscle Laminopathy
2.6. Derived Myogenic or Cardiac Cells from Patients’ Induced iPSCs Replicated Previously Demonstrated Muscular Laminopathy Cellular Phenotypes with 3D Culturing: A Promising Method of Identifying and Examining More Subtle Morphological Defects in Mutant Cells
3. Animal Models
3.1. Caenorhabditis elegans (Worm) Models
Worm Models Mimicked Patient Cellular Phenotypes, Highlighted the Role of Lamin A/C in Germ Cells and Reproduction, and Further Confirmed Results from Cell Lines and Mice Models
3.2. Drosophila melanogaster (Fruit Fly) Models
Fruit Fly Models Phenocopied Early Lethality in LMNA Null Patients, Demonstrated Cytosketal Disturbance, Presented with Muscle and Mobility Defects, and Associated the Oxidative/Reductive Stress Signalling Pathway (via Nrf2) to Striated Muscle Laminopathies
3.3. Danio rerio (Zebrafish) Models
Zebrafish Models Mirrored Patient and Mice Model Cardiac and Skeletal Muscle Phenotypes, and Presented a Feasible Avenue to Facilitate High Throughput Drug and Therapeutic Target Screening
3.4. Mus musculus (Mice) Models
3.4.1. Lmna Null Mice Models Proved the Causal Link between Lamin and Human Laminopathy Phenotypes, Resulting in Lethality in the Young—But Heterozygous Mice Failed to Show the Laminopathy Phenotype
3.4.2. Homozygous C-Terminal Truncated Lamin A Mice Model Recapitulated the Lmna Null Mice Phenotype and the Heterozygous Mice Displayed a Late-Onset Cardiac Phenotype
3.4.3. Mice Expressing Non-Farnesylated Prelamin A Only, Mature Lamin A Only or Lamin C Only Showed the Importance of Lamin A Processing and the Respective Roles of Lamins A and C in the Pathogenesis of Laminopathies
3.4.4. Lmna Knock-In and Transgenic Mice Further Established the Genotype and Phenotype Correlation and Revealed Several Signalling Pathways Involved in the Development of Myopathies Caused by LMNA Mutations
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACE | angiotensin II converting enzyme |
| Akt | Protein Kinase B |
| ANP | atrial natriuretic peptide |
| BNP | brain natriuretic peptide |
| CD | conduction defect |
| CMT2B1 | Charcot‒Marie‒Tooth disease type 2B1 |
| CRISPR/Cas9 | Clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 |
| Cx 40/43 | Connexin 40/43 |
| DCM | Dilated Cardiomyopathy |
| DMD | Duchenne Muscular Dystrophy |
| EDMD | Emery‒Dreifuss Muscular Dystrophy |
| ERK 1/2 | Extracellular signal-regulated kinases 1/2 |
| FHL2 | four-and-a half LIM protein-2 |
| FPLD2 | Familial partial lipodystrophy Dunnigan-type 2 |
| Hf1b/Sp4 | Transcription factor Sp4 |
| HGPS | Hutchison Gilford Progeria Syndrome |
| hpf | hours post-fertilization |
| iPSCs | induced pluripotent stem cells |
| JNK | c-Jun N-terminal kinases |
| Keap1 | Kelch-like ECH-associated protein 1 |
| LAP2 | Lamina-associated polypeptide 2 |
| L-CMD | LMNA-related Congenital Muscular Dystrophy |
| LGMD1B | 1B Limb-girdle muscular dystrophy |
| MAPK | Mitogen-activated protein kinase |
| MEFs | Mouse embryonic fibroblasts |
| MEK 1/2 | Dual-specificity mitogen-activated protein kinase kinase ½ |
| MO | Morpholino |
| mTor | mechanistic target of rapamycin |
| Myh6 | myosin heavy-chain alpha |
| Myh7 | myosin heavy-chain beta |
| NAT10 | N-Acetyltransferase 10 |
| NFκB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NPCs | Nuclear pore complexes |
| Nppb | Natriuretic peptide B |
| Nrf2 | Nuclear factor erythroid-2-related factor 2 |
| Nup153/154 | Nucleoporin 153/154 |
| p62/SQSTM1 | Sequestosome-1 (ubiquitin-binding protein p62) |
| PCNA | Proliferating cell nuclear antigen |
| PPAR-gamma | Peroxisome proliferator-activated receptors gamma |
| Smad 2/3 | Mothers against decapentaplegic homolog 2/3 |
| SREBF-1 | Sterol regulatory element binding transcription factor 1 |
| SRF | Serum response factor |
| SUMO1/2 | Small ubiquitin-related modifier 1/2 |
| syne1 | Spectrin repeat-containing nuclear envelope protein 1 |
| TF | Transcription factor |
| TGF-β | Transforming growth factor beta |
| WNT | Wingless/Integrated |
References
- Mercuri, E.; Brown, S.C.; Nihoyannopoulos, P.; Poulton, J.; Kinali, M.; Richard, P.; Piercy, R.J.; Messina, S.; Sewry, C.; Burke, M.M.; et al. Extreme variability of skeletal and cardiac muscle involvement in patients with mutations in exon 11 of the lamin A/C gene. Muscle Nerve 2005, 31, 602–609. [Google Scholar] [CrossRef]
- Francisco, A.R.G.; Santos Gonçalves, I.; Veiga, F.; Mendes Pedro, M.; Pinto, F.J.; Brito, D. Complex phenotype linked to a mutation in exon 11 of the lamin A/C gene: Hypertrophic cardiomyopathy, atrioventricular block, severe dyslipidemia and diabetes. Rev. Portug. Cardiol. 2017, 36, 699.e1–699.e4. [Google Scholar] [CrossRef]
- Forleo, C.; Carmosino, M.; Resta, N.; Rampazzo, A.; Valecce, R.; Sorrentino, S.; Iacoviello, M.; Pisani, F.; Procino, G.; Gerbino, A.; et al. Clinical and functional characterization of a novel mutation in lamin a/c gene in a multigenerational family with arrhythmogenic cardiac laminopathy. PLoS ONE 2015, 10, e0121723. [Google Scholar] [CrossRef]
- Kato, K.; Takahashi, N.; Fujii, Y.; Umehara, A.; Nishiuchi, S.; Makiyama, T.; Ohno, S.; Horie, M. LMNA cardiomyopathy detected in Japanese arrhythmogenic right ventricular cardiomyopathy cohort. J. Cardiol. 2016, 68, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Quarta, G.; Syrris, P.; Ashworth, M.; Jenkins, S.; Zuborne Alapi, K.; Morgan, J.; Muir, A.; Pantazis, A.; McKenna, W.J.; Elliott, P.M. Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2012, 33, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Worman, H.J. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J. Biol. Chem. 1993, 268, 16321–16326. [Google Scholar] [PubMed]
- Stewart, C.; Burke, B. Teratocarcinoma stem cells and early mouse embryos contain only a single major lamin polypeptide closely resembling lamin B. Cell 1987, 51, 383–392. [Google Scholar] [CrossRef]
- McKeon, F.D.; Kirschner, M.W.; Caput, D. Homologies in both primary and secondary structure between nuclear envelope and intermediate filament proteins. Nature 1986, 319, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.K.; Fryns, J.-P.; Auchus, R.J.; Garg, A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum. Mol. Genet. 2003, 12, 1995–2001. [Google Scholar] [CrossRef]
- Machiels, B.M.; Zorenc, A.H.; Endert, J.M.; Kuijpers, H.J.; van Eys, G.J.; Ramaekers, F.C.; Broers, J.L. An alternative splicing product of the lamin A/C gene lacks exon 10. J. Biol. Chem. 1996, 271, 9249–9253. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, K.; Inagaki, H.; Hotta, Y. Identification and cloning of an mRNA coding for a germ cell-specific A-type lamin in mice. Exp. Cell Res. 1994, 212, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Alsheimer, M.; Benavente, R. Change of karyoskeleton during mammalian spermatogenesis: Expression pattern of nuclear lamin C2 and its regulation. Exp. Cell Res. 1996, 228, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Stuurman, N.; Heins, S.; Aebi, U. Nuclear lamins: Their structure, assembly, and interactions. J. Struct. Biol. 1998, 122, 42–66. [Google Scholar] [CrossRef] [PubMed]
- Broers, J.L.; Ramaekers, F.C.; Bonne, G.; Yaou, R.B.; Hutchison, C.J. Nuclear lamins: Laminopathies and their role in premature ageing. Physiol. Rev. 2006, 86, 967–1008. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, D.J.; Trembath, R.C.; Shackleton, S. A novel interaction between lamin A and SREBP1: Implications for partial lipodystrophy and other laminopathies. Hum. Mol. Genet. 2002, 11, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Clements, L.; Manilal, S.; Love, D.R.; Morris, G.E. Direct interaction between emerin and lamin A. Biochem. Biophys. Res. Commun. 2000, 267, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Sakaki, M.; Koike, H.; Takahashi, N.; Sasagawa, N.; Tomioka, S.; Arahata, K.; Ishiura, S. Interaction between emerin and nuclear lamins. J. Biochem. (Tokyo) 2001, 129, 321–327. [Google Scholar] [CrossRef]
- Cartegni, L.; di Barletta, M.R.; Barresi, R.; Squarzoni, S.; Sabatelli, P.; Maraldi, N.; Mora, M.; Di Blasi, C.; Cornelio, F.; Merlini, L. Heart-specific localization of emerin: New insights into Emery-Dreifuss muscular dystrophy. Hum. Mol. Genet. 1997, 6, 2257–2264. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Meta, M.; Qiao, X.; Frost, D.; Bauch, J.; Coffinier, C.; Majumdar, S.; Bergo, M.O.; Young, S.G.; Fong, L.G. A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J. Clin. Investig. 2006, 116, 2115–2121. [Google Scholar] [CrossRef] [PubMed]
- Muchir, A.; Pavlidis, P.; Decostre, V.; Herron, A.J.; Arimura, T.; Bonne, G.; Worman, H.J. Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J. Clin. Investig. 2007, 117, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, C.; Wang, X.; Briggs, M.R.; Admon, A.; Wu, J.; Hua, X.; Goldstein, J.L.; Brown, M.S. SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell 1993, 75, 187–197. [Google Scholar] [CrossRef]
- Tontonoz, P.; Kim, J.B.; Graves, R.A.; Spiegelman, B.M. ADD1: A novel helix-loop-helix transcription factor associated with adipocyte determination and differentiation. Mol. Cell Biol. 1993, 13, 4753–4759. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Spiegelman, B.M. ADD1/SREBP1 promotes adipocyte differentiation and gene expression linked to fatty acid metabolism. Genes Dev. 1996, 10, 1096–1107. [Google Scholar] [CrossRef] [PubMed]
- Diraison, F.; Ravier, M.A.; Richards, S.K.; Smith, R.M.; Shimano, H.; Rutter, G.A. SREBP1 is required for the induction by glucose of pancreatic beta-cell genes involved in glucose sensing. J. Lipid Res. 2008, 49, 814–822. [Google Scholar] [CrossRef]
- Hasty, A.H.; Shimano, H.; Yahagi, N.; Amemiya-Kudo, M.; Perrey, S.; Yoshikawa, T.; Osuga, J.; Okazaki, H.; Tamura, Y.; Iizuka, Y.; et al. Sterol regulatory element-binding protein-1 is regulated by glucose at the transcriptional level. J. Biol. Chem. 2000, 275, 31069–31077. [Google Scholar] [CrossRef] [PubMed]
- Boguslavsky, R.L.; Stewart, C.L.; Worman, H.J. Nuclear lamin A inhibits adipocyte differentiation: Implications for Dunnigan-type familial partial lipodystrophy. Hum. Mol. Genet. 2006, 15, 653–663. [Google Scholar] [CrossRef]
- Wojtanik, K.M.; Edgemon, K.; Viswanadha, S.; Lindsey, B.; Haluzik, M.; Chen, W.; Poy, G.; Reitman, M.; Londos, C. The role of LMNA in adipose: A novel mouse model of lipodystrophy based on the Dunnigan-type familial partial lipodystrophy mutation. J. Lipid Res. 2009, 50, 1068–1079. [Google Scholar] [CrossRef] [PubMed]
- Lanzicher, T.; Martinelli, V.; Puzzi, L.; Del Favero, G.; Codan, B.; Long, C.S.; Mestroni, L.; Taylor, M.R.; Sbaizero, O. The Cardiomyopathy Lamin A/C D192G Mutation Disrupts Whole-Cell Biomechanics in Cardiomyocytes as Measured by Atomic Force Microscopy Loading-Unloading Curve Analysis. Sci. Rep. 2015, 5, 13388. [Google Scholar] [CrossRef]
- Zwerger, M.; Jaalouk, D.E.; Lombardi, M.L.; Isermann, P.; Mauermann, M.; Dialynas, G.; Herrmann, H.; Wallrath, L.L.; Lammerding, J. Myopathic lamin mutations impair nuclear stability in cells and tissue and disrupt nucleo-cytoskeletal coupling. Hum. Mol. Genet. 2013, 22, 2335–2349. [Google Scholar] [CrossRef]
- Lammerding, J.; Schulze, P.C.; Takahashi, T.; Kozlov, S.; Sullivan, T.; Kamm, R.D.; Stewart, C.L.; Lee, R.T. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J. Clin. Investig. 2004, 113, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.H.; Hale, C.M.; Panorchan, P.; Khatau, S.B.; George, J.P.; Tseng, Y.; Stewart, C.L.; Hodzic, D.; Wirtz, D. Nuclear lamin A/C deficiency induces defects in cell mechanics, polarization, and migration. Biophys. J. 2007, 93, 2542–2552. [Google Scholar] [CrossRef] [PubMed]
- Lammerding, J.; Fong, L.G.; Ji, J.Y.; Reue, K.; Stewart, C.L.; Young, S.G.; Lee, R.T. Lamins A and C but not lamin B1 regulate nuclear mechanics. J. Biol. Chem. 2006, 281, 25768–25780. [Google Scholar] [CrossRef] [PubMed]
- Laurini, E.; Martinelli, V.; Lanzicher, T.; Puzzi, L.; Borin, D.; Chen, S.N.; Long, C.S.; Lee, P.; Mestroni, L.; Taylor, M.R.G.; et al. Biomechanical defects and rescue of cardiomyocytes expressing pathologic nuclear lamins. Cardiovasc. Res. 2018, 114, 846–857. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, C.; Fischer, M.; Mamchaoui, K.; Bigot, A.; Lok, T.; Verdier, C.; Duperray, A.; Michel, R.; Holt, I.; Voit, T.; et al. Lamins and nesprin-1 mediate inside-out mechanical coupling in muscle cell precursors through FHOD1. Sci. Rep. 2017, 7, 1253. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, A.T.; Ziaei, S.; Ehret, C.; Duchemin, H.; Mamchaoui, K.; Bigot, A.; Mayer, M.; Quijano-Roy, S.; Desguerre, I.; Lainé, J.; et al. Cellular microenvironments reveal defective mechanosensing responses and elevated YAP signaling in LMNA-mutated muscle precursors. J. Cell Sci. 2014, 127 Pt 13, 2873–2884. [Google Scholar] [CrossRef]
- Tesson, F.; Saj, M.; Uvaize, M.M.; Nicolas, H.; Płoski, R.; Bilińska, Z. Lamin A/C mutations in dilated cardiomyopathy. Cardiol. J. 2014, 21, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Sylvius, N.; Bilinska, Z.T.; Veinot, J.P.; Fidzianska, A.; Bolongo, P.M.; Poon, S.; McKeown, P.; Davies, R.A.; Chan, K.L.; Tang, A.S.; et al. In vivo and in vitro examination of the functional significances of novel lamin gene mutations in heart failure patients. J. Med. Genet. 2005, 42, 639–647. [Google Scholar] [CrossRef]
- Taylor, M.R.; Fain, P.R.; Sinagra, G.; Robinson, M.L.; Robertson, A.D.; Carniel, E.; Di Lenarda, A.; Bohlmeyer, T.J.; Ferguson, D.A.; Brodsky, G.L.; et al. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J. Am. Coll. Cardiol. 2003, 41, 771–780. [Google Scholar] [CrossRef]
- Hasselberg, N.E.; Haland, T.F.; Saberniak, J.; Brekke, P.H.; Berge, K.E.; Leren, T.P.; Edvardsen, T.; Haugaa, K.H. Lamin A/C cardiomyopathy: Young onset, high penetrance, and frequent need for heart transplantation. Eur. Heart J. 2018, 39, 853–860. [Google Scholar] [CrossRef]
- Bonne, G.; Mercuri, E.; Muchir, A.; Urtizberea, A.; Bécane, H.M.; Recan, D.; Merlini, L.; Wehnert, M.; Boor, R.; Reuner, U.; et al. Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann. Neurol. 2000, 48, 170–180. [Google Scholar] [CrossRef]
- Quijano-Roy, S.; Mbieleu, B.; Bönnemann, C.G.; Jeannet, P.Y.; Colomer, J.; Clarke, N.F.; Cuisset, J.M.; Roper, H.; De Meirleir, L.; D’Amico, A.; et al. De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann. Neurol. 2008, 64, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Muchir, A.; Bonne, G.; van der Kooi, A.J.; van Meegen, M.; Baas, F.; Bolhuis, P.A.; de Visser, M.; Schwartz, K. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum. Mol. Genet. 2000, 9, 1453–1459. [Google Scholar] [CrossRef]
- Bonne, G.; Barletta, M.R.D.; Varnous, S.; Bécane, H.M.; Hammouda, E.H.; Merlini, L.; Muntoni, F.; Greenberg, C.R.; Gary, F.; Urtizberea, J.A.; et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat. Genet. 1999, 21, 285–288. [Google Scholar] [CrossRef]
- van Engelen, B.G.M.; Muchir, A.; Hutchison, C.J.; van der Kooi, A.J.; Bonne, G.; Lammens, M. The lethal phenotype of a homozygous nonsense mutation in the lamin A/C gene. Neurology 2005, 64, 374–376. [Google Scholar] [CrossRef] [PubMed]
- Muchir, A.; van Engelen, B.G.; Lammens, M.; Mislow, J.M.; McNally, E.; Schwartz, K.; Bonne, G. Nuclear envelope alterations in fibroblasts from LGMD1B patients carrying nonsense Y259X heterozygous or homozygous mutation in lamin A/C gene. Exp. Cell Res. 2003, 291, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, C.J. B-type lamins in health and disease. Semin. Cell Dev. Biol. 2014, 29, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Harborth, J.; Elbashir, S.M.; Bechert, K.; Tuschl, T.; Weber, K. Identification of essential genes in cultured mammalian cells using small interfering RNAs. J. Cell Sci. 2001, 114 Pt 24, 4557–4565. [Google Scholar]
- Raharjo, W.H.; Enarson, P.; Sullivan, T.; Stewart, C.L.; Burke, B. Nuclear envelope defects associated with LMNA mutations cause dilated cardiomyopathy and Emery-Dreifuss muscular dystrophy. J. Cell Sci. 2001, 114 Pt 24, 4447–4457. [Google Scholar]
- Holt, I.; Ostlund, C.; Stewart, C.L.; thi Man, N.; Worman, H.J.; Morris, G.E. Effect of pathogenic mis-sense mutations in lamin A on its interaction with emerin in vivo. J. Cell Sci. 2003, 116 Pt 14, 3027–3035. [Google Scholar] [CrossRef]
- Muchir, A.; Medioni, J.; Laluc, M.; Massart, C.; Arimura, T.; van der Kooi, A.J.; Desguerre, I.; Mayer, M.; Ferrer, X.; Briault, S.; et al. Nuclear envelope alterations in fibroblasts from patients with muscular dystrophy, cardiomyopathy, and partial lipodystrophy carrying lamin A/C gene mutations. Muscle Nerve 2004, 30, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Fatkin, D.; MacRae, C.; Sasaki, T.; Wolff, M.R.; Porcu, M.; Frenneaux, M.; Atherton, J.; Vidaillet, H.J., Jr.; Spudich, S.; De Girolami, U.; et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med. 1999, 341, 1715–1724. [Google Scholar] [CrossRef] [PubMed]
- Ostlund, C.; Bonne, G.; Schwartz, K.; Worman, H.J. Properties of lamin A mutants found in Emery-Dreifuss muscular dystrophy, cardiomyopathy and Dunnigan-type partial lipodystrophy. J. Cell Sci. 2001, 114 Pt 24, 4435–4445. [Google Scholar] [PubMed]
- Sylvius, N.; Hathaway, A.; Boudreau, E.; Gupta, P.; Labib, S.; Bolongo, P.M.; Rippstein, P.; McBride, H.; Bilinska, Z.T.; Tesson, F. Specific contribution of lamin A and lamin C in the development of laminopathies. Exp. Cell Res. 2008, 314, 2362–2375. [Google Scholar] [CrossRef] [PubMed]
- Motsch, I.; Kaluarachchi, M.; Emerson, L.J.; Brown, C.A.; Brown, S.C.; Dabauvalle, M.C.; Ellis, J.A. Lamins A and C are differentially dysfunctional in autosomal dominant Emery-Dreifuss muscular dystrophy. Eur. J. Cell Biol. 2005, 84, 765–781. [Google Scholar] [CrossRef]
- Fong, L.G.; Ng, J.K.; Lammerding, J.; Vickers, T.A.; Meta, M.; Coté, N.; Gavino, B.; Qiao, X.; Chang, S.Y.; Young, S.R.; et al. Prelamin A and lamin A appear to be dispensable in the nuclear lamina. J. Clin. Investig. 2006, 116, 743–752. [Google Scholar] [CrossRef]
- Vigouroux, C.; Magré, J.; Vantyghem, M.C.; Bourut, C.; Lascols, O.; Shackleton, S.; Lloyd, D.J.; Guerci, B.; Padova, G.; Valensi, P.; et al. Lamin A/C gene: Sex-determined expression of mutations in Dunnigan-type familial partial lipodystrophy and absence of coding mutations in congenital and acquired generalized lipoatrophy. Diabetes 2000, 49, 1958–1962. [Google Scholar] [CrossRef]
- Simon, D.N.; Zastrow, M.S.; Wilson, K.L. Direct actin binding to A- and B-type lamin tails and actin filament bundling by the lamin A tail. Nucl. Austin Tex. 2010, 1, 264–272. [Google Scholar] [CrossRef]
- Buendia, B. LMNA p.R482W mutation related to FPLD2 alters SREBP1-A type lamin interactions in human fibroblasts and adipose stem cells. Orphanet J. Rare Dis. 2015, 10, O13. [Google Scholar] [CrossRef]
- Vadrot, N.; Duband-Goulet, I.; Cabet, E.; Attanda, W.; Barateau, A.; Vicart, P.; Gerbal, F.; Briand, N.; Vigouroux, C.; Oldenburg, A.R.; et al. The p.R482W substitution in A-type lamins deregulates SREBP1 activity in Dunnigan-type familial partial lipodystrophy. Hum. Mol. Genet. 2015, 24, 2096–2109. [Google Scholar] [CrossRef]
- Sullivan, T.; Escalante-Alcalde, D.; Bhatt, H.; Anver, M.; Bhat, N.; Nagashima, K.; Stewart, C.L.; Burke, B. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol. 1999, 147, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Liu, X.G.; Zheng, H.L.; Li, J.B.; Xiong, X.D.; Zhang, C.L.; Luo, C.Y.; Zhou, Z.J.; Shi, Q.; Weng, Y.G. Deletion of the lmna gene induces growth delay and serum biochemical changes in C57BL/6 mice. Asian-Australas J. Anim. Sci. 2014, 27, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, S.; Gilbert, N.; Perry, P.; Ostlund, C.; Worman, H.J.; Bickmore, W.A. Altered protein dynamics of disease-associated lamin A mutants. BMC Cell Biol. 2004, 5, 46. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Bilinska, Z.T.; Sylvius, N.; Boudreau, E.; Veinot, J.P.; Labib, S.; Bolongo, P.M.; Hamza, A.; Jackson, T.; Ploski, R.; et al. Genetic and ultrastructural studies in dilated cardiomyopathy patients: A large deletion in the lamin A/C gene is associated with cardiomyocyte nuclear envelope disruption. Basic Res. Cardiol. 2010, 105, 365–377. [Google Scholar] [CrossRef]
- Cattin, M.-E.; Bertrand, A.T.; Schlossarek, S.; Le Bihan, M.C.; Skov Jensen, S.; Neuber, C.; Crocini, C.; Maron, S.; Lainé, J.; Mougenot, N.; et al. Heterozygous LmnadelK32 mice develop dilated cardiomyopathy through a combined pathomechanism of haploinsufficiency and peptide toxicity. Hum. Mol. Genet. 2013, 22, 3152–3164. [Google Scholar] [CrossRef]
- Nikolova, V.; Leimena, C.; McMahon, A.C.; Tan, J.C.; Chandar, S.; Jogia, D.; Kesteven, S.H.; Michalicek, J.; Otway, R.; Verheyen, F.; et al. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J. Clin. Investig. 2004, 113, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Muchir, A.; Wu, W.; Choi, J.C.; Iwata, S.; Morrow, J.; Homma, S.; Worman, H.J. Abnormal p38alpha mitogen-activated protein kinase signaling in dilated cardiomyopathy caused by lamin A/C gene mutation. Hum. Mol. Genet. 2012, 21, 4325–4333. [Google Scholar] [CrossRef] [PubMed]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef]
- Cobb, A.M.; Larrieu, D.; Warren, D.T.; Liu, Y.; Srivastava, S.; Smith, A.J.O.; Bowater, R.P.; Jackson, S.P.; Shanahan, C.M. Prelamin A impairs 53BP1 nuclear entry by mislocalizing NUP153 and disrupting the Ran gradient. Aging Cell 2016, 15, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Larrieu, D.; Britton, S.; Demir, M.; Rodriguez, R.; Jackson, S.P. Chemical inhibition of NAT10 corrects defects of laminopathic cells. Science 2014, 344, 527–532. [Google Scholar] [CrossRef]
- Davidson, P.M.; Lammerding, J. Broken nuclei--lamins, nuclear mechanics, and disease. Trends Cell Biol. 2014, 24, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Kochin, V.; Shimi, T.; Torvaldson, E.; Adam, S.A.; Goldman, A.; Pack, C.G.; Melo-Cardenas, J.; Imanishi, S.Y.; Goldman, R.D.; Eriksson, J.E. Interphase phosphorylation of lamin A. J. Cell Sci. 2014, 127 Pt 12, 2683–2696. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, H.; Hayashi, Y.K.; Matsuda, C.; Noguchi, S.; Wakatsuki, S.; Araki, T.; Nishino, I. Specific phosphorylation of Ser458 of A-type lamins in LMNA-associated myopathy patients. J. Cell Sci. 2010, 123 Pt 22, 3893–3900. [Google Scholar] [CrossRef]
- Krimm, I.; Ostlund, C.; Gilquin, B.; Couprie, J.; Hossenlopp, P.; Mornon, J.P.; Bonne, G.; Courvalin, J.C.; Worman, H.J.; Zinn-Justin, S. The Ig-like structure of the C-terminal domain of lamin A/C, mutated in muscular dystrophies, cardiomyopathy, and partial lipodystrophy. Struct. Lond. Engl. 2002, 10, 811–823. [Google Scholar] [CrossRef]
- Swift, J.; Ivanovska, I.L.; Buxboim, A.; Harada, T.; Dingal, P.C.; Pinter, J.; Pajerowski, J.D.; Spinler, K.R.; Shin, J.W.; Tewari, M.; et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 2013, 341, 1240104. [Google Scholar] [CrossRef] [PubMed]
- Cenni, V.; Sabatelli, P.; Mattioli, E.; Marmiroli, S.; Capanni, C.; Ognibene, A.; Squarzoni, S.; Maraldi, N.M.; Bonne, G.; Columbaro, M.; et al. Lamin A N-terminal phosphorylation is associated with myoblast activation: Impairment in Emery-Dreifuss muscular dystrophy. J. Med. Genet. 2005, 42, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-Q.; Sarge, K.D. Sumoylation regulates lamin A function and is lost in lamin A mutants associated with familial cardiomyopathies. J. Cell Biol. 2008, 182, 35–39. [Google Scholar] [CrossRef]
- Boudreau, E.; Labib, S.; Bertrand, A.T.; Decostre, V.; Bolongo, P.M.; Sylvius, N.; Bonne, G.; Tesson, F. Lamin A/C mutants disturb sumo1 localization and sumoylation in vitro and in vivo. PLoS ONE 2012, 7, e45918. [Google Scholar] [CrossRef] [PubMed]
- Siu, C.-W.; Lee, Y.-K.; Ho, J.C.-Y.; Lai, W.H.; Chan, Y.C.; Ng, K.M.; Wong, L.Y.; Au, K.W.; Lau, Y.M.; Zhang, J.; et al. Modeling of lamin A/C mutation premature cardiac aging using patient-specific induced pluripotent stem cells. Aging 2012, 4, 803–822. [Google Scholar] [CrossRef]
- Ho, J.C.Y.; Zhou, T.; Lai, W.-H.; Huang, Y.; Chan, Y.C.; Li, X.; Wong, N.L.; Li, Y.; Au, K.W.; Guo, D.; et al. Generation of induced pluripotent stem cell lines from 3 distinct laminopathies bearing heterogeneous mutations in lamin A/C. Aging 2011, 3, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Steele-Stallard, H.B.; Pinton, L.; Sarcar, S.; Ozdemir, T.; Maffioletti, S.M.; Zammit, P.S.; Tedesco, F.S. Modeling Skeletal Muscle Laminopathies Using Human Induced Pluripotent Stem Cells Carrying Pathogenic LMNA Mutations. Front. Physiol. 2018, 9, 1332. [Google Scholar] [CrossRef]
- Riemer, D.; Dodemont, H.; Weber, K. A nuclear lamin of the nematode Caenorhabditis elegans with unusual structural features; cDNA cloning and gene organization. Eur. J. Cell Biol. 1993, 62, 214–223. [Google Scholar] [PubMed]
- Liu, J.; Rolef Ben-Shahar, T.; Riemer, D.; Treinin, M.; Spann, P.; Weber, K.; Fire, A.; Gruenbaum, Y. Essential roles for Caenorhabditis elegans lamin gene in nuclear organization, cell cycle progression, and spatial organization of nuclear pore complexes. Mol. Biol. Cell 2000, 11, 3937–3947. [Google Scholar] [CrossRef] [PubMed]
- Alsheimer, M.; Liebe, B.; Sewell, L.; Stewart, C.L.; Scherthan, H.; Benavente, R. Disruption of spermatogenesis in mice lacking A-type lamins. J. Cell Sci. 2004, 117 Pt 7, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Chen, W.; Shao, B.; Qi, Y.; Xia, Z.; Wang, F.; Wang, L.; Guo, X.; Huang, X.; Sha, J. Lamin A/C proteins in the spermatid acroplaxome are essential in mouse spermiogenesis. Reprod. Camb. Engl. 2014, 148, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Mounkes, L.C.; Kozlov, S.V.; Rottman, J.N.; Stewart, C.L. Expression of an LMNA-N195K variant of A-type lamins results in cardiac conduction defects and death in mice. Hum. Mol. Genet. 2005, 14, 2167–2180. [Google Scholar] [CrossRef] [PubMed]
- Bank, E.M.; Ben-Harush, K.; Wiesel-Motiuk, N.; Barkan, R.; Feinstein, N.; Lotan, O.; Medalia, O.; Gruenbaum, Y. A laminopathic mutation disrupting lamin filament assembly causes disease-like phenotypes in Caenorhabditis elegans. Mol. Biol. Cell 2011, 22, 2716–2728. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, A.; Haliloglu, G.; Richard, P.; Talim, B.; Maugenre, S.; Ferreiro, A.; Guicheney, P.; Menditto, I.; Benedetti, S.; Bertini, E.; et al. Two patients with “Dropped head syndrome” due to mutations in LMNA or SEPN1 genes. Neuromuscul. Disord. NMD 2005, 15, 521–524. [Google Scholar] [CrossRef]
- Vytopil, M.; Ricci, E.; Dello Russo, A.; Hanisch, F.; Neudecker, S.; Zierz, S.; Ricotti, R.; Demay, L.; Richard, P.; Wehnert, M.; et al. Frequent low penetrance mutations in the Lamin A/C gene, causing Emery Dreifuss muscular dystrophy. Neuromuscul. Disord. NMD 2002, 12, 958–963. [Google Scholar] [CrossRef]
- Wiesel, N.; Mattout, A.; Melcer, S.; Melamed-Book, N.; Herrmann, H.; Medalia, O.; Aebi, U.; Gruenbaum, Y. Laminopathic mutations interfere with the assembly, localization, and dynamics of nuclear lamins. Proc. Natl. Acad. Sci. USA 2008, 105, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Mattout, A.; Pike, B.L.; Towbin, B.D.; Bank, E.M.; Gonzalez-Sandoval, A.; Stadler, M.B.; Meister, P.; Gruenbaum, Y.; Gasser, S.M. An EDMD mutation in C. elegans lamin blocks muscle-specific gene relocation and compromises muscle integrity. Curr. Biol. 2011, 21, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Riemer, D.; Stuurman, N.; Berrios, M.; Hunter, C.; Fisher, P.A.; Weber, K. Expression of Drosophila lamin C is developmentally regulated: Analogies with vertebrate A-type lamins. J. Cell Sci. 1995, 108 Pt 10, 3189–3198. [Google Scholar] [PubMed]
- Schulze, S.R.; Curio-Penny, B.; Li, Y.; Imani, R.A.; Rydberg, L.; Geyer, P.K.; Wallrath, L.L. Molecular genetic analysis of the nested Drosophila melanogaster lamin C gene. Genetics 2005, 171, 185–196. [Google Scholar] [CrossRef]
- Dialynas, G.; Speese, S.; Budnik, V.; Geyer, P.K.; Wallrath, L.L. The role of Drosophila Lamin C in muscle function and gene expression. Dev. Camb. Engl. 2010, 137, 3067–3077. [Google Scholar] [CrossRef] [PubMed]
- Stuurman, N.; Delbecque, J.P.; Callaerts, P.; Aebi, U. Ectopic overexpression of Drosophila lamin C is stage-specific lethal. Exp. Cell Res. 1999, 248, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Schulze, S.R.; Curio-Penny, B.; Speese, S.; Dialynas, G.; Cryderman, D.E.; McDonough, C.W.; Nalbant, D.; Petersen, M.; Budnik, V.; Geyer, P.K.; et al. A comparative study of Drosophila and human A-type lamins. PLoS ONE 2009, 4, e7564. [Google Scholar] [CrossRef]
- Dialynas, G.; Shrestha, O.K.; Ponce, J.M.; Zwerger, M.; Thiemann, D.A.; Young, G.H.; Moore, S.A.; Yu, L.; Lammerding, J.; Wallrath, L.L. Myopathic lamin mutations cause reductive stress and activate the nrf2/keap-1 pathway. PLoS Genet. 2015, 11, e1005231. [Google Scholar] [CrossRef]
- Dialynas, G.; Flannery, K.M.; Zirbel, L.N.; Nagy, P.L.; Mathews, K.D.; Moore, S.A.; Wallrath, L.L. LMNA variants cause cytoplasmic distribution of nuclear pore proteins in Drosophila and human muscle. Hum. Mol. Genet. 2012, 21, 1544–1556. [Google Scholar] [CrossRef]
- Shi, X.; Chen, R.; Zhang, Y.; Yun, J.; Brand-Arzamendi, K.; Liu, X.; Wen, X.Y. Zebrafish heart failure models: Opportunities and challenges. Amino Acids. 2018, 50, 787–798. [Google Scholar] [CrossRef]
- Vogel, B.; Meder, B.; Just, S.; Laufer, C.; Berger, I.; Weber, S.; Katus, H.A.; Rottbauer, W. In-vivo characterization of human dilated cardiomyopathy genes in zebrafish. Biochem. Biophys. Res. Commun. 2009, 390, 516–522. [Google Scholar] [CrossRef]
- Koshimizu, E.; Imamura, S.; Qi, J.; Toure, J.; Valdez, D.M., Jr.; Carr, C.E.; Hanai, J.; Kishi, S. Embryonic senescence and laminopathies in a progeroid zebrafish model. PLoS ONE 2011, 6, e17688. [Google Scholar] [CrossRef]
- Verma, A.D.; Parnaik, V.K. Heart-specific expression of laminopathic mutations in transgenic zebrafish. Cell Biol. Int. 2017, 41, 809–819. [Google Scholar] [CrossRef]
- Kubben, N.; Voncken, J.W.; Konings, G.; van Weeghel, M.; van den Hoogenhof, M.M.; Gijbels, M.; van Erk, A.; Schoonderwoerd, K.; van den Bosch, B.; Dahlmans, V.; et al. Post-natal myogenic and adipogenic developmental: Defects and metabolic impairment upon loss of A-type lamins. Nucl. Austin Tex. 2011, 2, 195–207. [Google Scholar] [CrossRef]
- Kim, Y.; Zheng, Y. Generation and characterization of a conditional deletion allele for Lmna in mice. Biochem. Biophys. Res. Commun. 2013, 440, 8–13. [Google Scholar] [CrossRef]
- Jahn, D.; Schramm, S.; Schnolzer, M.; Heilmann, C.J.; de Koster, C.G.; Schütz, W.; Benavente, R.; Alsheimer, M. A truncated lamin A in the Lmna -/- mouse line: Implications for the understanding of laminopathies. Nucl. Austin Tex. 2012, 3, 463–474. [Google Scholar] [CrossRef]
- Cutler, D.A.; Sullivan, T.; Marcus-Samuels, B.; Stewart, C.L.; Reitman, M.L. Characterization of adiposity and metabolism in Lmna-deficient mice. Biochem. Biophys. Res. Commun. 2002, 291, 522–527. [Google Scholar] [CrossRef]
- Wolf, C.M.; Wang, L.; Alcalai, R.; Pizard, A.; Burgon, P.G.; Ahmad, F.; Sherwood, M.; Branco, D.M.; Wakimoto, H.; Fishman, G.I.; et al. Lamin A/C haploinsufficiency causes dilated cardiomyopathy and apoptosis-triggered cardiac conduction system disease. J. Mol. Cell. Cardiol. 2008, 44, 293–303. [Google Scholar] [CrossRef]
- Davies, B.S.J.; Barnes, R.H.; Tu, Y.; Ren, S.; Andres, D.A.; Spielmann, H.P.; Lammerding, J.; Wang, Y.; Young, S.G.; Fong, L.G. An accumulation of non-farnesylated prelamin A causes cardiomyopathy but not progeria. Hum. Mol. Genet. 2010, 19, 2682–2694. [Google Scholar] [CrossRef]
- Coffinier, C.; Jung, H.-J.; Li, Z.; Nobumori, C.; Yun, U.J.; Farber, E.A.; Davies, B.S.; Weinstein, M.M.; Yang, S.H.; Lammerding, J.; et al. Direct synthesis of lamin A, bypassing prelamin a processing, causes misshapen nuclei in fibroblasts but no detectable pathology in mice. J. Biol. Chem. 2010, 285, 20818–20826. [Google Scholar] [CrossRef]
- Benedetti, S.; Menditto, I.; Degano, M.; Rodolico, C.; Merlini, L.; D’Amico, A.; Palmucci, L.; Berardinelli, A.; Pegoraro, E.; Trevisan, C.P.; et al. Phenotypic clustering of lamin A/C mutations in neuromuscular patients. Neurology 2007, 69, 1285–1292. [Google Scholar] [CrossRef]
- Sébillon, P.; Bouchier, C.; Bidot, L.D.; Bonne, G.; Ahamed, K.; Charron, P.; Drouin-Garraud, V.; Millaire, A.; Desrumeaux, G.; Benaïche, A.; et al. Expanding the phenotype of LMNA mutations in dilated cardiomyopathy and functional consequences of these mutations. J. Med. Genet. 2003, 40, 560–567. [Google Scholar] [CrossRef]
- Gordon, L.B.; Kleinman, M.E.; Miller, D.T.; Neuberg, D.S.; Giobbie-Hurder, A.; Gerhard-Herman, M.; Smoot, L.B.; Gordon, C.M.; Cleveland, R.; Snyder, B.D.; et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2012, 109, 16666–16671. [Google Scholar] [CrossRef]
- Markandeya, Y.S.; Tsubouchi, T.; Hacker, T.A.; Wolff, M.R.; Belardinelli, L.; Balijepalli, R.C. Inhibition of late sodium current attenuates ionic arrhythmia mechanism in ventricular myocytes expressing LaminA-N195K mutation. Heart Rhythm 2016, 13, 2228–2236. [Google Scholar] [CrossRef]
- Nguyên-Trân, V.T.; Kubalak, S.W.; Minamisawa, S.; Fiset, C.; Wollert, K.C.; Brown, A.B.; Ruiz-Lozano, P.; Barrere-Lemaire, S.; Kondo, R.; Norman, L.W.; et al. A novel genetic pathway for sudden cardiac death via defects in the transition between ventricular and conduction system cell lineages. Cell 2000, 102, 671–682. [Google Scholar] [CrossRef]
- Lu, D.; Lian, H.; Zhang, X.; Shao, H.; Huang, L.; Qin, C.; Zhang, L. LMNA E82K mutation activates FAS and mitochondrial pathways of apoptosis in heart tissue specific transgenic mice. PLoS ONE 2010, 5, e15167. [Google Scholar] [CrossRef]
- Wu, X.; Wang, Q.K.; Gui, L.; Liu, M.; Zhang, X.; Jin, R.; Li, W.; Yan, L.; Du, R.; Wang, Q. Identification of a new lamin A/C mutation in a Chinese family affected with atrioventricular block as the prominent phenotype. J. Huazhong Univ. Sci. Technol. Med. Sci. 2010, 30, 103–107. [Google Scholar] [CrossRef]
- Wang, H.; Zheng, W.; Wang, J.; Wang, X.J.; Zhen, Y.S.; Song, L.; Zou, Y.B.; Hui, R.T. A novel LMNA gene mutation E82K associated with familial dilated cardiomyopathy. Zhonghua Xin Xue Guan Bing Za Zhi 2005, 33, 875–879. [Google Scholar]
- Arimura, T.; Helbling-Leclerc, A.; Massart, C.; Varnous, S.; Niel, F.; Lacène, E.; Fromes, Y.; Toussaint, M.; Mura, A.M.; Keller, D.I.; et al. Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum. Mol. Genet. 2005, 14, 155–169. [Google Scholar] [CrossRef]
- Wang, Y.; Herron, A.J.; Worman, H.J. Pathology and nuclear abnormalities in hearts of transgenic mice expressing M371K lamin A encoded by an LMNA mutation causing Emery-Dreifuss muscular dystrophy. Hum. Mol. Genet. 2006, 15, 2479–2489. [Google Scholar] [CrossRef]
- van Rijsingen, I.A.W.; Nannenberg, E.A.; Arbustini, E.; Elliott, P.M.; Mogensen, J.; Hermans-van Ast, J.F.; van der Kooi, A.J.; van Tintelen, J.P.; van den Berg, M.P.; Grasso, M.; et al. Gender-specific differences in major cardiac events and mortality in lamin A/C mutation carriers. Eur. J. Heart Fail. 2013, 15, 376–384. [Google Scholar] [CrossRef]
- Arimura, T.; Onoue, K.; Takahashi-Tanaka, Y.; Ishikawa, T.; Kuwahara, M.; Setou, M.; Shigenobu, S.; Yamaguchi, K.; Bertrand, A.T.; Machida, N.; et al. Nuclear accumulation of androgen receptor in gender difference of dilated cardiomyopathy due to lamin A/C mutations. Cardiovasc. Res. 2013, 99, 382–394. [Google Scholar] [CrossRef]
- Muchir, A.; Reilly, S.A.; Wu, W.; Iwata, S.; Homma, S.; Bonne, G.; Worman, H.J. Treatment with selumetinib preserves cardiac function and improves survival in cardiomyopathy caused by mutation in the lamin A/C gene. Cardiovasc. Res. 2012, 93, 311–319. [Google Scholar] [CrossRef]
- Muchir, A.; Kim, Y.J.; Reilly, S.A.; Wu, W.; Choi, J.C.; Worman, H.J. Inhibition of extracellular signal-regulated kinase 1/2 signaling has beneficial effects on skeletal muscle in a mouse model of Emery-Dreifuss muscular dystrophy caused by lamin A/C gene mutation. Skelet. Muscle 2013, 3, 17. [Google Scholar] [CrossRef]
- Muchir, A.; Shan, J.; Bonne, G.; Lehnart, S.E.; Worman, H.J. Inhibition of extracellular signal-regulated kinase signaling to prevent cardiomyopathy caused by mutation in the gene encoding A-type lamins. Hum. Mol. Genet. 2009, 18, 241–247. [Google Scholar] [CrossRef]
- Wu, W.; Shan, J.; Bonne, G.; Worman, H.J.; Muchir, A. Pharmacological inhibition of c-Jun N-terminal kinase signaling prevents cardiomyopathy caused by mutation in LMNA gene. Biochim. Biophys. Acta 2010, 1802, 632–638. [Google Scholar] [CrossRef]
- Wu, W.; Muchir, A.; Shan, J.; Bonne, G.; Worman, H.J. Mitogen-activated protein kinase inhibitors improve heart function and prevent fibrosis in cardiomyopathy caused by mutation in lamin A/C gene. Circulation 2011, 123, 53–61. [Google Scholar] [CrossRef]
- Muchir, A.; Wu, W.; Sera, F.; Homma, S.; Worman, H.J. Mitogen-activated protein kinase kinase 1/2 inhibition and angiotensin II converting inhibition in mice with cardiomyopathy caused by lamin A/C gene mutation. Biochem. Biophys. Res. Commun. 2014, 452, 958–961. [Google Scholar] [CrossRef]
- Choi, J.C.; Muchir, A.; Wu, W.; Iwata, S.; Homma, S.; Morrow, J.P.; Worman, H.J. Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci. Transl. Med. 2012, 4, 144ra102. [Google Scholar] [CrossRef]
- Le Dour, C.; Macquart, C.; Sera, F.; Homma, S.; Bonne, G.; Morrow, J.P.; Worman, H.J.; Muchir, A. Decreased WNT/β-catenin signalling contributes to the pathogenesis of dilated cardiomyopathy caused by mutations in the lamin a/C gene. Hum. Mol. Genet. 2017, 26, 333–343. [Google Scholar] [CrossRef]
- Bertrand, A.T.; Renou, L.; Papadopoulos, A.; Beuvin, M.; Lacène, E.; Massart, C.; Ottolenghi, C.; Decostre, V.; Maron, S.; Schlossarek, S.; et al. DelK32-lamin A/C has abnormal location and induces incomplete tissue maturation and severe metabolic defects leading to premature death. Hum. Mol. Genet. 2012, 21, 1037–1048. [Google Scholar] [CrossRef]
- Mounkes, L.C.; Kozlov, S.; Hernandez, L.; Sullivan, T.; Stewart, C.L. A progeroid syndrome in mice is caused by defects in A-type lamins. Nature 2003, 423, 298–301. [Google Scholar] [CrossRef]
- Yamanaka, S. Induced pluripotent stem cells: Past, present, and future. Cell Stem Cell 2012, 10, 678–684. [Google Scholar] [CrossRef]
- Yoshihara, M.; Hayashizaki, Y.; Murakawa, Y. Genomic Instability of iPSCs: Challenges Towards Their Clinical Applications. Stem Cell Rev. 2017, 13, 7–16. [Google Scholar] [CrossRef]
- Davis, J.; Maillet, M.; Miano, J.M.; Molkentin, J.D. Lost in transgenesis: A user’s guide for genetically manipulating the mouse in cardiac research. Circ. Res. 2012, 111, 761–777. [Google Scholar] [CrossRef]
- Di Barletta, R.M.; Ricci, E.; Galluzzi, G.; Tonali, P.; Mora, M.; Morandi, L.; Romorini, A.; Voit, T.; Orstavik, K.H.; Merlini, L.; et al. Different mutations in the LMNA gene cause autosomal dominant and autosomal recessive Emery-Dreifuss muscular dystrophy. Am. J. Hum. Genet. 2000, 66, 1407–1412. [Google Scholar] [CrossRef]
- Ben Yaou, R.; Toutain, A.; Arimura, T.; Demay, L.; Massart, C.; Peccate, C.; Muchir, A.; Llense, S.; Deburgrave, N.; Leturcq, F.; et al. Multitissular involvement in a family with LMNA and EMD mutations: Role of digenic mechanism? Neurology 2007, 68, 1883–1894. [Google Scholar] [CrossRef]
- De Sandre-Giovannoli, A.; Chaouch, M.; Kozlov, S.; Vallat, J.M.; Tazir, M.; Kassouri, N.; Szepetowski, P.; Hammadouche, T.; Vandenberghe, A.; Stewart, C.L.; et al. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am. J. Hum. Genet. 2002, 70, 726–736. [Google Scholar] [CrossRef]
- Chaouch, M.; Allal, Y.; De Sandre-Giovannoli, A.; Vallat, J.M.; Amer-el-Khedoud, A.; Kassouri, N.; Chaouch, A.; Sindou, P.; Hammadouche, T.; Tazir, M.; et al. The phenotypic manifestations of autosomal recessive axonal Charcot-Marie-Tooth due to a mutation in Lamin A/C gene. Neuromuscul. Disord. NMD 2003, 13, 60–67. [Google Scholar] [CrossRef]
- Tazir, M.; Azzedine, H.; Assami, S.; Sindou, P.; Nouioua, S.; Zemmouri, R.; Hamadouche, T.; Chaouch, M.; Feingold, J.; Vallat, J.M.; et al. Phenotypic variability in autosomal recessive axonal Charcot-Marie-Tooth disease due to the R298C mutation in lamin A/C. Brain J. Neurol. 2004, 127 Pt 1, 154–163. [Google Scholar] [CrossRef]
- Bouhouche, A.; Birouk, N.; Azzedine, H.; Benomar, A.; Durosier, G.; Ente, D.; Muriel, M.P.; Ruberg, M.; Slassi, I.; Yahyaoui, M.; et al. Autosomal recessive axonal Charcot-Marie-Tooth disease (ARCMT2): Phenotype-genotype correlations in 13 Moroccan families. Brain J. Neurol. 2007, 130 Pt 4, 1062–1075. [Google Scholar] [CrossRef]
- MacRae, C. Phase 2 Study of A797, an Oral, Selective p38 Mitogen-Activated Protein Kinase Inhibitor, in Patients with Lamin A/C-Related Dilated Cardiomyopathy. In Proceedings of the European Society of Cardiology Congress, Rome, Italy, 27 August 2016. [Google Scholar]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2018, 46, 2699. [Google Scholar] [CrossRef]
- Chojnacki, S.; Cowley, A.; Lee, J.; Foix, A.; Lopez, R. Programmatic access to bioinformatics tools from EMBL-EBI update: 2017. Nucleic Acids Res. 2017, 45, W550–W553. [Google Scholar] [CrossRef]
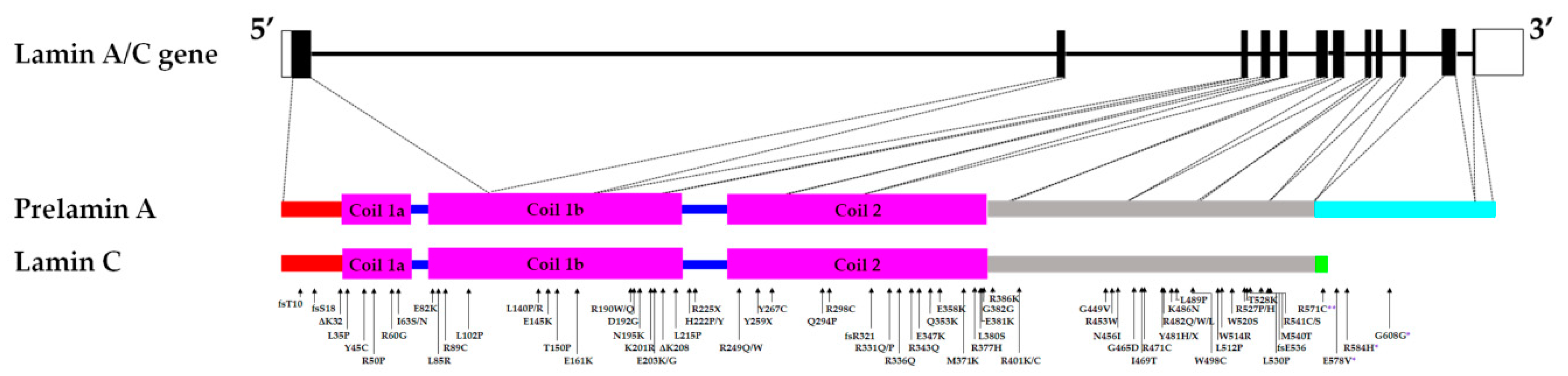

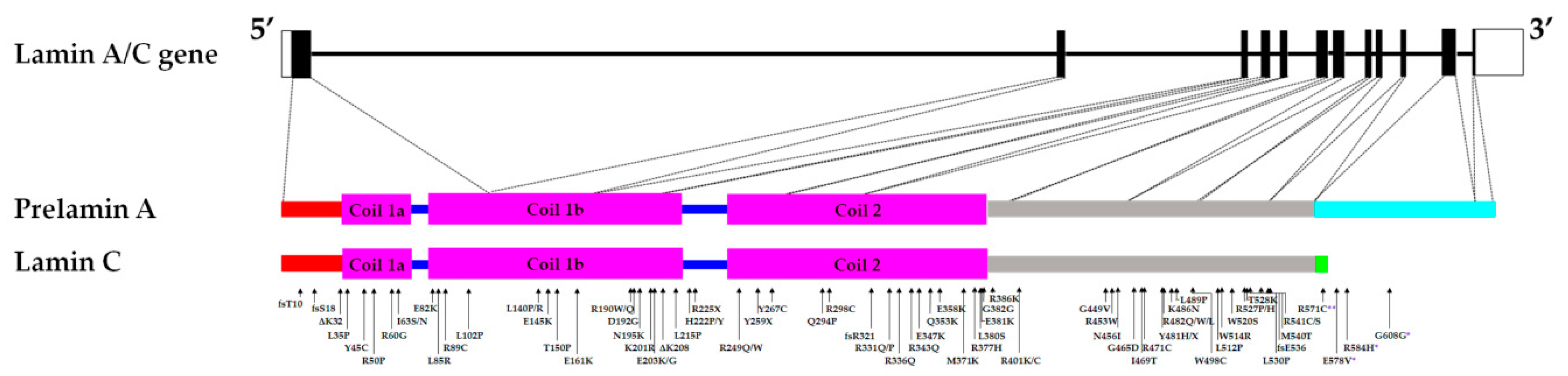{kind=link}
| Mislocalized Proteins | Role | Mislocalization | Model(s)/Patient Sample(s) |
|---|---|---|---|
| 1. Lap2 (α, β) | regulation of nuclear architecture | absence in nuclear poles and/or lobules, honeycomb pattern in nuclear blebs | fibroblasts from homozygous p.Y259X patient [46], Lmna LAO mice [109], LmnaΔ8–11 mice MEFs [61] |
| 2. Emerin | anchorage to the cytoskeleton | cytoplasmic, concentration in one pole, sequestration within nuclear lamin foci, honeycomb pattern | fibroblasts from homozygous p.Y259X patient [46], RNAi LMNA knockdown in HeLa cells [48], various lamin A/C variants expressed in different cell lines [49,50], C. elegans [90], fibroblasts from patients with various lamin A/C mutations [51], myogenic cells derived from myopathies patients’ iPSCs [81], LmnaΔ8–11 mice [50,61], LmnaN195K/N195K mice [86], p.K46del lamin-1 variant C. elegans [87] |
| 3. Syne1 | anchorage to the cytoskeleton | cytoplasmic | fibroblasts from homozygous p.Y259X patient [46] |
| 4. B-type lamins | involved in a variety of functions including regulation of expression, mitosis, cellular senescence | absence in one pole (i.e., concentration in one pole only) and/or absence in nuclear lobules, honeycomb pattern | fibroblasts from homozygous p.Y259X patient [46], fibroblasts from patients with various lamin A/C mutations [51], myogenic cells derived from myopathies patients’ iPSCs [81], LamC null and Lamin-C N-terminal deleted D. melanogaster mutants [93], LmnaΔ8–11 mice MEFs [61], cardiac expressing p.M371K lamin A/C mice [119] |
| 5. Nup153, Nup154 | component of the nuclear pore complex | absence in nuclear poles and/or lobules, clustering | fibroblasts from homozygous p.Y259X patient [46], fibroblasts from p. R225X patient [79], RNAi lmn-1 knockdown in C. elegans [83], LmnaΔ8–11 mice [61], LmnaN195K/N195K mice [86], LamC null and various lamin A/C mutations expressed in D. melanogaster [96], LmnaGT−/− mice [103], LmnaΔ8–11 mice MEFs [61] |
| 6. SUMO1 | post-translational modifications | sequestration within nuclear lamin foci | various lamin A/C variants expressed in Cos7 cells [38], C2C12 cells [78], LmnaH222P/H222P mice primary myoblasts [78], LmnaH222P/H222P mice skeletal muscle tissue [78] |
| 7. Actin | cytoskeletal component | filament disorganization, increased nuclear localization, and decreased expression | neonatal rat ventricular myocytes expressing various mutant lamin A/C [34], LamC null D. melanogaster [96], Lamin-C N-terminal deleted and various lamin A/C mutations expressed in D. melanogaster [97], patient myoblasts expressing various L-CMD variants [35,36] |
| 8. ERK ½ (phosphorylated) | involved in a variety of cellular responses | increased nuclear localization | LmnaH222P/H222P mice, p.H222P lamin A expressing Cos7 and C2C12 cells [21] |
| 9. Smad2/3 (phosphorylated) | TGF-β signalling pathway | increased nuclear localization | LmnaH222P/H222P mice [118] |
| 10. Androgen receptors, SRF -FHL2 | mediating actions of androgens | nuclear accumulation | neonatal rat cardiomyocytes expressing p.H222P variant or p.R225X variant [121], LmnaH222P/H222P mice and cardiac tissue from DCM patients [121] |
| 11. Cx40, Cx43 | gap junction proteins | Diffused pattern and decreased expression in atria | LmnaN195K/N195K mice [86] |
| Models | Affected Proteins and Signalling Pathways | Reference(s) | |
|---|---|---|---|
| Cellular Models | 1. Fibroblasts from p.E203K DCM patient or HeLa transfected cells | ↓ sumoylation | [77] |
| 2. Various lamin A/C variants expressed in neonatal rat ventricular myocytes | inhibition of p38 = rescue of actin and mechanical phenotype | [34] | |
| 3. p. L530P (EDMD) | ↓ binding to SREBF1 | [15] | |
| 4. Fibroblasts from p. R225X patient | inhibition of MEK/ERK 1/2 = rescue (decreased apoptosis and senescence) | [79] | |
| 5. Myoblasts from patients expressing various L-CMD variants | deregulation of yes-associated protein and formins | [35,36] | |
| Animal Models | 6. Various lamin A/C variants expressed in D. melanogaster | ↑ Nrf2, p62/SQSTM1 | [97] |
| 7. D. rerio lmna morphants | ↓ pparγ, ↓ PCNA | [101] | |
| 8. D. rerio lmna transgenics | ↑ PCNA | [102] | |
| 9. LmnanPLAO/nPLAO mice | ↓ Myh6, ↑ Myh7 | [108] | |
| 10. LmnaN195K/N195K mice | reactivation of foetal genes (↑ANP, ↑ BNP), inhibition of Na+ channel = improve symptom, ↓ connexin 40, mis-expression of Hf1b/Sp4 | [86] | |
| 11. Mice cardiac tissue only expression of p.E82K lamin A/C variant | hypertrophy markers (↑ in BNP, actin alpha 1, collagen type III alpha 1), ↑ FAS, ↑ cytochrome C | [115] | |
| 12. LmnaH222P/H222P mice | TGFβ (↑ nuclear phos-Smad 2/3), ↑ MAPK members (ERK 1/2, p38, JNK),↑ AKT/mTor, ↓ WNT/β-catenin | [21,67,121,122,123,124,125,126,127,128,129] | |
| 13. LmnaΔK32/ΔK32 mice | repression of SREBF1 | [130] | |
| 14. Muscle tissues from patients with various myopathies | phos-lamin A/C (Ser458) by Akt | [73] | |
| 15. LmnaΔK32/+ mice | ↑ Nppb, ↑ Myh7 | [65] | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicolas, H.A.; Akimenko, M.-A.; Tesson, F. Cellular and Animal Models of Striated Muscle Laminopathies. Cells 2019, 8, 291. https://doi.org/10.3390/cells8040291
Nicolas HA, Akimenko M-A, Tesson F. Cellular and Animal Models of Striated Muscle Laminopathies. Cells. 2019; 8(4):291. https://doi.org/10.3390/cells8040291
Chicago/Turabian StyleNicolas, Hannah A., Marie-Andrée Akimenko, and Frédérique Tesson. 2019. "Cellular and Animal Models of Striated Muscle Laminopathies" Cells 8, no. 4: 291. https://doi.org/10.3390/cells8040291
APA StyleNicolas, H. A., Akimenko, M.-A., & Tesson, F. (2019). Cellular and Animal Models of Striated Muscle Laminopathies. Cells, 8(4), 291. https://doi.org/10.3390/cells8040291