Membrane Protein Identification in Rodent Brain Tissue Samples and Acute Brain Slices


Abstract
:1. Introduction
2. Material and Methods
2.1. Animals
2.2. Tissue Preparation
2.3. Sample Preparation for Whole Protein Analysis
2.4. Plasma Membrane Enrichment
2.5. Measurement of Protein Concentration
2.6. Two-Dimensional (2D) Gel Electrophoresis
2.7. Silver Staining of 2D Gels
2.8. In-Solution Digestion of Proteins
2.9. Analysis by nanoLC-HDMSE
2.10. NanoLC-HDMSE Data Processing, Protein Identification, and Quantification
2.11. Statistical Analysis
3. Results
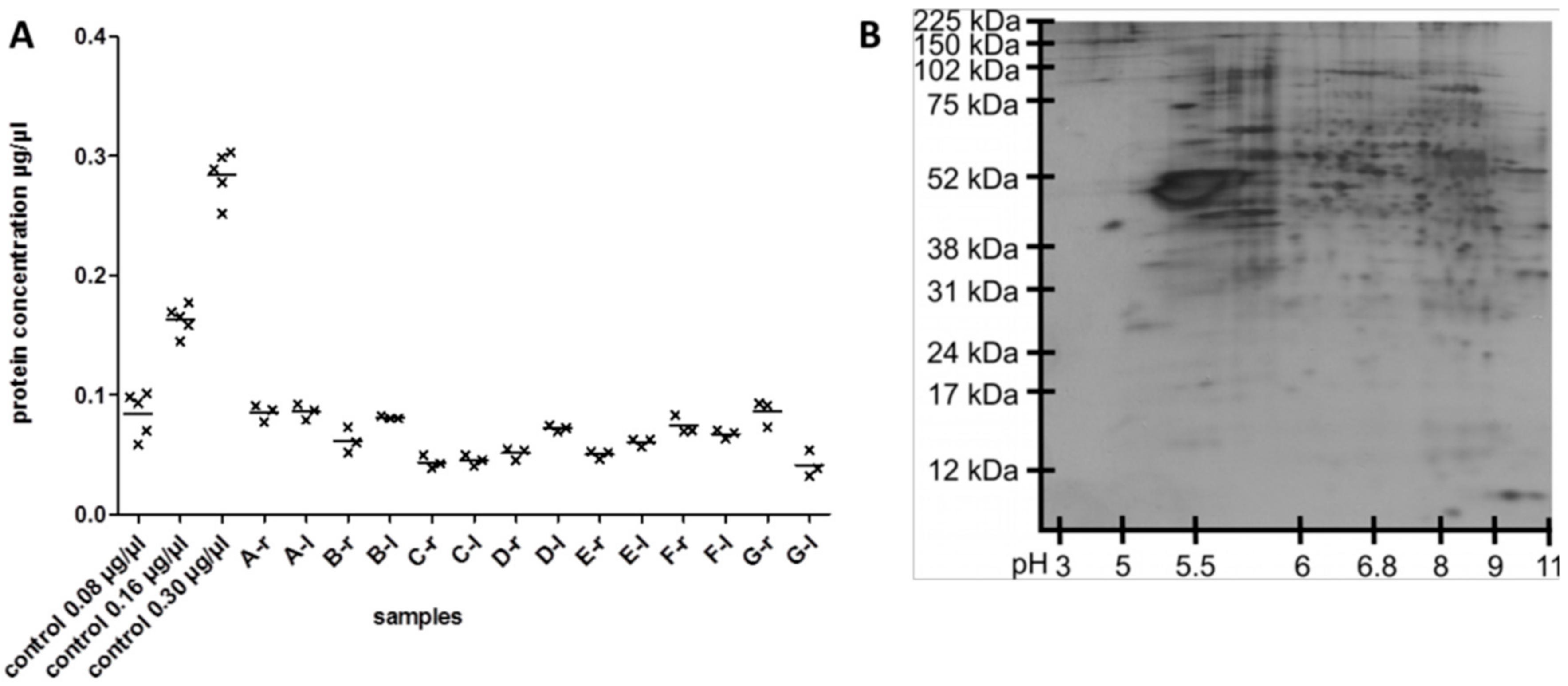
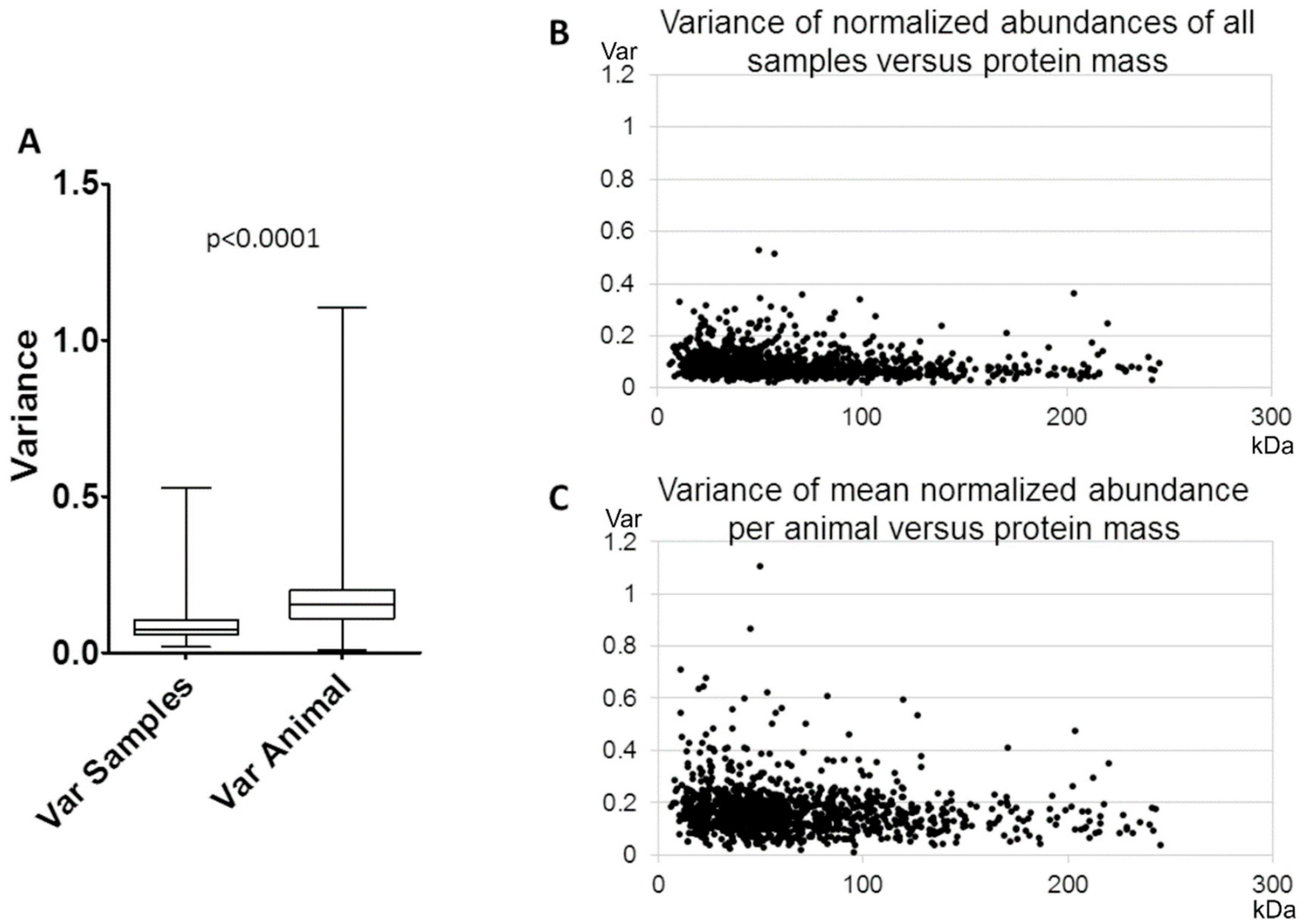
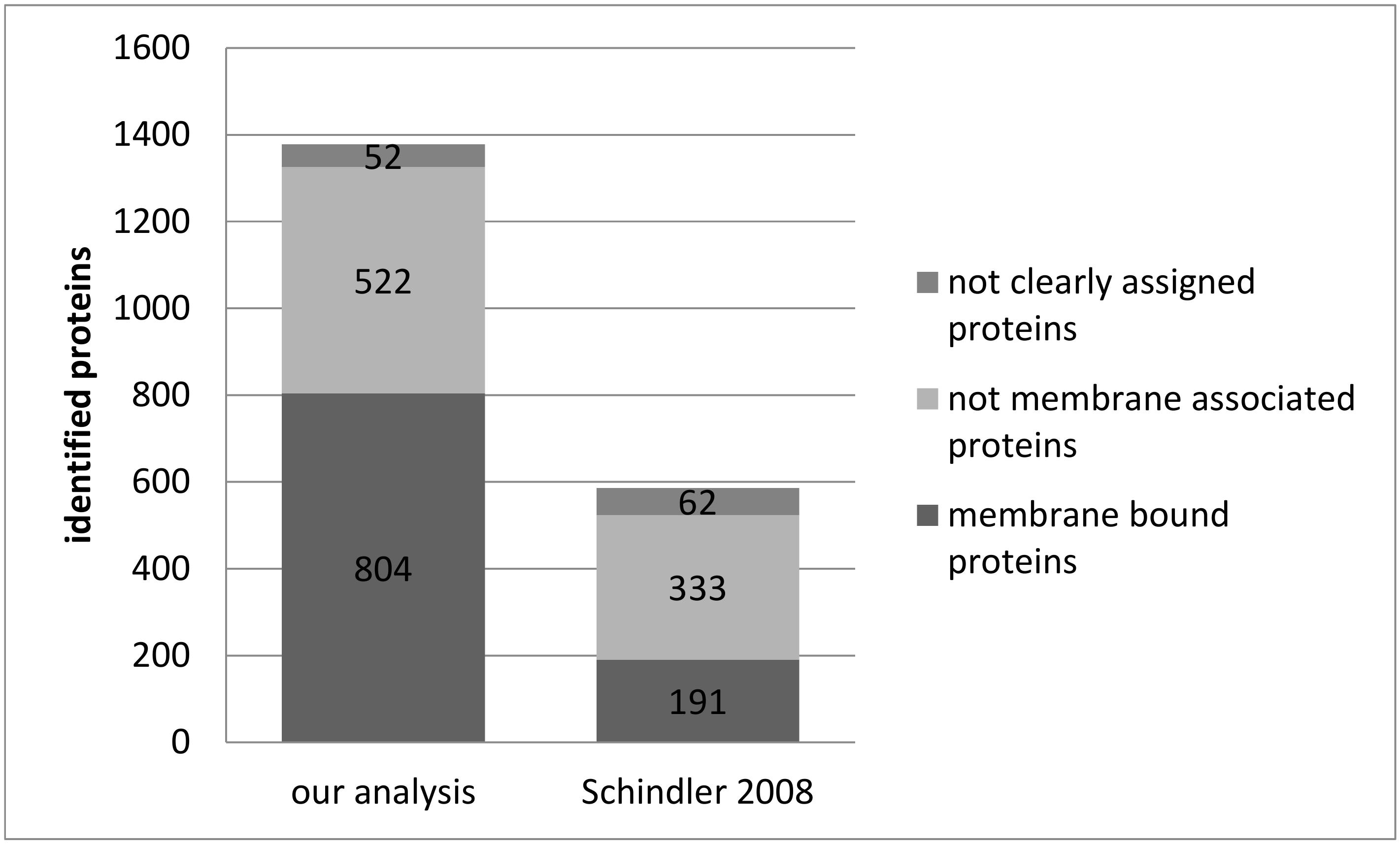
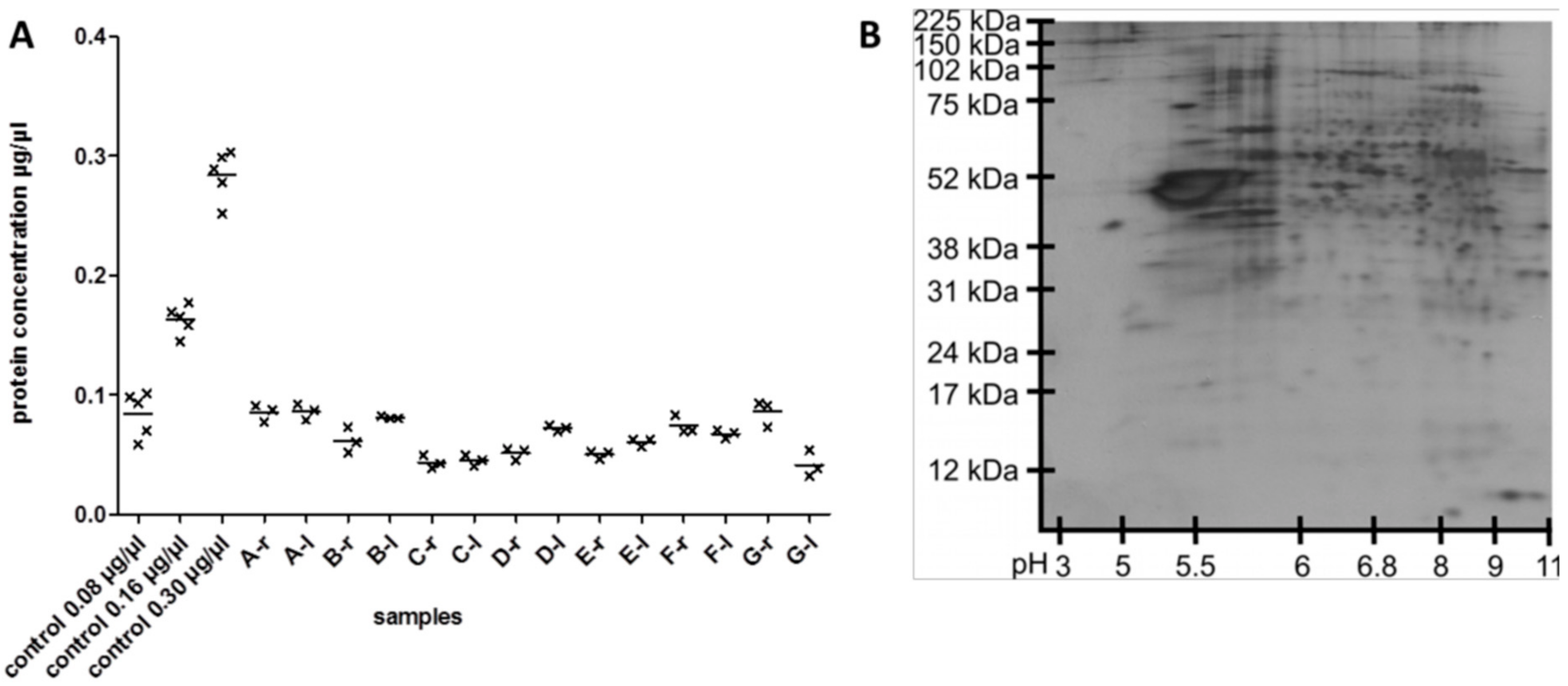
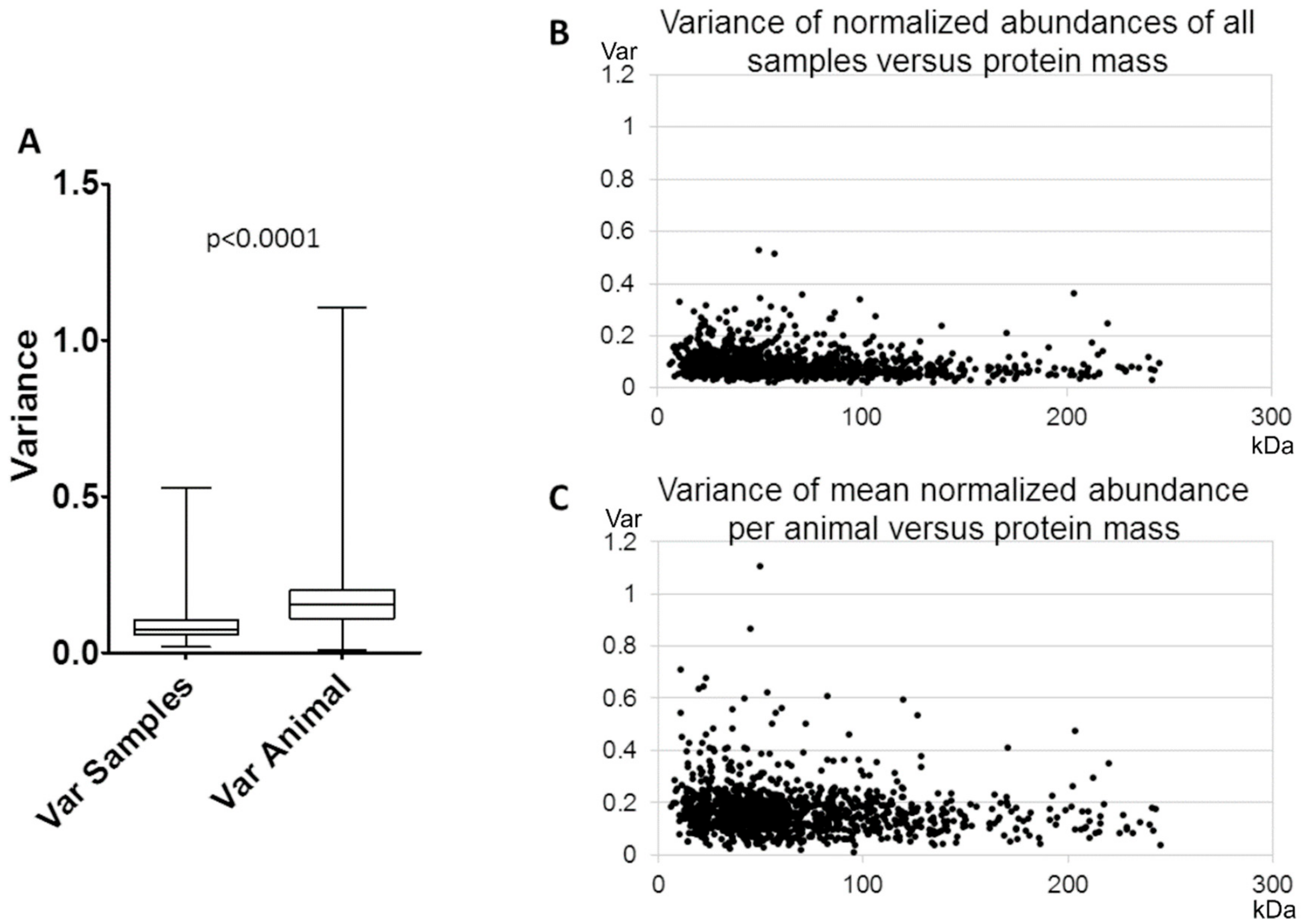
3.1. Reproducibility of Plasma Membrane Enrichment Protocol
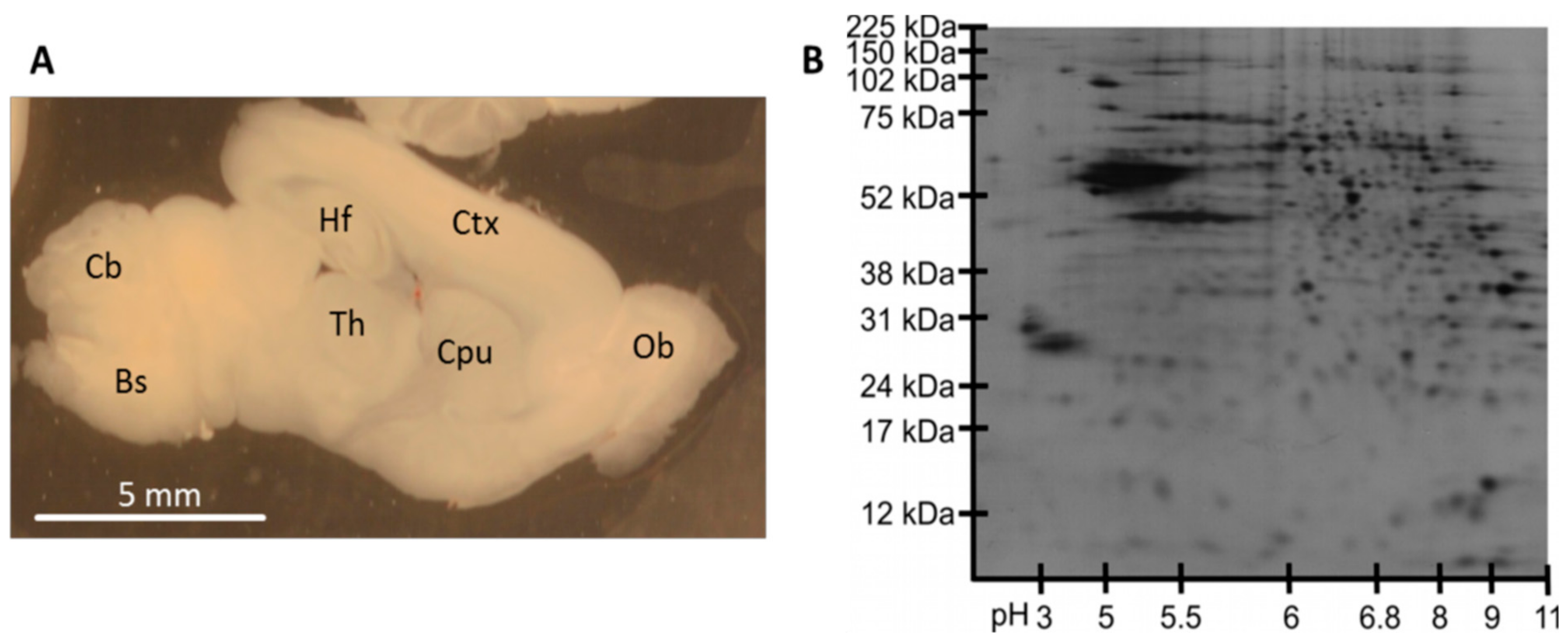
3.2. Protein Analysis in Single Acute Slices
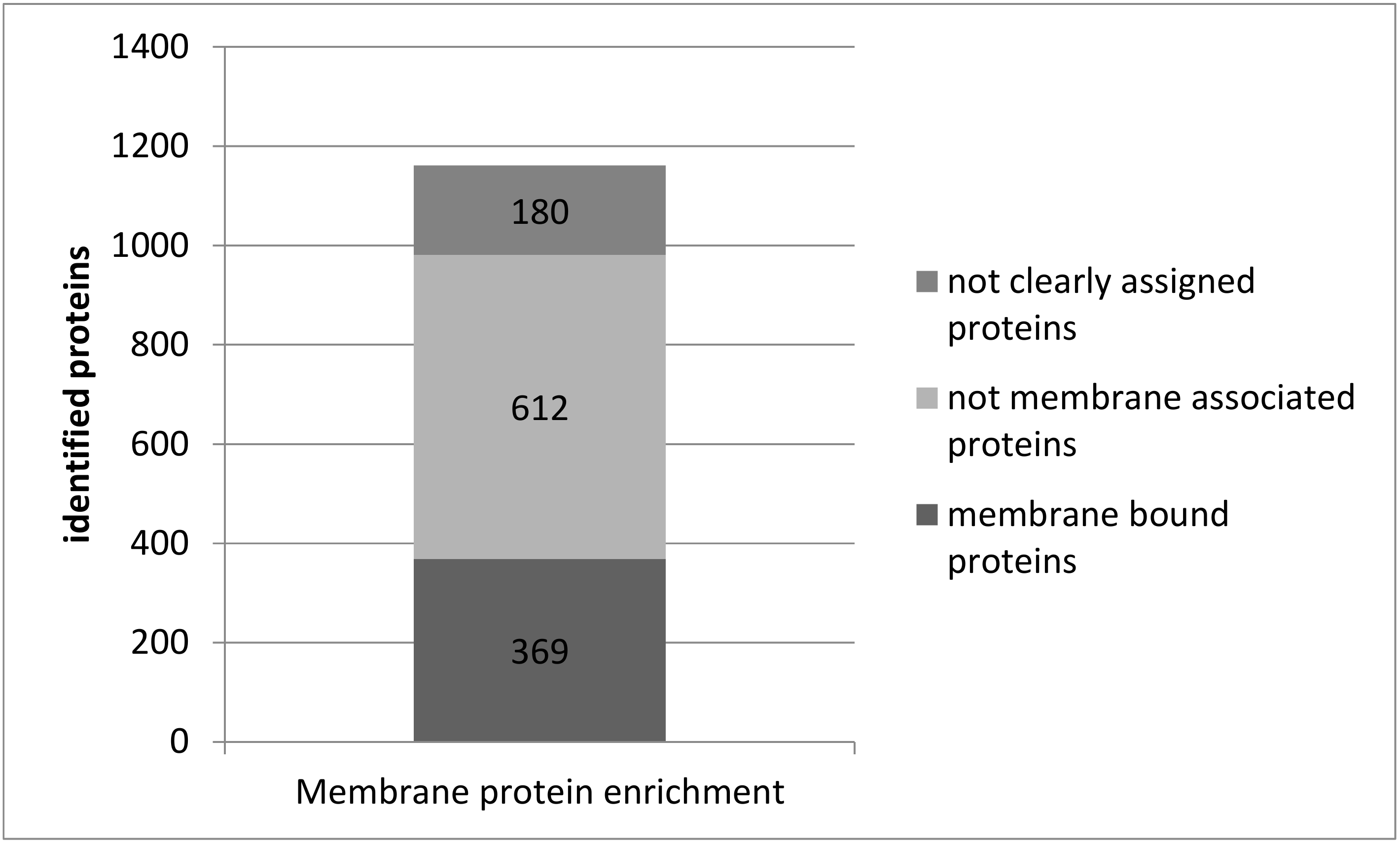
3.3. Plasma Membrane Protein Separation in Single Acute Slices
4. Discussion
4.1. Reproducibility of the Plasma Membrane Enrichment Protocol
4.2. Protein Analysis in Single Acute Slices
4.3. Membrane Proteins in Single Acute Slices
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lossi, L.; Merighi, A. The Use of ex Vivo Rodent Platforms in Neuroscience Translational Research with Attention to the 3Rs Philosophy. Front. Vet. Sci. 2018, 5, 164. [Google Scholar] [CrossRef] [PubMed]
- Humpel, C. Organotypic brain slice cultures: A review. Neuroscience 2015, 305, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Phillips, W.S.; Herly, M.; Del Negro, C.A.; Rekling, J.C. Organotypic slice cultures containing the preBötzinger complex generate respiratory-like rhythms. J. Neurophysiol. 2016, 115, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Joost, S.; Kobayashi, K.; Wree, A.; Haas, S.J.-P. Optimisation of murine organotypic slice culture preparation for a novel sagittal-frontal co-culture system. J. Neurosci. Methods 2017. [Google Scholar] [CrossRef]
- Bowling, H.; Bhattacharya, A.; Zhang, G.; Lebowitz, J.Z.; Alam, D.; Smith, P.T.; Kirshenbaum, K.; Neubert, T.A.; Vogel, C.; Chao, M.V.; et al. BONLAC: A combinatorial proteomic technique to measure stimulus-induced translational profiles in brain slices. Neuropharmacology 2016, 100, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Smolders, K.; Lombaert, N.; Valkenborg, D.; Baggerman, G.; Arckens, L. An effective plasma membrane proteomics approach for small tissue samples. Sci. Rep. 2015, 5, 10917. [Google Scholar] [CrossRef] [PubMed]
- Qiao, R.; Li, S.; Zhou, M.; Chen, P.; Liu, Z.; Tang, M.; Zhou, J. In-depth analysis of the synaptic plasma membrane proteome of small hippocampal slices using an integrated approach. Neuroscience 2017, 353, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Grant, K.J.; Wu, C.C. Advances in neuromembrane proteomics: Efforts towards a comprehensive analysis of membrane proteins in the brain. Brief. Funct. Genom. Proteom. 2007, 6, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Volknandt, W.; Karas, M. Proteomic analysis of the presynaptic active zone. Exp. Brain Res. 2012, 217, 449–461. [Google Scholar] [CrossRef]
- Vuckovic, D.; Dagley, L.F.; Purcell, A.W.; Emili, A. Membrane proteomics by high performance liquid chromatography-tandem mass spectrometry: Analytical approaches and challenges. Proteomics 2013, 13, 404–423. [Google Scholar] [CrossRef] [PubMed]
- Weekes, M.P.; Antrobus, R.; Lill, J.R.; Duncan, L.M.; Hör, S.; Lehner, P.J. Comparative Analysis of Techniques to Purify Plasma Membrane Proteins. J. Biomol. Tech. 2010, 21, 108–115. [Google Scholar] [PubMed]
- Hörmann, K.; Stukalov, A.; Müller, A.C.; Heinz, L.X.; Superti-Furga, G.; Colinge, J.; Bennett, K.L. A Surface Biotinylation Strategy for Reproducible Plasma Membrane Protein Purification and Tracking of Genetic and Drug-Induced Alterations. J. Proteome Res. 2016, 15, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Schindler, J.; Lewandrowski, U.; Sickmann, A.; Friauf, E. Aqueous polymer two-phase systems for the proteomic analysis of plasma membranes from minute brain samples. J. Proteome Res. 2008, 7, 432–442. [Google Scholar] [CrossRef]
- Schindler, J.; Nothwang, H.G. Aqueous polymer two-phase systems: Effective tools for plasma membrane proteomics. Proteomics 2006, 6, 5409–5417. [Google Scholar] [CrossRef] [PubMed]
- Wille, M.; Schumann, A.; Wree, A.; Kreutzer, M.; Glocker, M.O.; Mutzbauer, G.; Schmitt, O. The Proteome Profiles of the Cerebellum of Juvenile, Adult and Aged Rats—An Ontogenetic Study. Int. J. Mol. Sci. 2015, 16, 21454–21485. [Google Scholar] [CrossRef]
- Wille, M.; Schumann, A.; Kreutzer, M.; Glocker, M.O.; Wree, A.; Mutzbauer, G.; Schmitt, O. The proteome profiles of the olfactory bulb of juvenile, adult and aged rats—An ontogenetic study. Proteome Sci. 2015, 13, 8. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Pappesch, R.; Warnke, P.; Mikkat, S.; Normann, J.; Wisniewska-Kucper, A.; Huschka, F.; Wittmann, M.; Khani, A.; Schwengers, O.; Oehmcke-Hecht, S.; et al. The Regulatory Small RNA MarS Supports Virulence of Streptococcus pyogenes. Sci. Rep. 2017, 7, 12241. [Google Scholar] [CrossRef]
- Distler, U.; Kuharev, J.; Navarro, P.; Levin, Y.; Schild, H.; Tenzer, S. Drift time-specific collision energies enable deep-coverage data-independent acquisition proteomics. Nat. Methods 2014, 11, 167–170. [Google Scholar] [CrossRef]
- Silva, J.C.; Gorenstein, M.V.; Li, G.-Z.; Vissers, J.P.C.; Geromanos, S.J. Absolute quantification of proteins by LCMSE: A virtue of parallel MS acquisition. Mol. Cell Proteom. 2006, 5, 144–156. [Google Scholar] [CrossRef]
- Jahn, O.; Tenzer, S.; Werner, H.B. Myelin proteomics: Molecular anatomy of an insulating sheath. Mol. Neurobiol. 2009, 40, 55–72. [Google Scholar] [CrossRef]
- Lenz, M.; Galanis, C.; Kleidonas, D.; Fellenz, M.; Deller, T.; Vlachos, A. Denervated mouse dentate granule cells adjust their excitatory but not inhibitory synapses following in vitro entorhinal cortex lesion. Exp. Neurol. 2019, 312, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.A.; Furber, K.L.; Thangaraj, M.P.; Sobchishin, L.; Doucette, J.R.; Nazarali, A.J. Organotypic Cultures from the Adult CNS: A Novel Model to Study Demyelination and Remyelination Ex Vivo. Cell Mol. Neurobiol. 2018, 38, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Birgbauer, E.; Rao, T.S.; Webb, M. Lysolecithin induces demyelination in vitro in a cerebellar slice culture system. J. Neurosci. Res. 2004, 78, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Llufriu-Dabén, G.; Carrete, A.; Chierto, E.; Mailleux, J.; Camand, E.; Simon, A.; Vanmierlo, T.; Rose, C.; Allinquant, B.; Hendriks, J.J.A.; et al. Targeting demyelination via α-secretases promoting sAPPα release to enhance remyelination in central nervous system. Neurobiol. Dis. 2018, 109, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Nyamoya, S.; Leopold, P.; Becker, B.; Beyer, C.; Hustadt, F.; Schmitz, C.; Michel, A.; Kipp, M. G-Protein-Coupled Receptor Gpr17 Expression in Two Multiple Sclerosis Remyelination Models. Mol. Neurobiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kipp, M.; Nyamoya, S.; Hochstrasser, T.; Amor, S. Multiple sclerosis animal models: A clinical and histopathological perspective. Brain Pathol. 2017, 27, 123–137. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession | TMH | Schindler et al. | Our Analysis | ||
|---|---|---|---|---|---|
| Neurotransmitter Release | pep | A | B | ||
| P61765 | Syntaxin-binding protein 1 | 0 | 30 | 43 | 43 |
| Q9WU70 | Syntaxin-binding protein 5 | 1 | 1 | 4 | 4 |
| P32851 | Syntaxin-1A | 1 | 6 | 4 | 4 |
| P61265 | Syntaxin-1B2 | 1 | 21 | 17 | 16 |
| P60881 | Synaptosomal-associated protein 25 | 0 | 19 | 19 | 19 |
| Neurotransmitter Receptors | |||||
| P19490 | Glutamate receptor 1 | 5 | 3 | 9 | 9 |
| Q63226 | Glutamate receptor delta-2 subunit | 3 | 6 | 28 | 29 |
| P23385 | Metabotropic glutamate receptor 1 | 8 | 6 | 23 | 17 |
| Q9Z0U4 | Gamma-aminobutyric acid type B receptor, subunit 1 | 8 | 3 | 8 | 6 |
| O88871 | Gamma-aminobutyric acid type B receptor, subunit 2 | 8 | 2 | 10 | 10 |
| P62813 | Gamma-aminobutyric-acid receptor alpha-1 subunit | 5 | 5 | 4 | 4 |
| P30191 | Gamma-aminobutyric-acid receptor alpha-6 subunit | 4 | 0 | 8 | 7 |
| P63138 | Gamma-aminobutyric-acid receptor subunit beta-2 | 5 | 5 | 3 | 3 |
| P18506 | Gamma-aminobutyric-acid receptor delta subunit | 5 | 2 | 3 | 3 |
| P18508 | Gamma-aminobutyric-acid receptor gamma-2 subunit | 5 | 3 | 0 | 1 |
| Neurotransmitter Reuptake | |||||
| P31662 | Orphan sodium- and chloride-dependent neurotransmitter transporter NTT4 | 11 | 12 | 5 | 5 |
| P23978 | Sodium- and chloride-dependent GABA transporter 1 | 12 | 5 | 5 | 7 |
| P31647 | Sodium- and chloride-dependent GABA transporter 3 | 11 | 8 | 6 | 5 |
| P28572 | Sodium- and chloride-dependent glycine transporter 1 | 12 | 4 | 4 | 4 |
| P24942 | Excitatory amino acid transporter 1 | 10 | 14 | 8 | 7 |
| P31596 | Excitatory amino acid transporter 2 | 11 | 12 | 9 | 7 |
| O35921 | Excitatory amino acid transporter 4 | 8 | 7 | 7 | 6 |
| Ion Channels | |||||
| Q9Z2L0 | Voltage-dependent anion-selective channel protein 1 | 0 | 9 | 12 | 11 |
| P10499 | Potassium voltage-gated channel subfamily A member 1 | 6 | 4 | 1 | 1 |
| P25122 | Potassium voltage-gated channel subfamily C member 1 | 7 | 4 | 3 | 3 |
| P04775 | Sodium channel protein type 2 subunit alpha | 24 | 8 | 3 | 3 |
| Q00954 | Sodium channel beta-1 subunit | 2 | 1 | 2 | 2 |
| P54900 | Sodium channel beta-2 subunit | 2 | 5 | 4 | 5 |
| Transporters | |||||
| Q9JHZ9 | System N amino acid transporter 1 | 10 | 2 | 0 | 1 |
| P11167 | Solute carrier family 2, facilitated glucose transporter member 1 | 11 | 3 | 3 | 2 |
| Q8VII6 | Choline transporter-like protein 1 | 10 | 1 | 1 | 0 |
| Q63016 | Large neutral amino acids transporter small subunit 1 | 14 | 3 | 5 | 5 |
| Q63633 | Solute carrier family 12 member 5 | 12 | 12 | 17 | 15 |
| P11505 | Plasma membrane calcium-transporting ATPase 1 | 9 | 23 | 13 | 19 |
| P11506 | Plasma membrane calcium-transporting ATPase 2 | 9 | 36 | 2 | 3 |
| Q64542 | Plasma membrane calcium-transporting ATPase 4 | 11 | 13 | 10 | 9 |
| P06685 | Sodium/potassium-transporting ATPase alpha-1 chain | 8 | 42 | 29 | 28 |
| P06686 | Sodium/potassium-transporting ATPase alpha-2 chain | 8 | 48 | 28 | 28 |
| P06687 | Sodium/potassium-transporting ATPase alpha-3 chain | 8 | 50 | 32 | 32 |
| P07340 | Sodium/potassium-transporting ATPase subunit beta-1 | 1 | 14 | 15 | 15 |
| P13638 | Sodium/potassium-transporting ATPase subunit beta-2 | 1 | 10 | 8 | 9 |
| Q63377 | Sodium/potassium-transporting ATPase subunit beta-3 | 1 | 5 | 7 | 7 |
| P53987 | Monocarboxylate transporter 1 | 12 | 2 | 4 | 5 |
| Q01728 | Sodium/calcium exchanger 1 | 11 | 3 | 4 | 4 |
| P48768 | Sodium/calcium exchanger 2 | 11 | 11 | 14 | 15 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joost, S.; Mikkat, S.; Wille, M.; Schümann, A.; Schmitt, O. Membrane Protein Identification in Rodent Brain Tissue Samples and Acute Brain Slices. Cells 2019, 8, 423. https://doi.org/10.3390/cells8050423
Joost S, Mikkat S, Wille M, Schümann A, Schmitt O. Membrane Protein Identification in Rodent Brain Tissue Samples and Acute Brain Slices. Cells. 2019; 8(5):423. https://doi.org/10.3390/cells8050423
Chicago/Turabian StyleJoost, Sarah, Stefan Mikkat, Michael Wille, Antje Schümann, and Oliver Schmitt. 2019. "Membrane Protein Identification in Rodent Brain Tissue Samples and Acute Brain Slices" Cells 8, no. 5: 423. https://doi.org/10.3390/cells8050423
APA StyleJoost, S., Mikkat, S., Wille, M., Schümann, A., & Schmitt, O. (2019). Membrane Protein Identification in Rodent Brain Tissue Samples and Acute Brain Slices. Cells, 8(5), 423. https://doi.org/10.3390/cells8050423