1. Introduction
Molecular targeted therapy is one of the most important paradigm shifts in the history of cancer therapy. Traditional anticancer chemotherapeutic agents block cell division and DNA replication, and reduce the size of tumors. Although chemotherapeutic agents lead to an extension of patients’ overall survival, they are not effective for all types of cancer and induce side effects. Recently, molecular targeted drugs have been developed that interfere with specific molecules to block cancer growth, progression, and metastasis [
1,
2,
3]. Many molecular targeted drugs have demonstrated remarkable clinical success in treating myriad types of cancer, including breast, leukemia, colorectal, lung, and ovarian cancer. In addition, targeting the immune system, which accelerates anti-tumor activity through immune checkpoint inhibition, is proving to be an increasingly effective method for treating various cancers, prolonging life, and increasing progression-free survival [
1,
2,
3]. However, molecular targeted approaches continue to be limited by wide variations in the degree and durability of patient responses and side effects, and numerous cancers remain completely refractory to such therapy. Thus, molecular targeted therapy needs further improvement for greater clinical efficacy.
Historically, human cancer cell lines have been widely used for studies as preclinical models to evaluate anticancer agents. However, these models may not reflect the characteristics of the source tumor tissues in vivo, as they are frequently passaged for long periods of time, which may lead to alterations in their genome sequences, gene-expression profiles, and morphologies. In addition, almost all cell lines are cultured under monolayer conditions or used as xenografts in mice, which is not physically representative of tumor tissues [
4,
5]. Therefore, the results of evaluations performed with cancer cell lines do not accurate predict the clinical effects of anticancer drugs. Indeed, ~85% of preclinical agents entering oncology clinical trials fail to demonstrate sufficient safety or efficacy required to gain regulatory approval [
6,
7,
8]. In vitro systems, including patient-derived tumor cell, organoid, or spheroid models that accurately recapitulate tissue architecture and function, have been developed for various types of tumor tissues (e.g., colon, lung, pancreatic, prostate, endometrial, liver, bladder, breast, brain, kidney, endometrium, and stomach), as have high-throughput assay systems for using these systems [
9,
10,
11,
12,
13,
14,
15,
16,
17,
18,
19,
20]. These models are promising in terms of facilitating a better understanding of cancer biology and for evaluating drug efficacy in vitro.
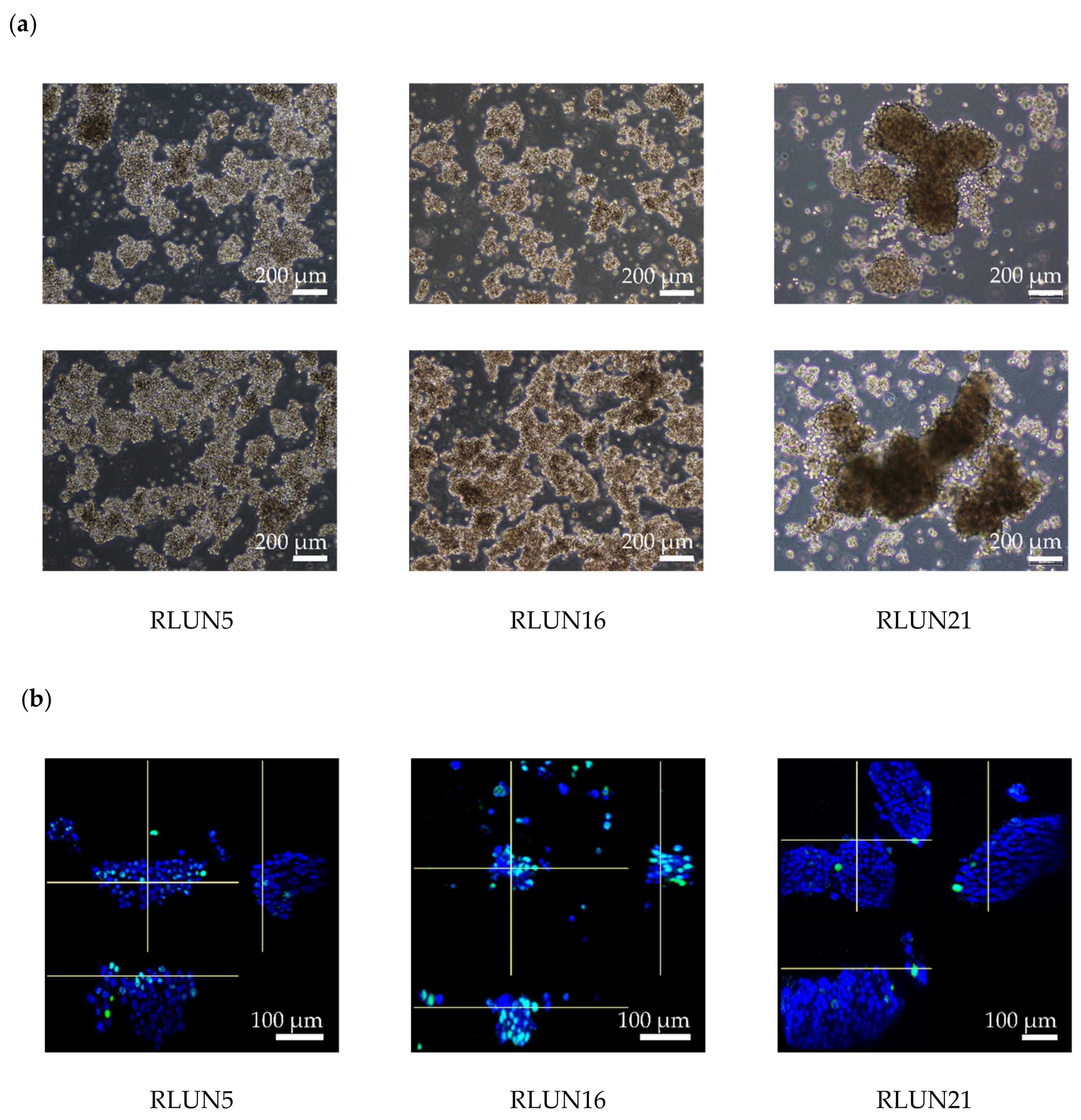
Previously, we established a novel series of patient-derived tumor organoids (PDOs) from various types of tumor tissues from the Fukushima Translational Research Project, which are designated as Fukushima (F)-PDOs. F-PDOs could be cultured for >6 months and formed cell clusters with similar morphologies to their source tumors [
21]. Comparative histological and comprehensive gene-expression analyses also demonstrated that the characteristics of PDOs were similar to those of their source tumors, even following long-term expansion in culture. In addition, suitable high-throughput assay systems were constructed for each F-PDO in 96- and 384-well plate formats. We suggest that assay systems based on F-PDOs may be utilized to evaluate anticancer agents under conditions that better reflect clinical conditions (compared with conventional methods using cancer cell lines) and to discover markers of the pharmacological effects of anticancer agents.
Although several cell-based assay systems using cancer cells have been developed for evaluating molecular targeted drugs, more efficient and simple cell-based assay systems for identifying clinically efficacious therapy potency are desired. To address this issue, we have attempted to construct efficient cell-based assays for evaluating molecular targeted drugs including small molecules, monoclonal antibodies, and immune-checkpoint inhibitors using F-PDOs, which maintain the characteristics of their source tumors. In this study, epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2) inhibitors, including small molecules, monoclonal antibodies, and antibody-drug conjugates (ADCs) in clinical use, were evaluated using lung F-PDOs. EGFR is a tyrosine kinase receptor, and its activation triggers the activation several downstream pathways including the RAS/mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinase (PI3K)/Akt, and Janus kinase (JAK)/signal transducer and activator of transcription protein (STAT) pathways that regulate cell proliferation, survival, adhesion, migration, and differentiation [
22,
23,
24,
25]. EGFR overexpression and EGFR-mediated signaling-pathway dysregulation have been observed in tumors from patients with several cancers, especially non-small cell lung cancer. Thus, several anti-EGFR drugs have been developed for clinical use in cancer therapy, including the small-molecule tyrosine kinase inhibitors (TKIs) gefitinib, erlotinib, afatinib, neratinib, dacomitinib, and osimertinib, as well as the monoclonal antibodies cetuximab and necitumumab [
22,
23,
24,
25]. In addition, HER2 can activate the same key signaling pathways as EGFR. Thus, HER2 is an ideal target for anticancer agents, and several HER2 inhibitors including trastuzumab and pertuzumab (monoclonal antibodies); afatinib, lapatinib, and neratinib (TKIs); and trastuzumab emtansine (an ADC) have been developed and approved for clinical use [
24,
26]. Therefore, we first evaluated the functional potency of several EGFR and HER2 inhibitors by high-throughput screening (HTS) using three lung F-PDOs (RLUN5, RLUN16, and RLUN21). Moreover, the antibody-dependent cellular cytotoxicity (ADCC) activity of trastuzumab was evaluated using three lung F-PDOs, and the complex interactions of immune cells with F-PDOs during ADCC responses to trastuzumab were visualized.
In addition, a system for evaluating immune-checkpoint inhibitors using lung F-PDOs was developed, based on non-invasive, label-free, and real-time cellular impedance monitoring technology (xCELLigence) [
27] to measure the potencies of biologics. Recently, Cerignoli et al. developed the real-time, impedance-monitoring system to assess immune-checkpoint inhibitors using prostate cancer PC3 cells, in combination with an anti- programmed cell death-1 (PD-1) antibody and peripheral blood mononuclear cells (PBMCs) [
27]. Therefore, we utilized the xCELLigence platform to investigate whether F-PDOs are also suitable for assessing the effects of checkpoint inhibitors on the immune system. Interactions between PD-1 expressed on the surface of activated T cells and its ligand PD-L1 on cancer cells inhibit the ability of T cells to attack the target cells. Immune-checkpoint inhibitors remove inhibitory signals of T-cell activation, which enables tumor-reactive T cells to overcome regulatory mechanisms and mount an effective antitumor response; thus, they are now used clinically for treating a broad range of tumor types [
28,
29]. We selected the anti-PD-1 monoclonal antibodies, nivolumab and pembrolizumab, as representative immune-checkpoint inhibitors in this study.
2. Materials and Methods
2.1. Antibodies, Compounds, and Reagents
Trastuzumab, pertuzumab, and trastuzumab emtansine were purchased from Roche (Basel, Switzerland). Cetuximab and nivolumab were obtained from Bristol-Myers Squibb (New York, NY, USA). Pembrolizumab was provided by Merck & Co. (Kenilworth, NJ, USA). An anti-Ki-67 antibody (ab16667) was purchased from Abcam (Cambridge, UK).
Seventy-eight anticancer agents were tested in this study (
Table S1). All compounds were dissolved in dimethyl sulfoxide at a concentration of 20 mM and stored at −80 °C until use. The purity and integrity of the compounds were measured via ultra-performance liquid chromatography-mass spectrometry (Waters Corporation, Milford, MA, USA), using a 1-µL injection volume, as follows. A Waters CORTECS C
18 column (particle size: 1.6 µm; column size: 2.1 × 50 mm; Waters Corporation) was developed with a linear aqueous acetonitrile (MeCN) gradient containing a 0.1% formic acid (5–90% MeCN, 1.6 min; flow rate, 1 mL/min), separation was performed at 40 °C, and the components of the major ultraviolet (UV) adsorption peaks were verified by mass spectrometry (
Table S1).
Epidermal growth factor (EGF) and interferon-γ (IFN-γ) were obtained from Fujifilm Wako Pure Chemical, Ltd. (Osaka, Japan). Staphylococcal enterotoxin B (SEB) was obtained from Sigma-Aldrich (St. Louis, MO, USA).
2.2. Cells
All experiments with human material were performed in accordance with the guidelines of the Declaration of Helsinki and were approved in advance by the ethics committee of the Fukushima Medical University (approval number: 1953; approval date: 21 October 2018). Clinical specimens used for the PDOs were acquired from cancer patients at Fukushima Medical University Hospital after providing informed consent. Previously, we reported the establishment of 53 F-PDOs from human tumor tissues [
21]. Out of those PDOs, three lung F-PDOs (RLUN5, RLUN16, and RLUN21) were selected for use in this study. Briefly, RLUN5, RLUN16, and RLUN21 were each established from lung cancer tissues removed from the lung by surgery. As described previously [
21], the cancer tissues were washed with Hank’s balanced salt solution (Fujifilm Wako Pure Chemical, Ltd.) supplemented with penicillin-streptomycin solution (Fujifilm Wako Pure Chemical, Ltd.), cut with a scalpel into small pieces (approximately 1 mm
3), and cultured in suspension for 3–6 months to establish the F-PDOs.
THP-1 cells were obtained from Japanese Collection of Research Bioresources Cell Bank (Osaka, Japan). PBMCs were provided by Precision Bioservices (Frederick, MD, USA).
2.3. Cell Culture
F-PDOs were cultured at 37 °C in 15 mL FBIM001 [
21] using ultra-low attachment 75-cm
2 flasks (Corning, Inc., NY, USA) in a humidified incubator with 5% CO
2. Because accurate cell numbers of F-PDOs could not be determined using a cell counter, the volumes of cell pellets after centrifuging cell suspensions in 15-mL tubes were visually estimated by comparison with tubes marked at volumes of 75, 100, and 150 µL. The 80% medium was changed twice weekly. When F-PDOs reached their maximum saturation density, the cells were passaged at a 1:2 ratio. The procedure for handling F-PDOs was described in detail elsewhere [
21]. F-PDOs were cultured in flasks until sufficient cell-pellet volumes were obtained for the assay. For all experiments, the F-PDO culture media were replaced at 1 day prior to seeding, as previously described [
21].
THP-1 cells were cultured in vendor-recommended growth media supplemented with fetal bovine serum (FSA; Sigma-Aldrich, St. Louis, MO, USA) and penicillin-streptomycin solution (Fujifilm Wako Pure Chemical, Ltd.) at final concentrations of 10 and 1%, respectively, at 37 °C in a humidified incubator with 5% CO2. Cell numbers and viabilities were automatically measured using trypan blue dye exclusion with a Vi-Cell XR Cell Viability Analyzer (Beckman Coulter, Inc., Brea, CA, USA), according to the manufacturer’s protocol.
2.4. Real-Time PCR
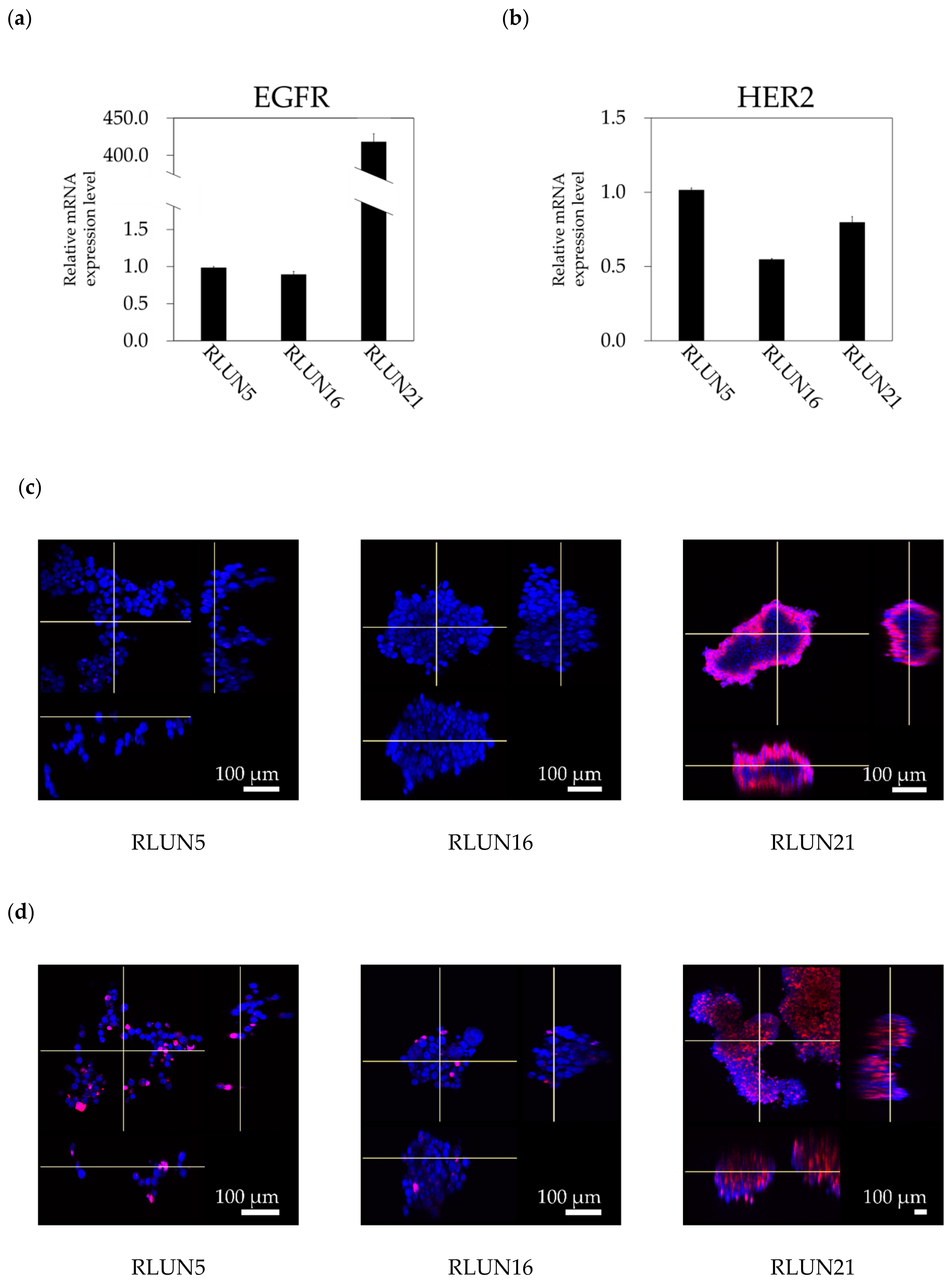
Relative levels of mRNA were quantified with the StepOnePlus Real-Time Polymerase Chain Reaction System (Thermo Fisher Scientific, Waltham, MA, USA) using TaqMan Fast Virus 1-Step Master Mix (Thermo Fisher Scientific) and TaqMan Gene Expression Assays (assay #Hs01076090_m1 for EGFR, assay #Hs01001580_m1 for HER2, and assay #Hs99999901_s1 for the 18S ribosomal gene as the internal control; Thermo Fisher Scientific) according to the manufacturer’s protocols.
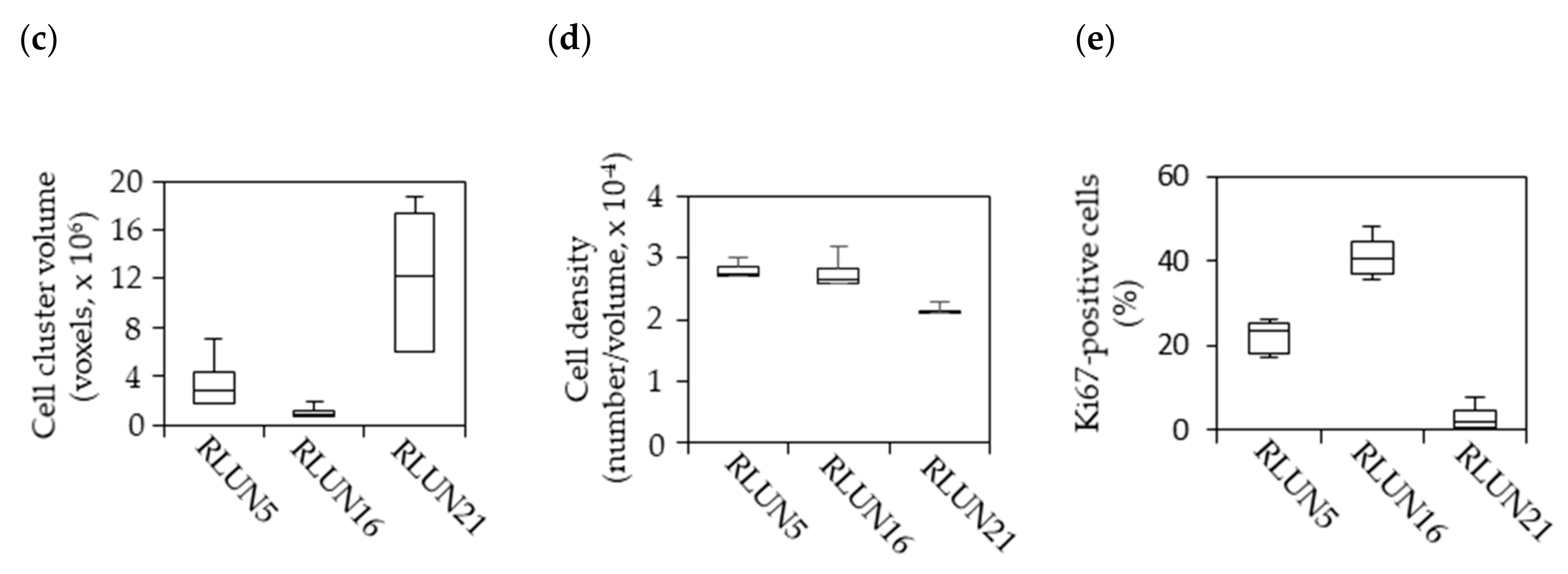
2.5. Three-Dimensional Cell Analysis
Images were captured using the FLUOVIEW FV3000 Confocal Laser Scanning Microscope (Olympus, Tokyo, Japan). All imaging data were analyzed with the NoviSight 3D Cell Analysis System (Olympus). Immunofluorescence staining of F-PDOs was performed using an anti-Ki-67 antibody (1:250 dilution) and 4′,6-diamidino-2-phenylindole (DAPI) after fixation in phosphate buffer solution containing 4% paraformaldehyde (PFA; Fujifilm Wako Pure Chemical, Ltd.) and 1% Triton X-100 (Fujifilm Wako Pure Chemical, Ltd.). The F-PDOs were then incubated for 5 h at room temperature in AbScale solution, comprised of Dulbecco’s phosphate-buffered saline (D-PBS (–); Fujifilm Wako Pure Chemical, Ltd.) containing 0.33 M urea and 0.1–0.5% Triton X-100 [
30], to wash the cells. Next, the F-PDOs were incubated for 30 min at room temperature in AbScale rinse solution, comprised of 0.1× D-PBS (–) containing 2.5% BSA and 0.05% (
w/v) Tween-20 [
30]. The rinse solution was exchanged with 4% PFA, and the F-PDOs were incubated at room temperature for 30 min.
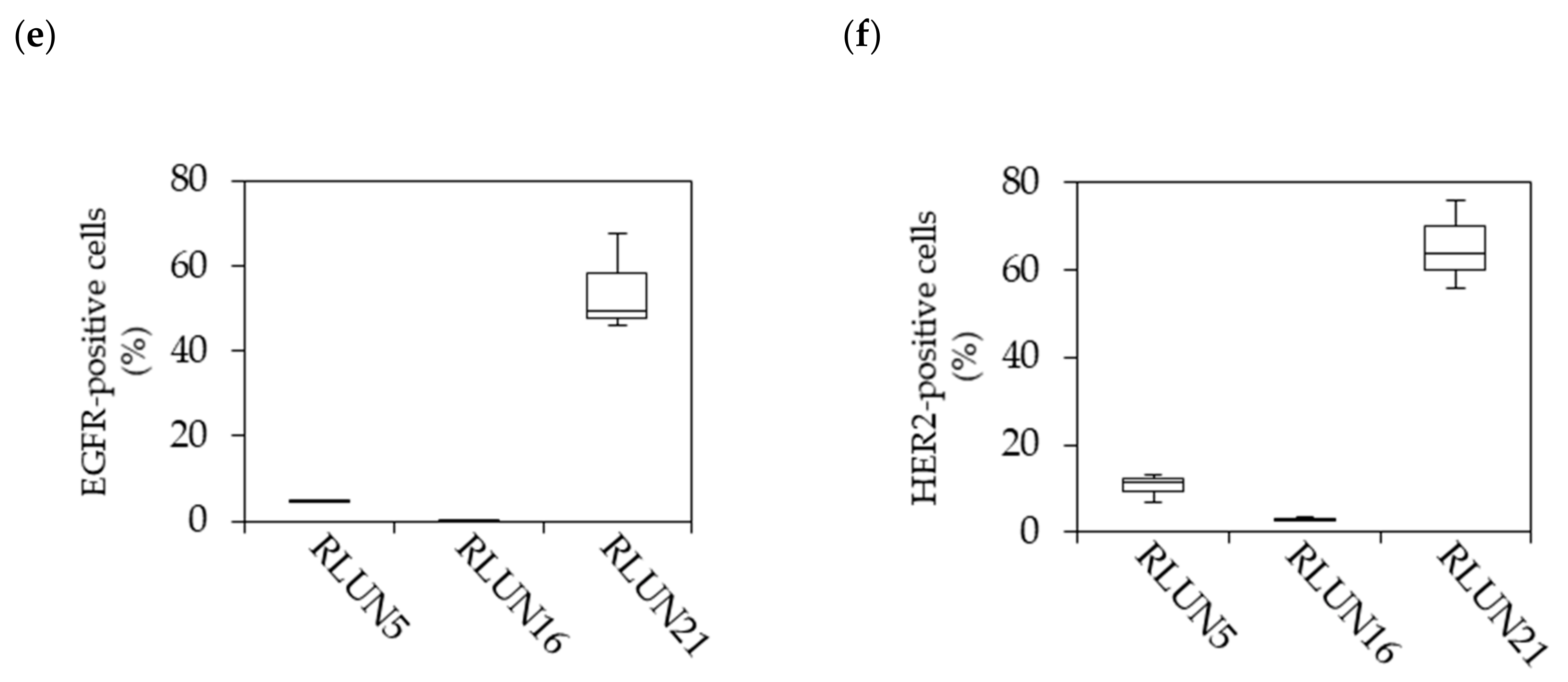
Cetuximab and trastuzumab were labeled using the HiLyte Fluor 555 Labeling Kit-NH2 (Dojindo Laboratories, Kumamoto, Japan) and used to quantify the cellular expression levels of EGFR and HER2. In detail, F-PDOs were incubated for 3 h with 10 µg/mL fluorophore-labeled antibodies in 24-well, flat-bottomed, ultra-low-attachment microplates (Corning, Inc.). Cells were then washed with D-PBS (–) and fixed overnight at 4 °C in 4% PFA. Subsequently, the F-PDOs were washed twice with D-PBS (–) and imbedded with 7.5% acrylamide gel. The F-PDOs were washed with D-PBS (–) and stained overnight at 4 °C with DAPI. Finally, the F-PDOs were incubated overnight at room temperature in SCALEVIEW-S4 (Fujifilm Wako Pure Chemical, Ltd.).
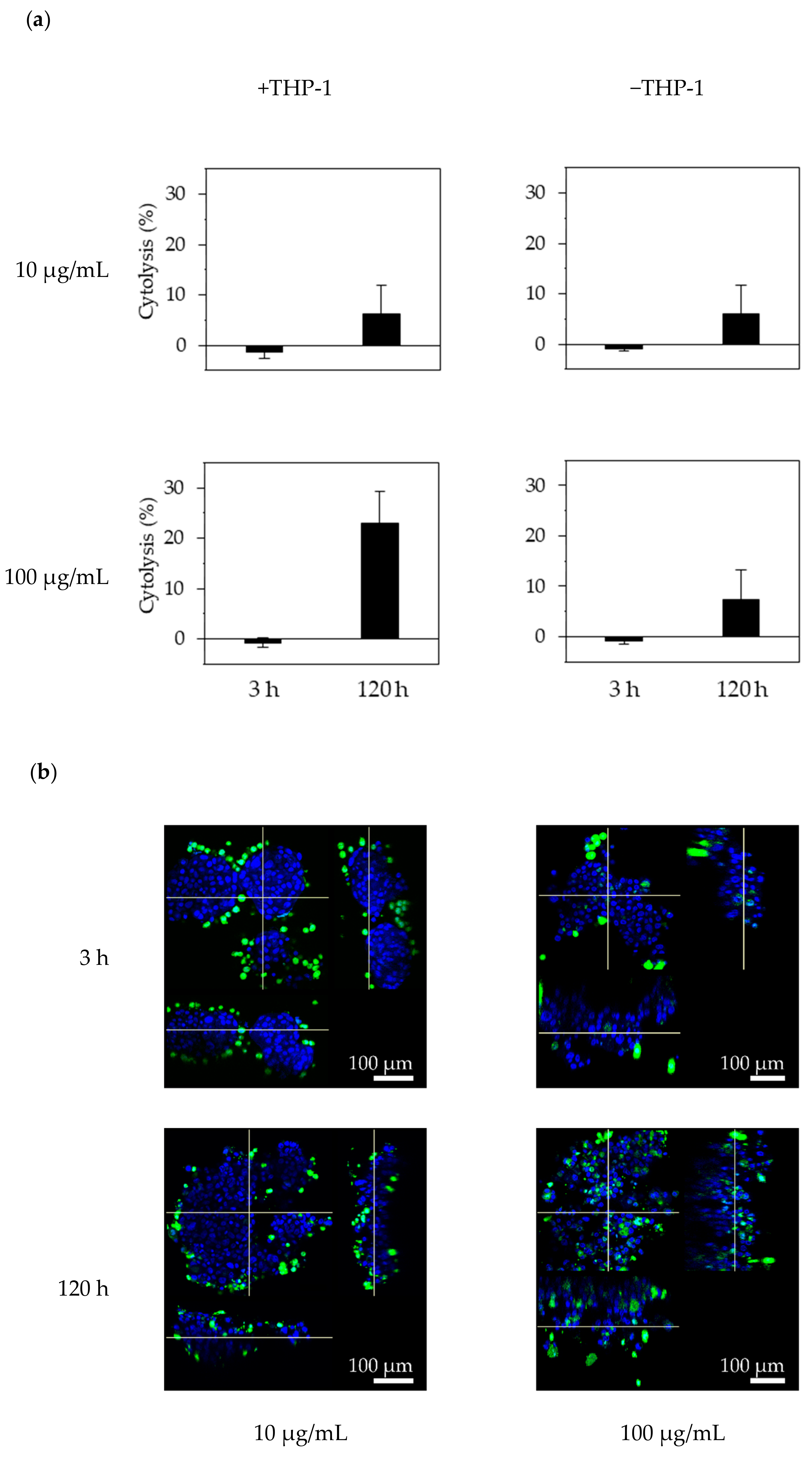
To determine whether the effector cells could access the F-PDOs via trastuzumab, F-PDOs were treated with antibodies and then co-cultured with THP-1 effector cells that were pre-stimulated with IFN-γ. Twenty-four hours before treating F-PDOs with antibodies, IFN-γ (10 ng/mL) was added to the THP-1 cells, and the F-PDOs were seeded in 24-well plates, as described above. On the following day, the F-PDOs were treated with 10 or 100 µg/mL of HiLyte Fluor 555-labeled antibodies for 30 min. Next, the THP-1 cells were rinsed with medium to wash away the IFN-γ and stained using the Cell Explore Fixable Live Cell Tracking Kit Green Fluorescence (AAT Bioquest, Inc., Sunnyvale, CA, USA). The fluorescently labelled THP-1 cells were added to the F-PDO target cells at a density of 1 × 105 cells per well and co-cultured for 3 or 120 h. The cells were fixed overnight with 4% PFA at 4 °C, washed three times with D-PBS (–), imbedded with 7.5% acrylamide gel, and stained with DAPI in D-PBS (–) containing 0.1% Triton X-100. Finally, the F-PDOs were incubated overnight at room temperature in SCALEVIEW-S4.
2.6. Cell-Viability Assay
The cell viability of F-PDOs was assayed using a previously reported method [
21]. Briefly, each F-PDO was minced using a CellPet FT (JTEC Corporation, Osaka, Japan) equipped with a filter holder containing a 70- or 100-µm mesh filter. Each F-PDO suspension was diluted 20-fold and seeded into 384-well, round-bottomed, ultra-low-attachment microplates (Corning, Inc.) in 40 μL medium, using a Multidrop Combi Reagent Dispenser (Thermo Fisher Scientific, Inc.). At 24 h after seeding, the F-PDOs were treated with 0.04-μL solutions of different compounds using an Echo 555 Liquid Handler (Labcyte, Inc., San Jose, CA, USA) or 0.8-μL solutions of different antibodies using an ADS-348-8 Multistage-Dispense Station (Biotec Co., Ltd., Tokyo, Japan). The cells were treated with compounds or antibodies at final concentrations ranging from 1.0 nM to 20 µM or 0.195 to 100 µg/mL, respectively, using a series of 10 concentrations (serially diluted 3-fold) in each case. After 144 h, 10 μL CellTiter-Glo 3D solution (Promega Corporation, Madison, WI, USA) was added to F-PDOs in each well, and the plates were mixed using a mixer and incubated for 15 min at 25°C. Luminescence was measured using an EnSpire Plate Reader (PerkinElmer, Inc., Waltham, MA, USA). Cell viability was calculated by dividing the amount of ATP in the test wells by that in the vehicle-control wells, after subtracting the background levels. The growth rate over 6 days was calculated by dividing the amount of ATP in the wells without anticancer agents by those in the vehicle-control wells 24 h after seeding.
The half-maximal inhibitory concentration (IC
50) and area under the activity curve measuring dose response (AUC) values (used to measure dose–response relationships) were calculated from the dose–response curves and analyzed using Morphit software, version 6.0 (The Edge Software Consultancy, Ltd., Guildford, UK). As the first approach, the response curves were fitted to the luminescence signal intensities using a 4-parameter sigmoid model. Alternatively, a sigmoidal fixed-slope model without a Hill equation was used. The data shown represent the mean ± standard deviation of triplicate experiments. The Z′ factor, a dimensionless parameter that ranges between 1 (infinite separation) and < 0, was defined as Z′ = 1 – (3σc
+ + 3σc
–)/|µc
+ – µc
–|, where σc
+, σc
−, µc
+, and µc
− are the standard deviations (σ) and averages (µ) of the high (c
+) and low (c
–) controls [
31].
2.7. Lactate Dehydrogenase (LDH) Assay
One day before cell seeding, 96-well microplates (Sumitomo Bakelite Co., Ltd., Tokyo, Japan) were coated with 50 µL fibronectin (50 µg/mL), and THP-1 cells were stimulated by adding 10 ng/mL IFN-γ. Each F-PDO was minced using the CellPet FT. Each F-PDO suspension was diluted 10-fold, and a 100-μL volume of each suspension was seeded into the pre-coated plates. At 24 h after seeding the F-PDOs, 60 µL of growth medium was removed and 10 µL of antibody solution was added at a final concentration of 10 or 100 µg/mL. THP-1 effector cells were added to each F-PDO at 2 × 104 cells/well 30 min after adding the antibody. Each well contained a final volume of 100 µL. After 3 or 120 h, 25 μL of supernatant was recovered, mixed with 25 μL of reagent solution from the Cytotoxicity Detection KitPLUS (Roche Diagnostics, Rotkreuz, Switzerland), and incubated for 15 min. Stop solution (12.5 μL) was added to the reaction mixture and mixed using a mixer for 10 s. Absorbance was measured at 490 and 690 nm using an EnSpire Plate Reader. Cytotoxicity was calculated by dividing the LDH activity in the test wells by that in the vehicle-control wells, after subtracting the background absorbance levels, expressed as a percentage.
2.8. Real-Time Potency Assessment Using the xCELLigence RCTA System
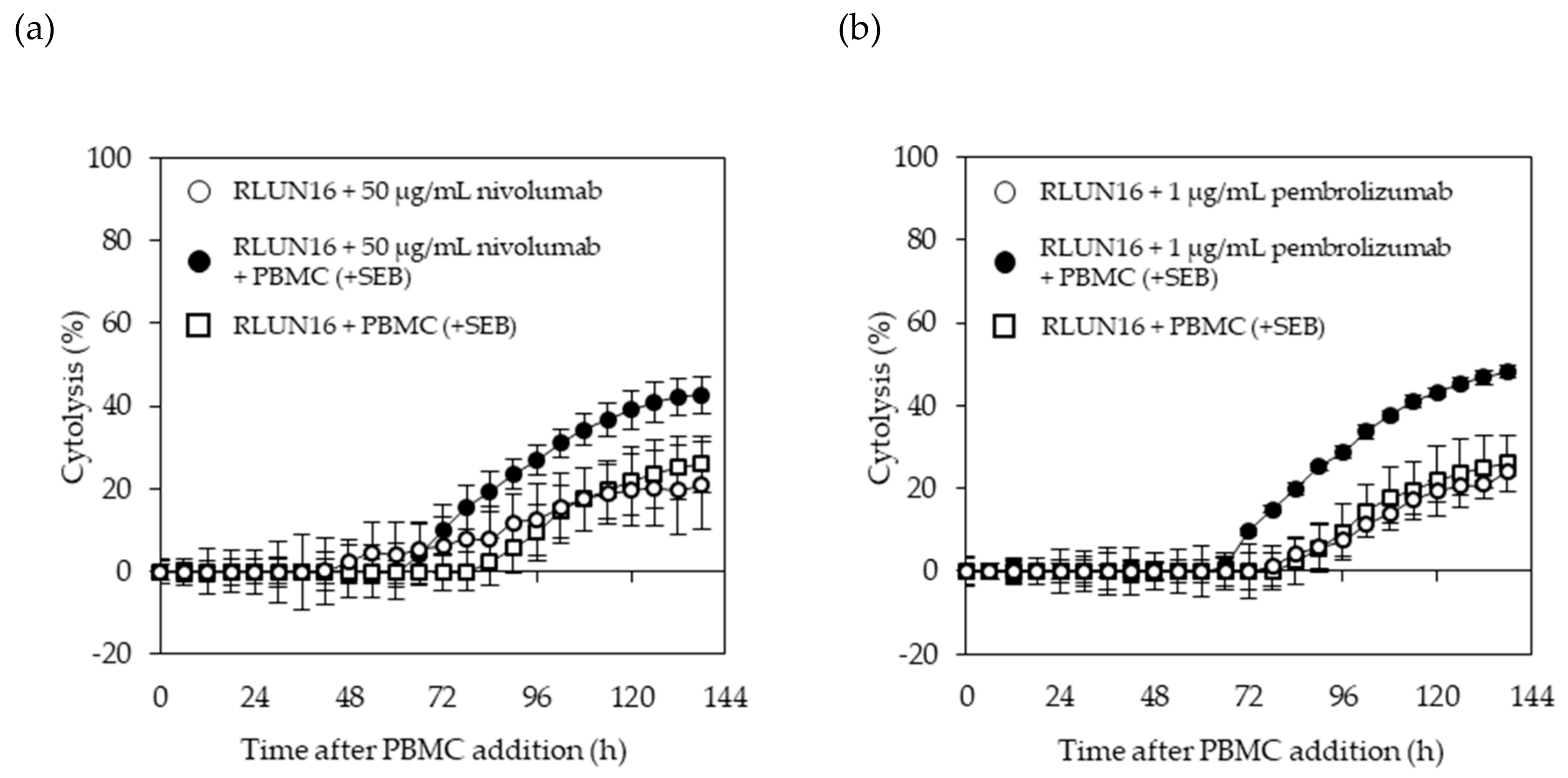
The xCELLigence RTCA System (ACEA Bioscience, San Diego, CA, USA) was used for evaluating the immune-checkpoint inhibitors. The E-plate 96 (ACEA Bioscience), which is a single-use 96-well plate specialized for performing cell-based assays with the xCELLigence RTCA System, was coated overnight with fibronectin (0.5 µg/well) at 4 °C. After removing the fibronectin, 50 µL of culturing medium was added to each well of the E-plate to measure the background impedance. Then, each F-PDO was minced using the CellPet FT. The F-PDO suspension was diluted 15-fold and 50 µL of each cell suspension was seeded into the wells. The plate was placed in a safety cabinet at room temperature for 30 min and transferred to the xCELLigence RTCA instrument at 37 °C in a CO2 incubator. At 24 h after seeding the F-PDOs into the E-plate, 60 µL of culture media was removed from each well, and 10 µL of antibody solution was added. PBMC were stimulated with 5 ng/mL SEB for 24 h and then added to F-PDOs 30 min post-antibody treatment. PBMCs were added at 1 × 104 cells per well to each F-PDO. Each well contained a final volume of 100 µL. The impedance was measured every 15 min. Changes in impedance signals were measured as the cell index and then converted to percent-cytolysis values using xCELLigence immunotherapy software (ACEA Bioscience). “Percent cytolysis” refers to the percentage of target cells that were killed by effector cells, checkpoint inhibitors, or both when compared to RLUN16 cells alone (as a control). The cell indexes of wells containing PBMCs alone was subtracted from the cell indexes of the sample wells, for each time point. Next, each value was normalized to the cell index at the time just before antibody addition. The normalized cell index was converted to % cytolysis using xCELLigence immunotherapy software according to the following equation: % cytolysis = (1 – normalized cell index [sample wells])/normalized cell index (target alone wells) × 100.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}