Regulating the BCL2 Family to Improve Sensitivity to Microtubule Targeting Agents



Abstract
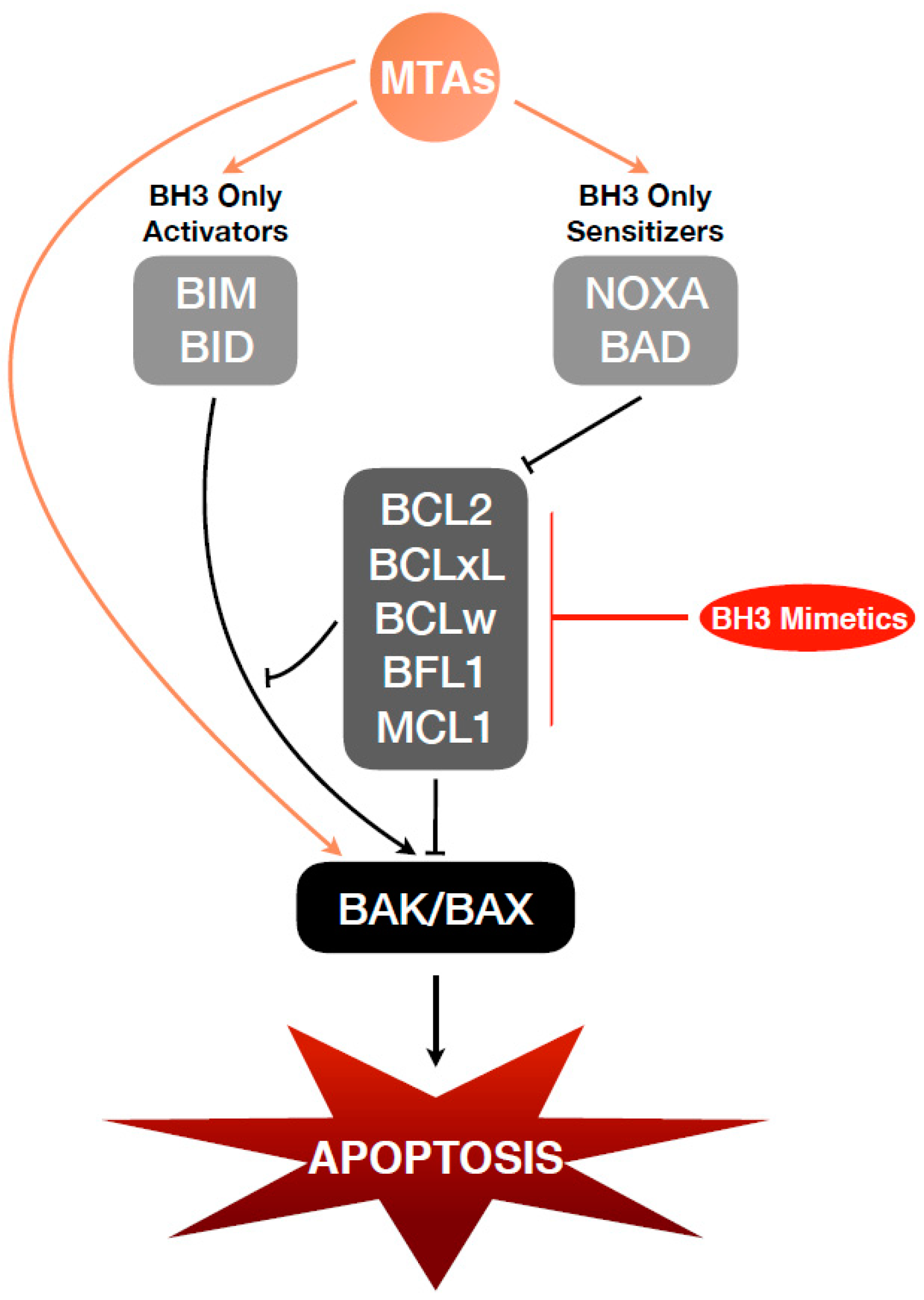
:1. Introduction
2. Microtubules and Tubulins
3. Microtubule Targeting Agents (MTAs)
3.1. Background
3.2. Vinca Alkaloids
3.3. Taxanes
3.4. MTA Chemoresistance
4. BCL2 Family of Proteins
4.1. Anti-Apoptotic BCL2 Family and MTAs
4.2. BCL2
4.3. BCLxL
4.4. BCLW and BFL1/A1
4.5. MCL1
5. Combination of BCL2 Family Inhibitors and MTAs
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Mukhtar, E.; Adhami, V.M.; Mukhtar, H. Targeting microtubules by natural agents for cancer therapy. Mol. Cancer Ther. 2014, 13, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.T.; Li, Z.L.; He, Z.X.; Qiu, J.X.; Zhou, S.F. Molecular mechanisms for tumour resistance to chemotherapy. Clin. Exp. Pharmacol. Physiol. 2016, 43, 723–737. [Google Scholar] [CrossRef]
- Bates, D.; Eastman, A. Microtubule destabilising agents: Far more than just antimitotic anticancer drugs. Br. J. Clin. Pharmacol. 2017, 83, 255–268. [Google Scholar] [CrossRef]
- Chaaban, S.; Brouhard, G.J. A microtubule bestiary: Structural diversity in tubulin polymers. Mol. Biol. Cell 2017, 28, 2924–2931. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef]
- Wiese, C.; Zheng, Y. Microtubule nucleation: Gamma-tubulin and beyond. J. Cell Sci. 2006, 119 Pt 20, 4143–4153. [Google Scholar] [CrossRef]
- McKean, P.G.; Vaughan, S.; Gull, K. The extended tubulin superfamily. J. Cell Sci. 2001, 114 Pt 15, 2723–2733. [Google Scholar]
- Burkhart, C.A.; Kavallaris, M.; Horwitz, S.B. The role of beta-tubulin isotypes in resistance to antimitotic drugs. Biochim. Biophys. Acta 2001, 2, O1–O9. [Google Scholar]
- Sullivan, K.F.; Cleveland, D.W. Identification of conserved isotype-defining variable region sequences for four vertebrate beta tubulin polypeptide classes. Proc. Natl. Acad. Sci. USA 1986, 83, 4327–4331. [Google Scholar] [CrossRef]
- Sullivan, K.F. Structure and utilization of tubulin isotypes. Annu. Rev. Cell Biol. 1988, 4, 687–716. [Google Scholar] [CrossRef]
- Roll-Mecak, A. How cells exploit tubulin diversity to build functional cellular microtubule mosaics. Curr. Opin. Cell Biol. 2019, 56, 102–108. [Google Scholar] [CrossRef]
- Cross, R.A. Microtubule lattice plasticity. Curr. Opin. Cell Biol. 2019, 56, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Saxton, W.M.; Stemple, D.L.; Leslie, R.J.; Salmon, E.D.; Zavortink, M.; McIntosh, J.R. Tubulin dynamics in cultured mammalian cells. J. Cell Biol. 1984, 99, 2175–2186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchison, T.J. Microtubule dynamics and kinetochore function in mitosis. Annu. Rev. Cell Biol. 1988, 4, 527–549. [Google Scholar] [CrossRef] [PubMed]
- Musacchio, A.; Salmon, E.D. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 2007, 8, 379–393. [Google Scholar] [CrossRef]
- Izawa, D.; Pines, J. How APC/C-Cdc20 changes its substrate specificity in mitosis. Nat. Cell Biol. 2011, 13, 223–233. [Google Scholar] [CrossRef]
- Maciejowski, J.; Drechsler, H.; Grundner-Culemann, K.; Ballister, E.R.; Rodriguez-Rodriguez, J.A.; Rodriguez-Bravo, V.; Jones, M.J.K.; Foley, E.; Lampson, M.A.; Daub, H.; et al. Mps1 Regulates Kinetochore-Microtubule Attachment Stability via the Ska Complex to Ensure Error-Free Chromosome Segregation. Dev. Cell 2017, 41, 143–156. [Google Scholar] [CrossRef]
- Le Roux, M.A.G. Francoise, From the Pacific Yew (Taxus brevifolia) to the English Yew (Taxus baccata): Steps Towards the Discovery of Docetaxel (Taxotere®). In Navelbine® and Taxotere®; Elsevier: Oxford, UK, 2017; pp. 151–212. [Google Scholar]
- Huizing, M.T.; Misser, V.H.; Pieters, R.C.; ten Bokkel Huinink, W.W.; Veenhof, C.H.; Vermorken, J.B.; Pinedo, H.M.; Beijnen, J.H. Taxanes: A new class of antitumor agents. Cancer Investig. 1995, 13, 381–404. [Google Scholar] [CrossRef]
- Akhmanova, A.; Steinmetz, M.O. Control of microtubule organization and dynamics: Two ends in the limelight. Nat. Rev. Mol. Cell Biol. 2015, 16, 711–726. [Google Scholar] [CrossRef]
- Haschka, M.; Karbon, G.; Fava, L.L.; Villunger, A. Perturbing mitosis for anti-cancer therapy: Is cell death the only answer? EMBO Rep. 2018, 19, e45440. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [Green Version]
- Seve, P.; Dumontet, C. Class III beta tubulin expression in nonsmall cell lung cancer. Rev. Mal. Respir. 2010, 27, 383–386. [Google Scholar] [CrossRef]
- Noble, R.L.; Beer, C.T.; Cutts, J.H. Role of chance observations in chemotherapy: Vinca rosea. Ann. N. Y. Acad. Sci. 1958, 76, 882–894. [Google Scholar] [CrossRef]
- DeVita, V.T., Jr.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef]
- Xie, S.; Zhou, J. Harnessing Plant Biodiversity for the Discovery of Novel Anticancer Drugs Targeting Microtubules. Front. Plant Sci. 2017, 8, 720. [Google Scholar] [CrossRef]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef]
- Barbuti, A.M.; Chen, Z.S. Paclitaxel Through the Ages of Anticancer Therapy: Exploring Its Role in Chemoresistance and Radiation Therapy. Cancers 2015, 7, 2360–2371. [Google Scholar] [CrossRef] [Green Version]
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Coggon, P.; McPhail, A.T. Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef]
- Schiff, P.B.; Fant, J.; Horwitz, S.B. Promotion of microtubule assembly in vitro by taxol. Nature 1979, 277, 665–667. [Google Scholar] [CrossRef]
- Yared, J.A.; Tkaczuk, K.H. Update on taxane development: New analogs and new formulations. Drug Des. Dev. Ther. 2012, 6, 371–384. [Google Scholar]
- Steinmetz, M.O.; Prota, A.E. Microtubule-Targeting Agents: Strategies to Hijack the Cytoskeleton. Trends Cell Biol. 2018, 28, 776–792. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, S.H.; Guo, X.L. New insights into Vinca alkaloids resistance mechanism and circumvention in lung cancer. Biomed. Pharmacother. 2017, 96, 659–666. [Google Scholar] [CrossRef]
- Thomas, H.; Coley, H.M. Overcoming multidrug resistance in cancer: An update on the clinical strategy of inhibiting p-glycoprotein. Cancer Control 2003, 10, 159–165. [Google Scholar] [CrossRef]
- Parker, A.L.; Kavallaris, M.; McCarroll, J.A. Microtubules and their role in cellular stress in cancer. Front. Oncol. 2014, 4, 153. [Google Scholar] [CrossRef]
- Lebok, P.; Ozturk, M.; Heilenkotter, U.; Jaenicke, F.; Muller, V.; Paluchowski, P.; Geist, S.; Wilke, C.; Burandt, E.; Lebeau, A.; et al. High levels of class III beta-tubulin expression are associated with aggressive tumor features in breast cancer. Oncol. Lett. 2016, 11, 1987–1994. [Google Scholar] [CrossRef]
- Savry, A.; Carre, M.; Berges, R.; Rovini, A.; Pobel, I.; Chacon, C.; Braguer, D.; Bourgarel-Rey, V. Bcl-2-enhanced efficacy of microtubule-targeting chemotherapy through Bim overexpression: Implications for cancer treatment. Neoplasia 2013, 15, 49–60. [Google Scholar] [CrossRef]
- Blagosklonny, M.V.; Giannakakou, P.; el-Deiry, W.S.; Kingston, D.G.; Higgs, P.I.; Neckers, L.; Fojo, T. Raf-1/bcl-2 phosphorylation: A step from microtubule damage to cell death. Cancer Res. 1997, 57, 130–135. [Google Scholar]
- Haldar, S.; Chintapalli, J.; Croce, C.M. Taxol induces bcl-2 phosphorylation and death of prostate cancer cells. Cancer Res. 1996, 56, 1253–1255. [Google Scholar]
- Pathan, N.; Aime-Sempe, C.; Kitada, S.; Haldar, S.; Reed, J.C. Microtubule-targeting drugs induce Bcl-2 phosphorylation and association with Pin1. Neoplasia 2001, 3, 70–79. [Google Scholar] [CrossRef]
- Montero, J.; Letai, A. Why do BCL-2 inhibitors work and where should we use them in the clinic? Cell Death Differ. 2018, 25, 56–64. [Google Scholar] [CrossRef]
- Huang, Y.; Ibrado, A.M.; Reed, J.C.; Bullock, G.; Ray, S.; Tang, C.; Bhalla, K. Co-expression of several molecular mechanisms of multidrug resistance and their significance for paclitaxel cytotoxicity in human AML HL-60 cells. Leukemia 1997, 11, 253–257. [Google Scholar] [CrossRef] [Green Version]
- Zasadil, L.M.; Andersen, K.A.; Yeum, D.; Rocque, G.B.; Wilke, L.G.; Tevaarwerk, A.J.; Raines, R.T.; Burkard, M.E.; Weaver, B.A. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci. Transl. Med. 2014, 6, 229ra43. [Google Scholar] [CrossRef]
- Haschka, M.D.; Soratroi, C.; Kirschnek, S.; Hacker, G.; Hilbe, R.; Geley, S.; Villunger, A.; Fava, L.L. The NOXA-MCL1-BIM axis defines lifespan on extended mitotic arrest. Nat. Commun. 2015, 6, 6891. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Mitotic arrest and cell fate: Why and how mitotic inhibition of transcription drives mutually exclusive events. Cell Cycle 2007, 6, 70–74. [Google Scholar] [CrossRef]
- Haldar, S.; Basu, A.; Croce, C.M. Bcl2 is the guardian of microtubule integrity. Cancer Res. 1997, 57, 229–233. [Google Scholar]
- Mollinedo, F.; Gajate, C. Microtubules, microtubule-interfering agents and apoptosis. Apoptosis 2003, 8, 413–450. [Google Scholar] [CrossRef] [Green Version]
- Wertz, I.E.; Kusam, S.; Lam, C.; Okamoto, T.; Sandoval, W.; Anderson, D.J.; Helgason, E.; Ernst, J.A.; Eby, M.; Liu, J.; et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011, 471, 110–114. [Google Scholar] [CrossRef]
- Fukuhara, S. Significance of 14q translocations in non-Hodgkin lymphomas. Virchows Arch. B 1978, 29, 99–106. [Google Scholar]
- Delbridge, A.R.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef]
- Reed, J.C.; Cuddy, M.; Slabiak, T.; Croce, C.M.; Nowell, P.C. Oncogenic potential of bcl-2 demonstrated by gene transfer. Nature 1988, 336, 259–261. [Google Scholar] [CrossRef]
- Tsujimoto, Y. Stress-resistance conferred by high level of bcl-2 alpha protein in human B lymphoblastoid cell. Oncogene 1989, 4, 1331–1336. [Google Scholar]
- Letai, A.; Bassik, M.C.; Walensky, L.D.; Sorcinelli, M.D.; Weiler, S.; Korsmeyer, S.J. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002, 2, 183–192. [Google Scholar] [CrossRef] [Green Version]
- Lanave, C.; Santamaria, M.; Saccone, C. Comparative genomics: The evolutionary history of the Bcl-2 family. Gene 2004, 333, 71–79. [Google Scholar] [CrossRef]
- Ke, F.F.S.; Vanyai, H.K.; Cowan, A.D.; Delbridge, A.R.D.; Whitehead, L.; Grabow, S.; Czabotar, P.E.; Voss, A.K.; Strasser, A. Embryogenesis and Adult Life in the Absence of Intrinsic Apoptosis Effectors BAX, BAK, and BOK. Cell 2018, 173, 1217–1230. [Google Scholar] [CrossRef]
- Knudson, C.M.; Tung, K.S.; Tourtellotte, W.G.; Brown, G.A.; Korsmeyer, S.J. Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science 1995, 270, 96–99. [Google Scholar] [CrossRef]
- Luke, J.J.; van de Wetering, C.I.; Knudson, C.M. Lymphoma development in Bax transgenic mice is inhibited by Bcl-2 and associated with chromosomal instability. Cell Death Differ. 2003, 10, 740–748. [Google Scholar] [CrossRef] [Green Version]
- Los, M.; Van de Craen, M.; Penning, L.C.; Schenk, H.; Westendorp, M.; Baeuerle, P.A.; Droge, W.; Krammer, P.H.; Fiers, W.; Schulze-Osthoff, K. Requirement of an ICE/CED-3 protease for Fas/APO-1-mediated apoptosis. Nature 1995, 375, 81–83. [Google Scholar] [CrossRef] [Green Version]
- Sabbatini, P.; Han, J.; Chiou, S.K.; Nicholson, D.W.; White, E. Interleukin 1 beta converting enzyme-like proteases are essential for p53-mediated transcriptionally dependent apoptosis. Cell Growth Differ. 1997, 8, 643–653. [Google Scholar]
- Sattler, M.; Liang, H.; Nettesheim, D.; Meadows, R.P.; Harlan, J.E.; Eberstadt, M.; Yoon, H.S.; Shuker, S.B.; Chang, B.S.; Minn, A.J.; et al. Structure of Bcl-xL-Bak peptide complex: Recognition between regulators of apoptosis. Science 1997, 275, 983–986. [Google Scholar] [CrossRef]
- Liu, X.; Dai, S.; Zhu, Y.; Marrack, P.; Kappler, J.W. The structure of a Bcl-xL/Bim fragment complex: Implications for Bim function. Immunity 2003, 19, 341–352. [Google Scholar] [CrossRef]
- Feng, W.; Huang, S.; Wu, H.; Zhang, M. Molecular basis of Bcl-xL’s target recognition versatility revealed by the structure of Bcl-xL in complex with the BH3 domain of Beclin-1. J. Mol. Biol. 2007, 372, 223–235. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Du, H.; Wolf, J.; Schafer, B.; Moldoveanu, T.; Chipuk, J.E.; Kuwana, T. BH3 domains other than Bim and Bid can directly activate Bax/Bak. J. Biol. Chem. 2011, 286, 491–501. [Google Scholar] [CrossRef]
- Hockings, C.; Anwari, K.; Ninnis, R.L.; Brouwer, J.; O’Hely, M.; Evangelista, M.; Hinds, M.G.; Czabotar, P.E.; Lee, E.F.; Fairlie, W.D.; et al. Bid chimeras indicate that most BH3-only proteins can directly activate Bak and Bax, and show no preference for Bak versus Bax. Cell Death Dis. 2015, 6, e1735. [Google Scholar] [CrossRef]
- Vela, L.; Gonzalo, O.; Naval, J.; Marzo, I. Direct interaction of Bax and Bak proteins with Bcl-2 homology domain 3 (BH3)-only proteins in living cells revealed by fluorescence complementation. J. Biol. Chem. 2013, 288, 4935–4946. [Google Scholar] [CrossRef]
- Willis, S.N.; Chen, L.; Dewson, G.; Wei, A.; Naik, E.; Fletcher, J.I.; Adams, J.M.; Huang, D.C. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005, 19, 1294–1305. [Google Scholar] [CrossRef] [Green Version]
- Willis, S.N.; Fletcher, J.I.; Kaufmann, T.; van Delft, M.F.; Chen, L.; Czabotar, P.E.; Ierino, H.; Lee, E.F.; Fairlie, W.D.; Bouillet, P.; et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 2007, 315, 856–859. [Google Scholar] [CrossRef]
- O’Neill, K.L.; Huang, K.; Zhang, J.; Chen, Y.; Luo, X. Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak through the outer mitochondrial membrane. Genes Dev. 2016, 30, 973–988. [Google Scholar] [CrossRef] [Green Version]
- Zong, W.X.; Li, C.; Hatzivassiliou, G.; Lindsten, T.; Yu, Q.C.; Yuan, J.; Thompson, C.B. Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J. Cell Biol. 2003, 162, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Thomenius, M.J.; Distelhorst, C.W. Bcl-2 on the endoplasmic reticulum: Protecting the mitochondria from a distance. J. Cell Sci. 2003, 116 Pt 22, 4493–4499. [Google Scholar] [CrossRef]
- Edlich, F.; Banerjee, S.; Suzuki, M.; Cleland, M.M.; Arnoult, D.; Wang, C.; Neutzner, A.; Tjandra, N.; Youle, R.J. Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell 2011, 145, 104–116. [Google Scholar] [CrossRef]
- Ruefli-Brasse, A.; Reed, J.C. Therapeutics targeting Bcl-2 in hematological malignancies. Biochem. J. 2017, 474, 3643–3657. [Google Scholar] [CrossRef]
- Tang, C.; Willingham, M.C.; Reed, J.C.; Miyashita, T.; Ray, S.; Ponnathpur, V.; Huang, Y.; Mahoney, M.E.; Bullock, G.; Bhalla, K. High levels of p26BCL-2 oncoprotein retard taxol-induced apoptosis in human pre-B leukemia cells. Leukemia 1994, 8, 1960–1969. [Google Scholar]
- Thomadaki, H.; Floros, K.V.; Scorilas, A. Molecular response of HL-60 cells to mitotic inhibitors vincristine and taxol visualized with apoptosis-related gene expressions, including the new member BCL2L12. Ann. N. Y. Acad. Sci. 2009, 1171, 276–283. [Google Scholar] [CrossRef]
- Ibrado, A.M.; Liu, L.; Bhalla, K. Bcl-xL overexpression inhibits progression of molecular events leading to paclitaxel-induced apoptosis of human acute myeloid leukemia HL-60 cells. Cancer Res. 1997, 57, 1109–1115. [Google Scholar]
- Huang, S.; Tang, R.; Poon, R.Y. BCL-W is a regulator of microtubule inhibitor-induced mitotic cell death. Oncotarget 2016, 7, 38718–38730. [Google Scholar] [CrossRef]
- Xia, L.; Wurmbach, E.; Waxman, S.; Jing, Y. Upregulation of Bfl-1/A1 in leukemia cells undergoing differentiation by all-trans retinoic acid treatment attenuates chemotherapeutic agent-induced apoptosis. Leukemia 2006, 20, 1009–1016. [Google Scholar] [CrossRef] [Green Version]
- Leverson, J.D.; Zhang, H.; Chen, J.; Tahir, S.K.; Phillips, D.C.; Xue, J.; Nimmer, P.; Jin, S.; Smith, M.; Xiao, Y.; et al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis. 2015, 6, e1590. [Google Scholar] [CrossRef]
- Puhr, M.; Hoefer, J.; Neuwirt, H.; Eder, I.E.; Kern, J.; Schafer, G.; Geley, S.; Heidegger, I.; Klocker, H.; Culig, Z. PIAS1 is a crucial factor for prostate cancer cell survival and a valid target in docetaxel resistant cells. Oncotarget 2014, 5, 12043–12056. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Placzek, W.J. PTBP1 modulation of MCL1 expression regulates cellular apoptosis induced by antitubulin chemotherapeutics. Cell Death Differ. 2016, 23, 1681–1690. [Google Scholar] [CrossRef] [Green Version]
- Chu, R.; Alford, S.E.; Hart, K.; Kothari, A.; Mackintosh, S.G.; Kovak, M.R.; Chambers, T.C. Mitotic arrest-induced phosphorylation of Mcl-1 revisited using two-dimensional gel electrophoresis and phosphoproteomics: Nine phosphorylation sites identified. Oncotarget 2016, 7, 78958–78970. [Google Scholar] [CrossRef]
- Eichhorn, J.M.; Sakurikar, N.; Alford, S.E.; Chu, R.; Chambers, T.C. Critical role of anti-apoptotic Bcl-2 protein phosphorylation in mitotic death. Cell Death Dis. 2013, 4, e834. [Google Scholar] [CrossRef] [PubMed]
- Kruczynski, A.; Etievant, C.; Perrin, D.; Chansard, N.; Duflos, A.; Hill, B.T. Characterization of cell death induced by vinflunine, the most recent Vinca alkaloid in clinical development. Br. J. Cancer 2002, 86, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Rodi, D.J.; Janes, R.W.; Sanganee, H.J.; Holton, R.A.; Wallace, B.A.; Makowski, L. Screening of a library of phage-displayed peptides identifies human bcl-2 as a taxol-binding protein. J. Mol. Biol. 1999, 285, 197–203. [Google Scholar] [CrossRef]
- Rodi, D.J.; Makowski, L. Similarity between the sequences of taxol-selected peptides and the disordered loop of the anti-apoptotic protein, Bcl-2. Pac. Symp. Biocomput. 1999, 4, 532–541. [Google Scholar]
- Ferlini, C.; Cicchillitti, L.; Raspaglio, G.; Bartollino, S.; Cimitan, S.; Bertucci, C.; Mozzetti, S.; Gallo, D.; Persico, M.; Fattorusso, C.; et al. Paclitaxel directly binds to Bcl-2 and functionally mimics activity of Nur77. Cancer Res. 2009, 69, 6906–6914. [Google Scholar] [CrossRef] [PubMed]
- Knipling, L.; Wolff, J. Direct interaction of Bcl-2 proteins with tubulin. Biochem. Biophys. Res. Commun. 2006, 341, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Esteve, M.A.; Carre, M.; Bourgarel-Rey, V.; Kruczynski, A.; Raspaglio, G.; Ferlini, C.; Braguer, D. Bcl-2 down-regulation and tubulin subtype composition are involved in resistance of ovarian cancer cells to vinflunine. Mol. Cancer Ther. 2006, 5, 2824–2833. [Google Scholar] [CrossRef] [Green Version]
- Bennett, A.; Sloss, O.; Topham, C.; Nelson, L.; Tighe, A.; Taylor, S.S. Inhibition of Bcl-xL sensitizes cells to mitotic blockers, but not mitotic drivers. Open Biol. 2016, 6, 160134. [Google Scholar] [CrossRef] [Green Version]
- Sloss, O.; Topham, C.; Diez, M.; Taylor, S. Mcl-1 dynamics influence mitotic slippage and death in mitosis. Oncotarget 2016, 7, 5176–5192. [Google Scholar] [CrossRef] [Green Version]
- Millman, S.E.; Pagano, M. MCL1 meets its end during mitotic arrest. EMBO Rep. 2011, 12, 384–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson II, R.S.; Placzek, W.; Fernandez, A.; Ziaee, S.; Chu, C.Y.; Wei, J.; Stebbins, J.; Kitada, S.; Fritz, G.; Reed, J.C.; et al. Sabutoclax, a Mcl-1 antagonist, inhibits tumorigenesis in transgenic mouse and human xenograft models of prostate cancer. Neoplasia 2012, 14, 656–665. [Google Scholar] [CrossRef]
- Rapino, F.; Naumann, I.; Fulda, S. Bortezomib antagonizes microtubule-interfering drug-induced apoptosis by inhibiting G2/M transition and MCL-1 degradation. Cell Death Dis. 2013, 4, e925. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Stebbins, J.L.; Kitada, S.; Dash, R.; Placzek, W.; Rega, M.F.; Wu, B.; Cellitti, J.; Zhai, D.; Yang, L.; et al. BI-97C1, an optically pure Apogossypol derivative as pan-active inhibitor of antiapoptotic B-cell lymphoma/leukemia-2 (Bcl-2) family proteins. J. Med. Chem. 2010, 53, 4166–4176. [Google Scholar] [CrossRef]
- Billard, C. BH3 mimetics: Status of the field and new developments. Mol. Cancer Ther. 2013, 12, 1691–1700. [Google Scholar] [CrossRef]
- Kutuk, O.; Letai, A. Alteration of the mitochondrial apoptotic pathway is key to acquired paclitaxel resistance and can be reversed by ABT-737. Cancer Res. 2008, 68, 7985–7994. [Google Scholar] [CrossRef] [Green Version]
- Parrondo, R.; de Las Pozas, A.; Reiner, T.; Perez-Stable, C. ABT-737, a small molecule Bcl-2/Bcl-xL antagonist, increases antimitotic-mediated apoptosis in human prostate cancer cells. PeerJ 2013, 1, e144. [Google Scholar] [CrossRef]
- Watanabe, M.; Umezawa, K.; Higashihara, M.; Horie, R. Combined inhibition of NF-kappaB and Bcl-2 triggers synergistic reduction of viability and induces apoptosis in melanoma cells. Oncol. Res. 2013, 21, 173–180. [Google Scholar] [CrossRef]
- Lieber, J.; Eicher, C.; Wenz, J.; Kirchner, B.; Warmann, S.W.; Fuchs, J.; Armeanu-Ebinger, S. The BH3 mimetic ABT-737 increases treatment efficiency of paclitaxel against hepatoblastoma. BMC Cancer 2011, 11, 362. [Google Scholar] [CrossRef]
- Bah, N.; Maillet, L.; Ryan, J.; Dubreil, S.; Gautier, F.; Letai, A.; Juin, P.; Barille-Nion, S. Bcl-xL controls a switch between cell death modes during mitotic arrest. Cell Death Dis. 2014, 5, e1291. [Google Scholar] [CrossRef]
- Wang, C.; Huang, S.B.; Yang, M.C.; Lin, Y.T.; Chu, I.H.; Shen, Y.N.; Chiu, Y.H.; Hung, S.H.; Kang, L.; Hong, Y.R.; et al. Combining paclitaxel with ABT-263 has a synergistic effect on paclitaxel resistant prostate cancer cells. PLoS ONE 2015, 10, e0120913. [Google Scholar] [CrossRef]
- Vogler, M.; Walter, H.S.; Dyer, M.J.S. Targeting anti-apoptotic BCL2 family proteins in haematological malignancies - from pathogenesis to treatment. Br. J. Haematol. 2017, 178, 364–379. [Google Scholar] [CrossRef]
- Kotschy, A.; Szlavik, Z.; Murray, J.; Davidson, J.; Maragno, A.L.; Le Toumelin-Braizat, G.; Chanrion, M.; Kelly, G.L.; Gong, J.N.; Moujalled, D.M.; et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016, 538, 477–482. [Google Scholar] [CrossRef]
- Merino, D.; Whittle, J.R.; Vaillant, F.; Serrano, A.; Gong, J.N.; Giner, G.; Maragno, A.L.; Chanrion, M.; Schneider, E.; Pal, B.; et al. Synergistic action of the MCL-1 inhibitor S63845 with current therapies in preclinical models of triple-negative and HER2-amplified breast cancer. Sci. Transl. Med. 2017, 9, 401. [Google Scholar] [CrossRef]
- Ackler, S.; Mitten, M.J.; Foster, K.; Oleksijew, A.; Refici, M.; Tahir, S.K.; Xiao, Y.; Tse, C.; Frost, D.J.; Fesik, S.W.; et al. The Bcl-2 inhibitor ABT-263 enhances the response of multiple chemotherapeutic regimens in hematologic tumors in vivo. Cancer Chemother. Pharmacol. 2010, 66, 869–880. [Google Scholar] [CrossRef]
- Dai, H.; Ding, H.; Meng, X.W.; Lee, S.H.; Schneider, P.A.; Kaufmann, S.H. Contribution of Bcl-2 phosphorylation to Bak binding and drug resistance. Cancer Res. 2013, 73, 6998–7008. [Google Scholar] [CrossRef]
- Castro, J.E.; Loria, O.J.; Aguillon, R.A.; James, D.; Llanos, C.A.; Rassenti, L.; Wood, B.A.; Homlund, J.T.; Kipps, T.J. A phase II, open label study of AT-101 in combination with rituximab in patients with relapsed or refractory chronic lymphocytic leukemia. Evaluation of two dose regimens. Blood 2007, 110, 3119. [Google Scholar]
- Cakar, B.; Gursoy, P.; Atmaca, H.; Kisim, A.; Bozhurt, E.; Uzunoglu, S.; Sezgin, C.; Sanli, U.A.; Karabulut, B.; Uslu, R.; et al. Paclitaxel in combination with AT-101 induces apoptosis via supressing Bcl-2, bcl-XL, mcl-1 proteins in human breast cancer cells. J. Clin. Oncol. 2017, 31 (Suppl. 15), e13578. [Google Scholar]
- Caenepeel, S.; Brown, S.P.; Belmontes, B.; Moody, G.; Keegan, K.S.; Chui, D.; Whittington, D.A.; Huang, X.; Poppe, L.; Cheng, A.C.; et al. AMG 176, a selective MCL1 inhibitor, is effective in hematologic cancer models alone and in combiantion with established therapies. Cancer Discov. 2018, 8, 1582–1597. [Google Scholar]
- Tron, A.E.; Belmonte, M.A.; Adam, A.; Aquila, B.M.; Boise, L.H.; Chiarparin, E.; Cidado, J.; Embrey, K.J.; Gangl, E.; Gibbons, F.D.; et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat. Commun. 2018, 9, 5341. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| MTA | Protein | Cell Type/Model | Effect | Reference |
|---|---|---|---|---|
| Paclitaxel | BCL2 | Breast, Lung, Prostate | * Presence sensitizes | [37,39] |
| BCL2 | Leukemia | Presence induces resistance; paclitaxel decreases BCL2 mRNA expression | [74,75] | |
| BCLxL | Leukemia, Colon | Upregulation induces resistance, inhibition sensitizes | [76] | |
| BCLW | Leukemia | Knockdown/out sensitizes | [77] | |
| BFL1/A1 | Leukemia | Upregulation induces resistance | [78] | |
| Docetaxel | BCLxL | Lung, Myeloma | Inhibition sensitizes | [79] |
| MCL1 | Prostate | * Inhibition or downregulation sensitizes | [80,81] | |
| Vincristine | BCL2 | Leukemia | Treatment decreases BCL2 mRNA expression | [75] |
| Vinblastine | MCL1 | HeLa | Treatment decreases MCL1 protein levels | [82,83] |
| Vinflunine | BFL1/A1 | Lymphoma | Increase linked with resistance | [84] |
| BH3 Mimetic | MCL1 | BFL1/A1 | BCLW | BCLxL | BCL2 | Cell Type/Model | Effect | Reference |
|---|---|---|---|---|---|---|---|---|
| ABT-737 | X | X | X | Melanoma, Breast, Prostate, Liver | Sensitizes cells to paclitaxel or docetaxel | [100,102,103] | ||
| ABT-263 | X | X | X | Prostate | Additive with vincristine; synergy with paclitaxel | [104,109] | ||
| ABT-199 | X | Leukemia, Lymphoma | Approved for CLL; sensitizes cells to paclitaxel | [95,110] | ||||
| WEHI-539 | X | Colon | Sensitizes to Paclitaxel | [90] | ||||
| Sabutoclax | X | X | X | X | X | Prostate | Sensitizes to docetaxel | [81,93] |
| AT-101 | X | X | X | X | Breast | Synergizes with paclitaxel | [111,112] | |
| S63845 | X | Breast | Synergizes with docetaxel | [107,108] | ||||
| A1210477 | X | [79] | ||||||
| AMG-176 | X | [113] | ||||||
| AZD-5991 | X | [114] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Whitaker, R.H.; Placzek, W.J. Regulating the BCL2 Family to Improve Sensitivity to Microtubule Targeting Agents. Cells 2019, 8, 346. https://doi.org/10.3390/cells8040346
Whitaker RH, Placzek WJ. Regulating the BCL2 Family to Improve Sensitivity to Microtubule Targeting Agents. Cells. 2019; 8(4):346. https://doi.org/10.3390/cells8040346
Chicago/Turabian StyleWhitaker, Robert H., and William J. Placzek. 2019. "Regulating the BCL2 Family to Improve Sensitivity to Microtubule Targeting Agents" Cells 8, no. 4: 346. https://doi.org/10.3390/cells8040346
APA StyleWhitaker, R. H., & Placzek, W. J. (2019). Regulating the BCL2 Family to Improve Sensitivity to Microtubule Targeting Agents. Cells, 8(4), 346. https://doi.org/10.3390/cells8040346