Functional Characterization of Clinically-Relevant Rare Variants in ABCG2 Identified in a Gout and Hyperuricemia Cohort

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Subjects
2.2. PCR Amplification of ABCG2 and Sequence Analysis
2.3. Materials
2.4. Preparation of ABCG2 Mutants’ Expression Vectors
2.5. Cell Culture
2.6. Preparation of Whole Cell Lysates
2.7. Preparation of ABCG2-Expressing Plasma Membrane Vesicles
2.8. Immunoblotting
2.9. Confocal Laser Scanning Microscopic Observation
2.10. Urate Transport Assay
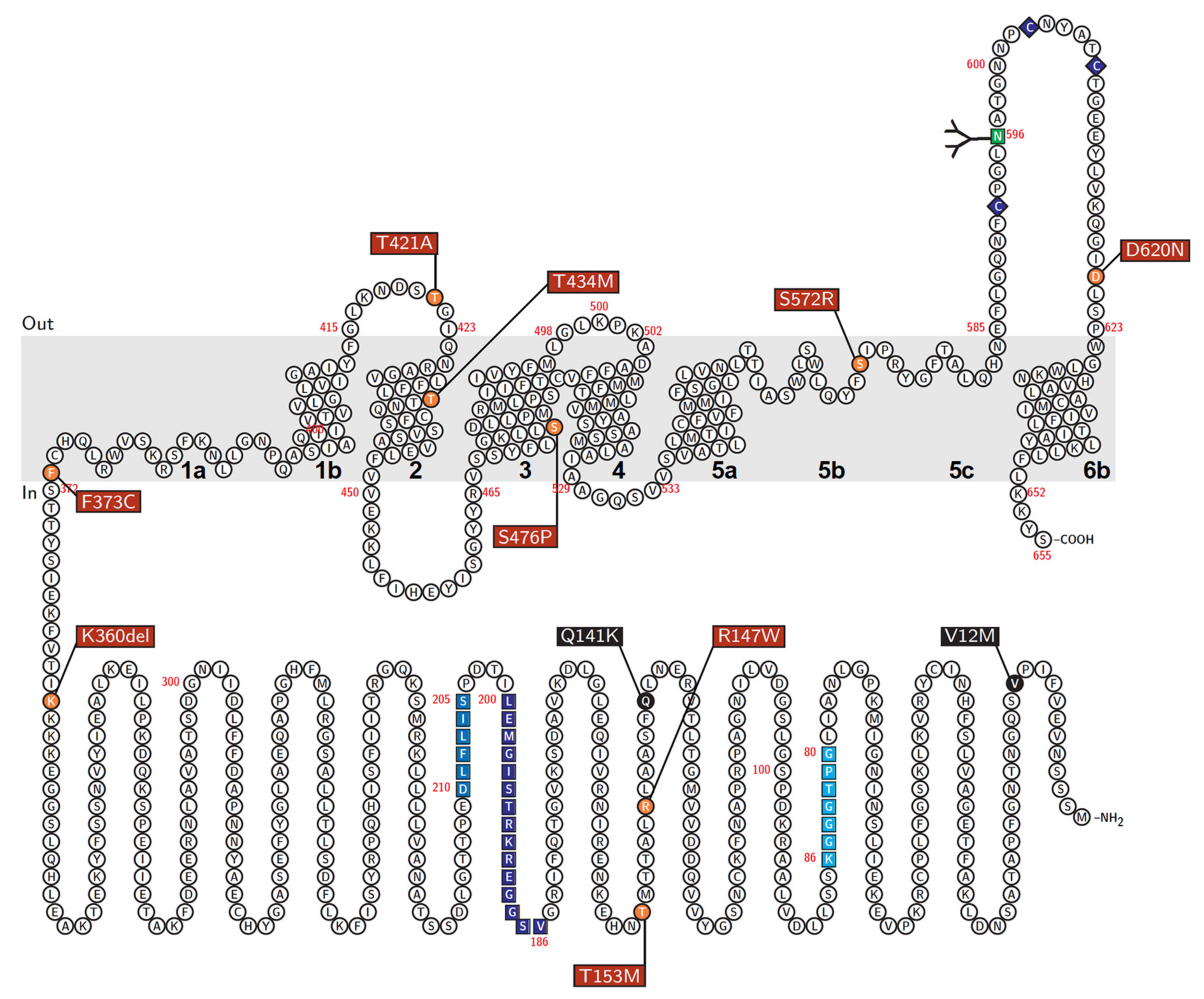
2.11. Schematic Illustration of ABCG2 Protein
2.12. Statistical Analysis
3. Results and Discussion
3.1. Subjects
3.2. Identification of ABCG2 Variants in a Gout/Hyperuricemia Cohort
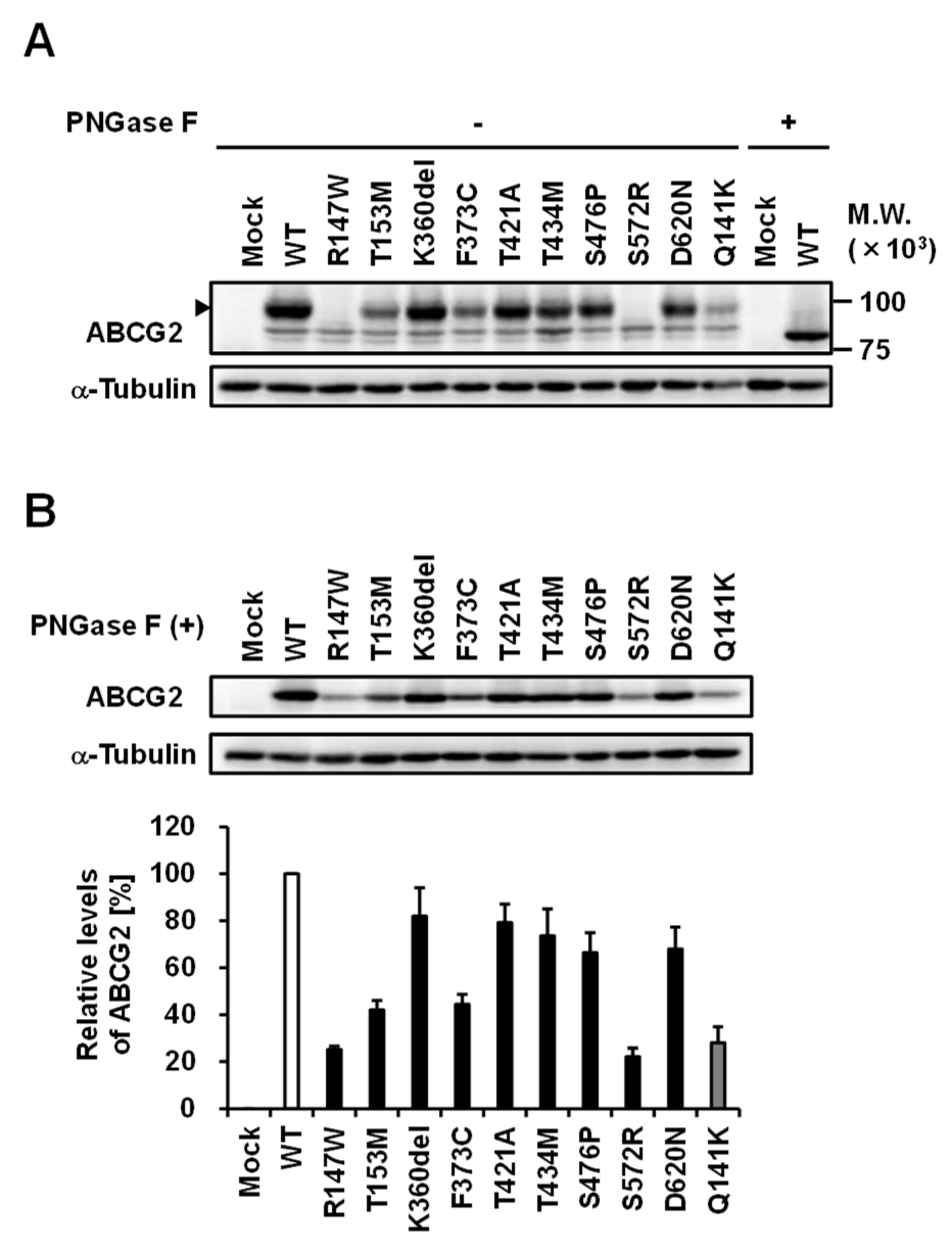
3.3. Effect of Each Mutation on the Glycosylation Status of ABCG2 Protein
3.4. Effect of Each Mutation on the Intracellular Localization of ABCG2 Protein
3.5. Effect of Each Mutation on the Urate Transport Activity of ABCG2 Protein
3.6. Integration of the Obtained Data
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABCG2 | ATP-binding cassette subfamily G member 2 |
| BCRP | Brest cancer resistance protein |
| CI | Confidence interval |
| GWAS | Genome-wide association study |
| IQR | Interquartile range |
| MAF | Minor allele frequency |
| PM | Plasma membrane |
| SNP | Single nucleotide polymorphism |
| SUA | Serum uric acid |
| TBST | Tris-buffered saline containing 0.05% Tween 20 |
| WT | Wild-type |
References
- Dalbeth, N.; Merriman, T.R.; Stamp, L.K. Gout. Lancet 2016, 388, 2039–2052. [Google Scholar] [CrossRef]
- Yeldandi, A.V.; Wang, X.D.; Alvares, K.; Kumar, S.; Rao, M.S.; Reddy, J.K. Human urate oxidase gene: Cloning and partial sequence analysis reveal a stop codon within the fifth exon. Biochem. Biophys. Res. Commun. 1990, 171, 641–646. [Google Scholar] [CrossRef]
- Enomoto, A.; Kimura, H.; Chairoungdua, A.; Shigeta, Y.; Jutabha, P.; Cha, S.H.; Hosoyamada, M.; Takeda, M.; Sekine, T.; Igarashi, T.; et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 2002, 417, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Vitart, V.; Rudan, I.; Hayward, C.; Gray, N.K.; Floyd, J.; Palmer, C.N.; Knott, S.A.; Kolcic, I.; Polasek, O.; Graessler, J.; et al. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat. Genet. 2008, 40, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, H.; Chiba, T.; Nagamori, S.; Nakayama, A.; Domoto, H.; Phetdee, K.; Wiriyasermkul, P.; Kikuchi, Y.; Oda, T.; Nishiyama, J.; et al. Mutations in glucose transporter 9 gene SLC2A9 cause renal hypouricemia. Am. J. Hum. Genet. 2008, 83, 744–751. [Google Scholar]
- Caulfield, M.J.; Munroe, P.B.; O’Neill, D.; Witkowska, K.; Charchar, F.J.; Doblado, M.; Evans, S.; Eyheramendy, S.; Onipinla, A.; Howard, P.; et al. SLC2A9 is a high-capacity urate transporter in humans. PLoS Med. 2008, 5, e197. [Google Scholar] [CrossRef]
- Ichida, K.; Matsuo, H.; Takada, T.; Nakayama, A.; Murakami, K.; Shimizu, T.; Yamanashi, Y.; Kasuga, H.; Nakashima, H.; Nakamura, T.; et al. Decreased extra-renal urate excretion is a common cause of hyperuricemia. Nat. Commun. 2012, 3, 764. [Google Scholar] [CrossRef] [PubMed]
- Woodward, O.M.; Kottgen, A.; Coresh, J.; Boerwinkle, E.; Guggino, W.B.; Kottgen, M. Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc. Natl. Acad. Sci. USA 2009, 106, 10338–10342. [Google Scholar] [CrossRef]
- Matsuo, H.; Takada, T.; Ichida, K.; Nakamura, T.; Nakayama, A.; Ikebuchi, Y.; Ito, K.; Kusanagi, Y.; Chiba, T.; Tadokoro, S.; et al. Common defects of ABCG2, a high-capacity urate exporter, cause gout: A function-based genetic analysis in a Japanese population. Sci. Transl. Med. 2009, 1, 5ra11. [Google Scholar] [CrossRef]
- Nakayama, A.; Matsuo, H.; Nakaoka, H.; Nakamura, T.; Nakashima, H.; Takada, Y.; Oikawa, Y.; Takada, T.; Sakiyama, M.; Shimizu, S.; et al. Common dysfunctional variants of ABCG2 have stronger impact on hyperuricemia progression than typical environmental risk factors. Sci. Rep. 2014, 4, 5227. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, H.; Nakayama, A.; Sakiyama, M.; Chiba, T.; Shimizu, S.; Kawamura, Y.; Nakashima, H.; Nakamura, T.; Takada, Y.; Oikawa, Y.; et al. ABCG2 dysfunction causes hyperuricemia due to both renal urate underexcretion and renal urate overload. Sci. Rep. 2014, 4, 3755. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.W.; To, K.K.; Polgar, O.; Dohse, M.; Fetsch, P.; Dean, M.; Bates, S.E. ABCG2: A perspective. Adv. Drug Deliv. Rev. 2009, 61, 3–13. [Google Scholar] [CrossRef]
- Knutsen, T.; Rao, V.K.; Ried, T.; Mickley, L.; Schneider, E.; Miyake, K.; Ghadimi, B.M.; Padilla-Nash, H.; Pack, S.; Greenberger, L.; et al. Amplification of 4q21-q22 and the MXR gene in independently derived mitoxantrone-resistant cell lines. Genes Chromosomes Cancer 2000, 27, 110–116. [Google Scholar] [CrossRef]
- Major, T.J.; Dalbeth, N.; Stahl, E.A.; Merriman, T.R. An update on the genetics of hyperuricaemia and gout. Nat. Rev. Rheumatol. 2018, 14, 341–353. [Google Scholar] [CrossRef]
- Nakayama, A.; Nakaoka, H.; Yamamoto, K.; Sakiyama, M.; Shaukat, A.; Toyoda, Y.; Okada, Y.; Kamatani, Y.; Nakamura, T.; Takada, T.; et al. GWAS of clinically defined gout and subtypes identifies multiple susceptibility loci that include urate transporter genes. Ann. Rheum. Dis. 2017, 76, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, H.; Yamamoto, K.; Nakaoka, H.; Nakayama, A.; Sakiyama, M.; Chiba, T.; Takahashi, A.; Nakamura, T.; Nakashima, H.; Takada, Y.; et al. Genome-wide association study of clinically defined gout identifies multiple risk loci and its association with clinical subtypes. Ann. Rheum. Dis. 2016, 75, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Kottgen, A.; Albrecht, E.; Teumer, A.; Vitart, V.; Krumsiek, J.; Hundertmark, C.; Pistis, G.; Ruggiero, D.; O’Seaghdha, C.M.; Haller, T.; et al. Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat. Genet. 2013, 45, 145–154. [Google Scholar] [CrossRef]
- Kolz, M.; Johnson, T.; Sanna, S.; Teumer, A.; Vitart, V.; Perola, M.; Mangino, M.; Albrecht, E.; Wallace, C.; Farrall, M.; et al. Meta-analysis of 28,141 individuals identifies common variants within five new loci that influence uric acid concentrations. PLoS Genet. 2009, 5, e1000504. [Google Scholar] [CrossRef] [PubMed]
- Dehghan, A.; Kottgen, A.; Yang, Q.; Hwang, S.J.; Kao, W.L.; Rivadeneira, F.; Boerwinkle, E.; Levy, D.; Hofman, A.; Astor, B.C.; et al. Association of three genetic loci with uric acid concentration and risk of gout: A genome-wide association study. Lancet 2008, 372, 1953–1961. [Google Scholar] [CrossRef]
- Heyes, N.; Kapoor, P.; Kerr, I.D. Polymorphisms of the Multidrug Pump ABCG2: A Systematic Review of Their Effect on Protein Expression, Function, and Drug Pharmacokinetics. Drug Metab. Dispos. 2018, 46, 1886–1899. [Google Scholar] [CrossRef]
- Matsuo, H.; Ichida, K.; Takada, T.; Nakayama, A.; Nakashima, H.; Nakamura, T.; Kawamura, Y.; Takada, Y.; Yamamoto, K.; Inoue, H.; et al. Common dysfunctional variants in ABCG2 are a major cause of early-onset gout. Sci. Rep. 2013, 3, 2014. [Google Scholar] [CrossRef] [PubMed]
- Higashino, T.; Takada, T.; Nakaoka, H.; Toyoda, Y.; Stiburkova, B.; Miyata, H.; Ikebuchi, Y.; Nakashima, H.; Shimizu, S.; Kawaguchi, M.; et al. Multiple common and rare variants of ABCG2 cause gout. RMD Open 2017, 3, e000464. [Google Scholar] [CrossRef]
- Stiburkova, B.; Pavelcova, K.; Zavada, J.; Petru, L.; Simek, P.; Cepek, P.; Pavlikova, M.; Matsuo, H.; Merriman, T.R.; Pavelka, K. Functional non-synonymous variants of ABCG2 and gout risk. Rheumatology (Oxford) 2017, 56, 1982–1992. [Google Scholar] [CrossRef] [PubMed]
- Stiburkova, B.; Miyata, H.; Zavada, J.; Tomcik, M.; Pavelka, K.; Storkanova, G.; Toyoda, Y.; Takada, T.; Suzuki, H. Novel dysfunctional variant in ABCG2 as a cause of severe tophaceous gout: Biochemical, molecular genetics and functional analysis. Rheumatology (Oxford) 2016, 55, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Stiburkova, B.; Pavelcova, K.; Pavlikova, M.; Jesina, P.; Pavelka, K. The impact of dysfunctional variants of ABCG2 on hyperuricemia and gout in pediatric-onset patients. Arthritis Res. Ther. 2019, 21, 77. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.L.; Robinson, H.; Masi, A.T.; Decker, J.L.; McCarty, D.J.; Yu, T.F. Preliminary criteria for the classification of the acute arthritis of primary gout. Arthritis Rheum. 1977, 20, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, Y.; Takada, T.; Miyata, H.; Ishikawa, T.; Suzuki, H. Regulation of the Axillary Osmidrosis-Associated ABCC11 Protein Stability by N-Linked Glycosylation: Effect of Glucose Condition. PLoS ONE 2016, 11, e0157172. [Google Scholar] [CrossRef]
- Toyoda, Y.; Sakurai, A.; Mitani, Y.; Nakashima, M.; Yoshiura, K.; Nakagawa, H.; Sakai, Y.; Ota, I.; Lezhava, A.; Hayashizaki, Y.; et al. Earwax, osmidrosis, and breast cancer: Why does one SNP (538G>A) in the human ABC transporter ABCC11 gene determine earwax type? FASEB J. 2009, 23, 2001–2013. [Google Scholar] [CrossRef]
- Toyoda, Y.; Takada, T.; Gomi, T.; Nakagawa, H.; Ishikawa, T.; Suzuki, H. Clinical and Molecular Evidence of ABCC11 Protein Expression in Axillary Apocrine Glands of Patients with Axillary Osmidrosis. Int. J. Mol. Sci. 2017, 18, 417. [Google Scholar] [CrossRef]
- Nakagawa, H.; Wakabayashi-Nakao, K.; Tamura, A.; Toyoda, Y.; Koshiba, S.; Ishikawa, T. Disruption of N-linked glycosylation enhances ubiquitin-mediated proteasomal degradation of the human ATP-binding cassette transporter ABCG2. FEBS J. 2009, 276, 7237–7252. [Google Scholar] [CrossRef]
- Miyata, H.; Takada, T.; Toyoda, Y.; Matsuo, H.; Ichida, K.; Suzuki, H. Identification of Febuxostat as a New Strong ABCG2 Inhibitor: Potential Applications and Risks in Clinical Situations. Front. Pharmacol. 2016, 7, 518. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, Y.; Takada, T.; Suzuki, H. Halogenated hydrocarbon solvent-related cholangiocarcinoma risk: Biliary excretion of glutathione conjugates of 1,2-dichloropropane evidenced by untargeted metabolomics analysis. Sci. Rep. 2016, 6, 24586. [Google Scholar] [CrossRef] [PubMed]
- Takada, T.; Yamamoto, T.; Matsuo, H.; Tan, J.K.; Ooyama, K.; Sakiyama, M.; Miyata, H.; Yamanashi, Y.; Toyoda, Y.; Higashino, T.; et al. Identification of ABCG2 as an Exporter of Uremic Toxin Indoxyl Sulfate in Mice and as a Crucial Factor Influencing CKD Progression. Sci. Rep. 2018, 8, 11147. [Google Scholar] [CrossRef] [PubMed]
- Hurba, O.; Mancikova, A.; Krylov, V.; Pavlikova, M.; Pavelka, K.; Stiburkova, B. Complex analysis of urate transporters SLC2A9, SLC22A12 and functional characterization of non-synonymous allelic variants of GLUT9 in the Czech population: No evidence of effect on hyperuricemia and gout. PLoS ONE 2014, 30, e107902. [Google Scholar] [CrossRef]
- Taylor, N.M.I.; Manolaridis, I.; Jackson, S.M.; Kowal, J.; Stahlberg, H.; Locher, K.P. Structure of the human multidrug transporter ABCG2. Nature 2017, 546, 504–509. [Google Scholar] [CrossRef]
- Beitz, E. T(E)Xtopo: Shaded membrane protein topology plots in LAT(E)X2epsilon. Bioinformatics 2000, 16, 1050–1051. [Google Scholar] [CrossRef]
- Tamura, A.; Watanabe, M.; Saito, H.; Nakagawa, H.; Kamachi, T.; Okura, I.; Ishikawa, T. Functional validation of the genetic polymorphisms of human ATP-binding cassette (ABC) transporter ABCG2: Identification of alleles that are defective in porphyrin transport. Mol. Pharmacol. 2006, 70, 287–296. [Google Scholar]
- Toyoda, Y.; Takada, T.; Umezawa, M.; Tomura, F.; Yamanashi, Y.; Takeda, K.; Suzuki, H. Identification of hepatic NPC1L1 as an NAFLD risk factor evidenced by ezetimibe-mediated steatosis prevention and recovery. FASEB BioAdv. 2019, in press. [Google Scholar] [CrossRef]
- Kondo, C.; Suzuki, H.; Itoda, M.; Ozawa, S.; Sawada, J.; Kobayashi, D.; Ieiri, I.; Mine, K.; Ohtsubo, K.; Sugiyama, Y. Functional analysis of SNPs variants of BCRP/ABCG2. Pharm. Res. 2004, 21, 1895–1903. [Google Scholar] [CrossRef]
- Nakagawa, H.; Toyoda, Y.; Wakabayashi-Nakao, K.; Tamaki, H.; Osumi, M.; Ishikawa, T. Ubiquitin-mediated proteasomal degradation of ABC transporters: A new aspect of genetic polymorphisms and clinical impacts. J. Pharm. Sci. 2011, 100, 3602–3619. [Google Scholar] [CrossRef]
- Zambo, B.; Bartos, Z.; Mozner, O.; Szabo, E.; Varady, G.; Poor, G.; Palinkas, M.; Andrikovics, H.; Hegedus, T.; Homolya, L.; et al. Clinically relevant mutations in the ABCG2 transporter uncovered by genetic analysis linked to erythrocyte membrane protein expression. Sci. Rep. 2018, 8, 7487. [Google Scholar] [CrossRef] [PubMed]
- Haider, A.J.; Cox, M.H.; Jones, N.; Goode, A.J.; Bridge, K.S.; Wong, K.; Briggs, D.; Kerr, I.D. Identification of residues in ABCG2 affecting protein trafficking and drug transport, using co-evolutionary analysis of ABCG sequences. Biosci. Rep. 2015, 35, e00241. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, Z.; Liu, S.; Wang, C.; Han, L.; Cui, L.; Zhou, J.; Zou, H.; Liu, Z.; Chen, J.; et al. Genome-wide association analysis identifies three new risk loci for gout arthritis in Han Chinese. Nat. Commun. 2015, 6, 7041. [Google Scholar] [CrossRef] [PubMed]
- Sakiyama, M.; Matsuo, H.; Takada, Y.; Nakamura, T.; Nakayama, A.; Takada, T.; Kitajiri, S.; Wakai, K.; Suzuki, H.; Shinomiya, N. Ethnic differences in ATP-binding cassette transporter, sub-family G, member 2 (ABCG2/BCRP): Genotype combinations and estimated functions. Drug Metab. Pharmacokinet. 2014, 29, 490–492. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [PubMed]
- Toyoda, Y.; Takada, T.; Suzuki, H. Inhibitors of human ABCG2: From technical background to recent updates with clinical implications. Front. Pharmacol. 2019, 10, 208. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | All Patients (N = 250) | Gout Patients (N = 182) | Hyperuricemia Patients (N = 68) | P-Value # | |||||||
| N | % | N | % | N | % | ||||||
| Gender | Male | 214 36 | 85.6 14.4 | 166 16 | 91.2 16.8 | 48 20 | 70.6 29.4 | 0.0002 | |||
| Female | |||||||||||
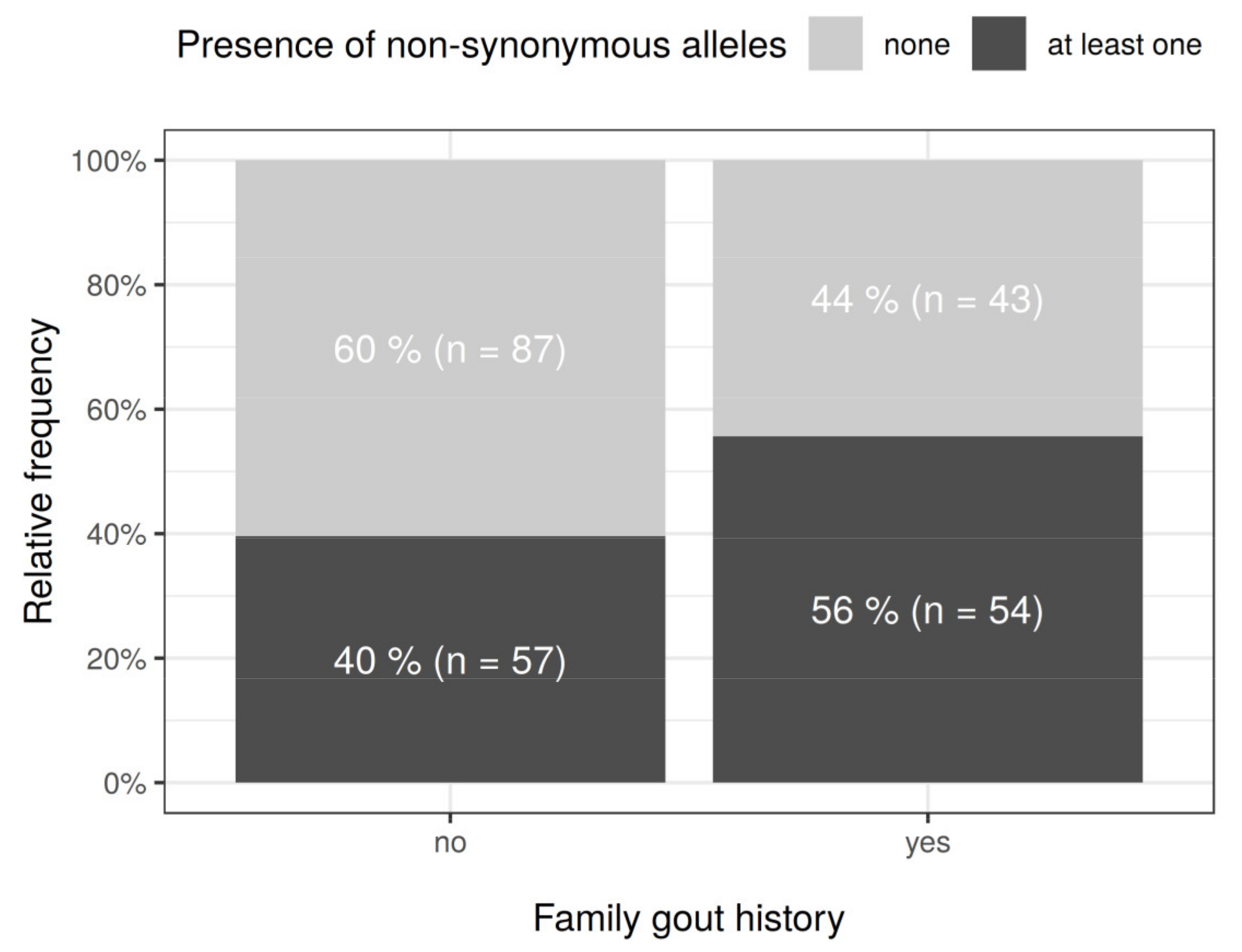
| Familial occurrence | 97 | 38.8 (40.2 *) | 66 | 36.3 (36.5 *) | 31 | 45.6 (51.7 *) | 0.0480 | ||||
| Characteristic | N | Median (IQR) | Range | N | Median (IQR) | Range | N | Median (IQR) | Range | P-Value † | |
| Age at examination [years] | 250 | 51.5 (25.0) | 3–90 | 182 | 54.0 (21.0) | 11–90 | 68 | 36.0 (42.0) | 3–78 | <0.0001 | |
| BMI at examination | 209 | 28.4 (5.8) | 16–50 | 151 | 28.4 (5.4) | 19.5–50 | 58 | 28.1 (6.4) | 16–41 | 0.0822 | |
| Gout/hyperuricemia onset $ [years] | 236 | 40.0 (28.0) | 1.2–84 | 181 | 40.0 (24.0) | 8–84 | 55 | 27.0 (40.5) | 1.2–76 | 0.0070 | |
| SUA at examination, with medication [µmol/L] | 201 | 375.0 (134.0) | 163–808 | 159 | 372.0 (128.0) | 163–808 | 42 | 424.0 (140.0) | 240–628 | 0.0515 | |
| FEUA at examination, with medication | 194 | 3.4 (2.0) | 0.9–14 | 158 | 3.4 (1.9) | 0.9–14 | 36 | 3.8 (2.1) | 1.3–8 | 0.5862 | |
| ABCG2 Variants (rs Number) | Gout (N = 182) | Hyperuricemia (N = 68) | All Patients (N = 250) | Normouricemia (N = 132) | Population MAF | P-Value # | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| N | MAF | N | MAF | N | MAF | 95% CI * | N | MAF | |||
| Common | |||||||||||
| V12M (rs2231137) | 8 | 0.0220 | 1 | 0.0074 | 9 | 0.0180 | 0.0083, 0.0339 | 5 | 0.0189 | 0.0610 | <0.0001 |
| Q141K (rs2231142) | 90 | 0.2473 | 29 | 0.2132 | 119 | 0.2380 | 0.2013, 0.2778 | 22 | 0.0833 | 0.0940 | <0.0001 |
| Rare | N | N | N | MAF | N | Population MAF | |||||
| R147W (rs372192400) | 1 | 0 | 1 | 0.0020 | 1 | 0.0001 | |||||
| T153M (rs753759474) | 1 | 0 | 1 | 0.0020 | 0 | 0.0001 | |||||
| F373C (rs752626614) | 1 | 0 | 1 | 0.0020 | 0 | 0.0000 | |||||
| T421A (rs199854112) | 0 | 1 | 1 | 0.0020 | 0 | 0.0001 | |||||
| T434M (rs769734146) | 1 | 1 | 2 | 0.0040 | 1 | 0.0000 | |||||
| S476P (Not annotated) | 1 | 0 | 1 | 0.0020 | 0 | No data | |||||
| S572R (rs200894058) | 1 | 0 | 1 | 0.0020 | 0 | 0.0002 | |||||
| D620N (rs34783571) | 2 | 0 | 2 | 0.0040 | 0 | 0.0040 | |||||
| K360del (rs750972998) | 1 | 0 | 1 | 0.0020 | 0 | 0.0001 | |||||
| rs Number | Nucleotide Change | AA Change | PM Localization | Protein Level on PM | Urate Transport | Effect on the Cellular ABCG2 Function |
|---|---|---|---|---|---|---|
| rs372192400 | 439C>T | R147W | − | N.D. | N.D. | Null |
| rs753759474 | 458C>T | T153M | + | + | + | Decrease to about a quarter |
| rs750972998 | 1079_1081delAGA | K360del | + | ++ | ++ | N.S. |
| rs752626614 | 1118T>G | F373C | + | + | ++ | Decrease to about half |
| rs199854112 | 1261A>G | T421A | + | ++ | ++ | N.S. |
| rs769734146 | 1301C>T | T434M | + | ++ | − | Almost null |
| Not annotated | 1426T>C | S476P | + | ++ | − | Almost null |
| rs200894058 | 1714A>C | S572R | − | N.D. | N.D. | Null |
| rs34783571 | 1858G>A | D620N | + | ++ | ++ | N.S. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toyoda, Y.; Mančíková, A.; Krylov, V.; Morimoto, K.; Pavelcová, K.; Bohatá, J.; Pavelka, K.; Pavlíková, M.; Suzuki, H.; Matsuo, H.; et al. Functional Characterization of Clinically-Relevant Rare Variants in ABCG2 Identified in a Gout and Hyperuricemia Cohort. Cells 2019, 8, 363. https://doi.org/10.3390/cells8040363
Toyoda Y, Mančíková A, Krylov V, Morimoto K, Pavelcová K, Bohatá J, Pavelka K, Pavlíková M, Suzuki H, Matsuo H, et al. Functional Characterization of Clinically-Relevant Rare Variants in ABCG2 Identified in a Gout and Hyperuricemia Cohort. Cells. 2019; 8(4):363. https://doi.org/10.3390/cells8040363
Chicago/Turabian StyleToyoda, Yu, Andrea Mančíková, Vladimír Krylov, Keito Morimoto, Kateřina Pavelcová, Jana Bohatá, Karel Pavelka, Markéta Pavlíková, Hiroshi Suzuki, Hirotaka Matsuo, and et al. 2019. "Functional Characterization of Clinically-Relevant Rare Variants in ABCG2 Identified in a Gout and Hyperuricemia Cohort" Cells 8, no. 4: 363. https://doi.org/10.3390/cells8040363
APA StyleToyoda, Y., Mančíková, A., Krylov, V., Morimoto, K., Pavelcová, K., Bohatá, J., Pavelka, K., Pavlíková, M., Suzuki, H., Matsuo, H., Takada, T., & Stiburkova, B. (2019). Functional Characterization of Clinically-Relevant Rare Variants in ABCG2 Identified in a Gout and Hyperuricemia Cohort. Cells, 8(4), 363. https://doi.org/10.3390/cells8040363