3D-Organotypic Cultures to Unravel Molecular and Cellular Abnormalities in Atopic Dermatitis and Ichthyosis Vulgaris


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. 3D Cultures to Study Cellular and Molecular Abnormalities in Ichthyosis Vulgaris and Beyond
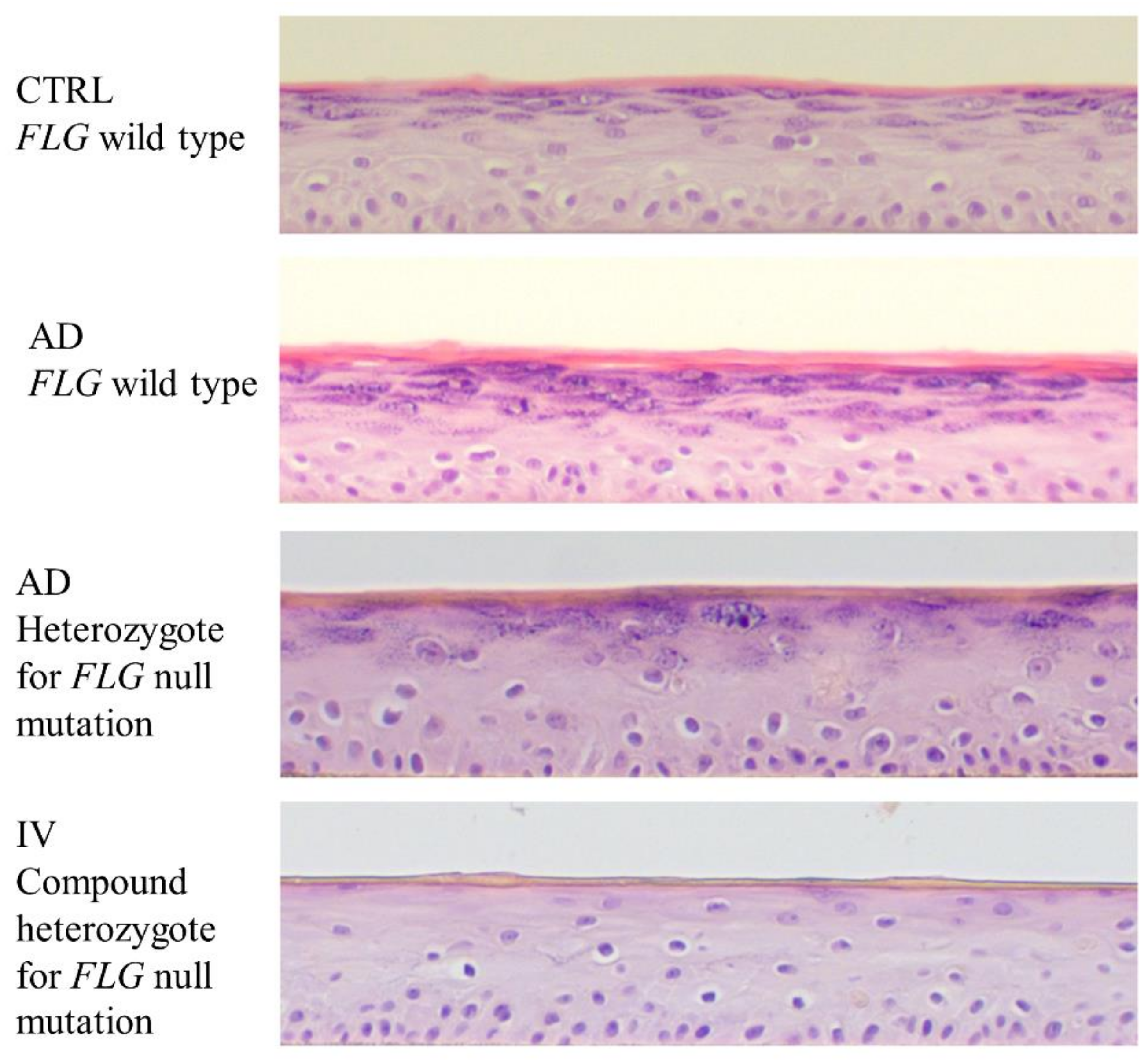
2.1. Absence of Keratohyalin Granules and Subtle Alterations in Lipid Composition in FLG-Deficient 3D Cultures
2.2. The Efficacy of FLG Knockdown in 3D Cultures Correlates with SC Barrier Fitness
2.3. Higher Porousness of FLG-Deficient 3D Cultures to Staphylococcus aureus
2.4. FLG Deficiency Has Contrasting Effects on Tight Junctions and KC Differentiation in 3D Cultures
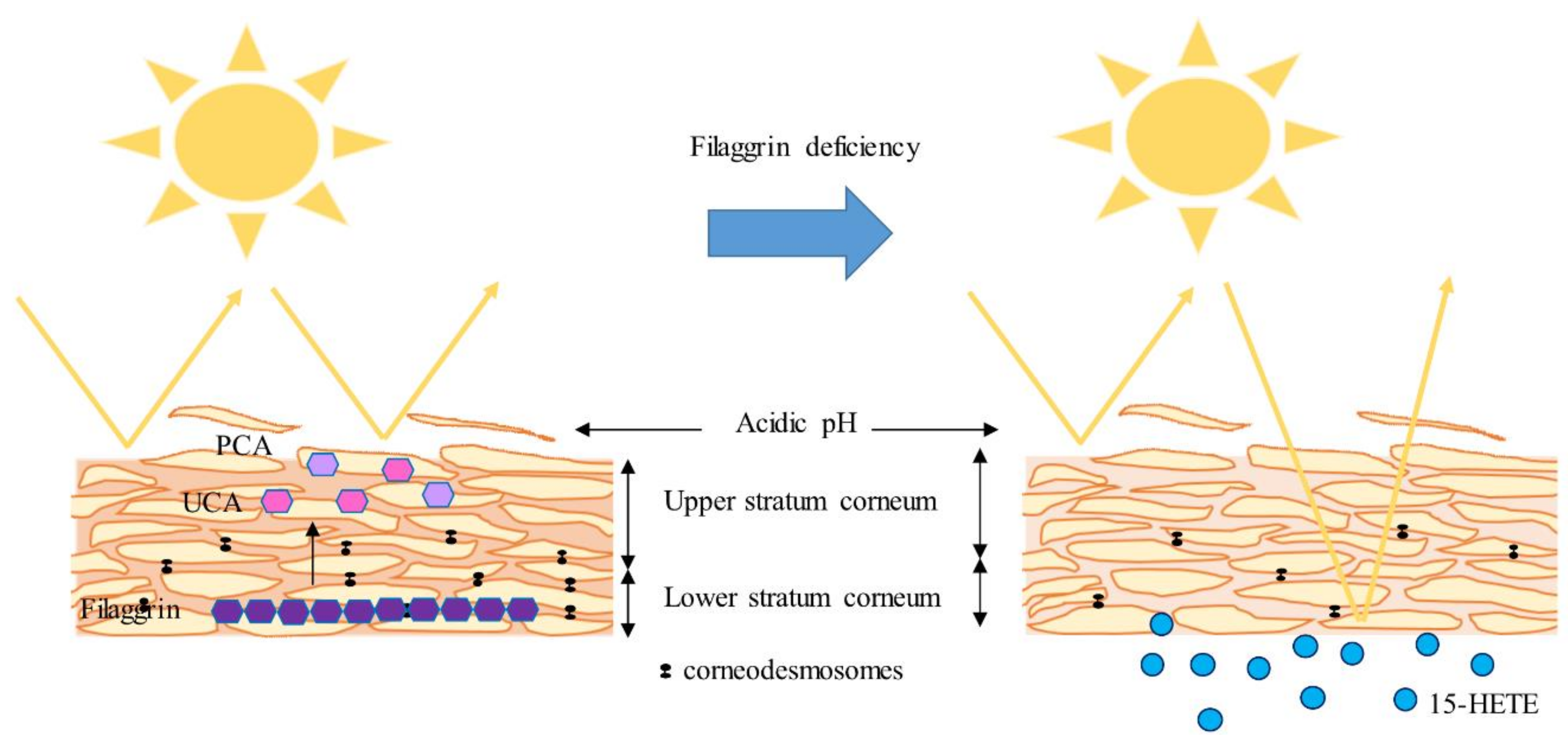
2.5. FLG-Deficient 3D Cultures: Reduced NMFs but Increased 15-HETE
2.6. Increased Secretion of TSLP in FLG-Deficient 3D Cultures
2.7. ARCI 3D Cultures to Test Enzyme Replacement Therapy
3. 3D Cultures Helped Unravel New Pathogenic Pathways in Atopic Dermatitis
3.1. Alarmins Affect the Proliferation/Differentiation Balance in Keratinocytes but do not Trigger Inflammation in 3D Cultures
3.2. JAK Inhibitors Restore FLG and Dampen Th2 Mediators in 3D AD Cultures
3.3. Elucidation of the Role of Microtubules and the F-Actin Cytoskeleton in AD by Using 3D Cultures
3.4. Histamine Induces Cellular Abnormalities in 3D Cultures Resembling Defects in AD
3.5. Coal Tar Alleviates Cellular Abnormalities in AD 3D Cultures
3.6. Toward More Complex 3D AD Cultures
3.7. New Pathogenic Pathways Related to Eicosanoids Illuminated with 3D AD Cultures
Funding
Conflicts of Interest
Abbreviations
| AA | arachidonic acid |
| AD | atopic dermatitis |
| AHR | aryl hydrocarbon receptor |
| AMP | antimicrobial peptide |
| ARCI | autosomal recessive congenital ichthyosis |
| CE | cornified envelope |
| CER(EOS) | ester-linked omega-hydroxy sphingosine |
| CER(EOdS) | ester-linked omega-hydroxy dihydrosphingosine |
| CGRP | calcitonin gene-related peptide |
| CLDN | claudin |
| DHA | docosahexaenoic acid |
| DPA | docosapentaenoic acid |
| EGF | epidermal growth factor |
| EPA | eicosapentaenoic acid |
| ERK | extracellular signal-regulated kinases |
| FFA | free fatty acid |
| FLG | filaggrin |
| HBD | human β-defensin |
| HEE | human epidermal equivalent |
| HMGB1 | high mobility group protein B1 |
| HPETE | hydroperoxyeicosatetraenoic acid |
| HRNR | hornerin |
| HSE | human skin equivalent |
| HIF-1α | hypoxia-inducible factor-1α |
| iPSC | pluripotent stem cell |
| IV | ichthyosis vulgaris |
| IVL | involucrin |
| IL | interleukin |
| JAK | janus kinase |
| JNK | c-Jun N-terminal kinases |
| KC | keratinocyte |
| KRT | keratin |
| LB | lamellar body |
| LCE | late cornified envelope |
| LOR | loricrin |
| LOX | lipoxygenase |
| LY | lucifer yellow |
| Myo5b | actin-based motor myosin Vb |
| NF-κB | nuclear factor ’kappa-light-chain-enhancer’ of activated B-cells |
| NHE-1 | sodium-hydrogen antiporter 1 |
| NMF | natural moisturizing factor |
| NRF2 | nuclear factor (erythroid-derived 2)-like 2 |
| OCLN | occludin |
| PCA | pyrrolidone carboxylic acid |
| PUFA | polyunsaturated fatty acid |
| SC | stratum corneum |
| SG | stratum granulosum |
| shRNA | short hairpin RNA |
| siRNA | small interfering RNA |
| SP | substance P |
| sPLA2 | secretory phospholipase A2 |
| TCDD | tetrachloro-dibenzo-dioxin |
| TEER | transepithelial electrical resistance |
| TEWL | transepidermal water loss |
| TGM | transglutaminase |
| TLR | toll-like receptor |
| TSLP | thymic stromal lymphopoietin |
| UCA | trans-urocanic acid |
| UV | ultraviolet |
| ZO-1 | zonula occludens-1 |
References
- Shahwan, K.T.; Kimball, A.B. Itch intensity in moderate-to-severe plaque psoriasis versus atopic dermatitis: A meta-analysis. J. Am. Acad. Dermatol. 2017, 76, 1198–1200.e1. [Google Scholar] [CrossRef] [PubMed]
- Feldman, S.; Behnam, S.M.; Behnam, S.E.; Koo, J.Y. Involving the patient: impact of inflammatory skin disease and patient-focused care. J. Am. Acad. Dermatol. 2005, 53, S78–S85. [Google Scholar] [CrossRef]
- Kelleher, M.; Dunn-Galvin, A.; Hourihane, J.O.; Murray, D.; Campbell, L.E.; McLean, W.H.; Irvine, A.D. Skin barrier dysfunction measured by transepidermal water loss at 2 days and 2 months predates and predicts atopic dermatitis at 1 year. J. Allergy Clin. Immunol. 2015, 135, 930–935.e1. [Google Scholar] [CrossRef]
- Hoffjan, S.; Stemmler, S. On the role of the epidermal differentiation complex in ichthyosis vulgaris, atopic dermatitis and psoriasis. Br. J. Dermatol. 2007, 157, 441–449. [Google Scholar] [CrossRef]
- Marenholz, I.; Esparza-Gordillo, J.; Rüschendorf, F.; Bauerfeind, A.; Strachan, D.P.; Spycher, B.D.; Baurecht, H.; Margaritte-Jeannin, P.; Sääf, A.; Kerkhof, M.; et al. Meta-analysis identifies seven susceptibility loci involved in the atopic march. Nat. Commun. 2015, 6, 8804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, D.Y.; Guttman-Yassky, E. Deciphering the complexities of atopic dermatitis: Shifting paradigms in treatment approaches. J. Allergy Clin. Immunol. 2014, 134, 769–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, J.N.; Palmer, C.N.; Zhao, Y.; Liao, H.; Hull, P.R.; Lee, S.P.; Allen, M.H.; Meggitt, S.J.; Reynolds, N.J.; Trembath, R.C.; et al. Null mutations in the filaggrin gene (FLG) determine major susceptibility to early-onset atopic dermatitis that persists into adulthood. J. Investig. Dermatol. 2007, 127, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Weidinger, S.; Rodríguez, E.; Stahl, C.; Wagenpfeil, S.; Klopp, N.; Illig, T.; Novak, N. Filaggrin mutations strongly predispose to early-onset and extrinsic atopic dermatitis. J. Investig. Dermatol. 2007, 127, 724–726. [Google Scholar] [CrossRef]
- Esaki, H.; Brunner, P.M.; Renert-Yuval, Y.; Czarnowicki, T.; Huynh, T.; Tran, G.; Lyon, S.; Rodriguez, G.; Immaneni, S.; Johnson, D.B.; et al. Early-onset pediatric atopic dermatitis is TH2 but also TH17 polarized in skin. J. Allergy Clin. Immunol. 2016, 138, 1639–1651. [Google Scholar] [CrossRef]
- Karimkhani, C.; Silverberg, J.I.; Dellavalle, R.P. Defining intrinsic vs. extrinsic atopic dermatitis. Dermatol. Online J. 2015, 21. [Google Scholar]
- Moyle, M.; Cevikbas, F.; Harden, J.L.; Guttman-Yassky, E. Understanding the immune landscape in atopic dermatitis: The era of biologics and emerging therapeutic approaches. Exp. Dermatol. 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- Moosbrugger-Martinz, V.; Schmuth, M.; Dubrac, S. A Mouse Model for Atopic Dermatitis Using Topical Application of Vitamin D3 or of Its Analog MC903. Methods Mol. Biol. 2017, 1559, 91–106. [Google Scholar] [PubMed]
- Byrd, A.L.; Belkaid, Y.J.A.S. The human skin microbiome. Nat. Rev. Microbiol. 2018, 16, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.H.; Oh, J.; Deming, C.; Conlan, S.; Grice, E.A.; Beatson, M.A.; Nomicos, E.; Polley, E.C.; Komarow, H.D.; NISC Comparative Sequence Program; et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. 2012, 22, 850–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrd, A.L.; Deming, C.; Cassidy, S.K.B.; Harrison, O.J.; Ng, W.I.; Conlan, S.; NISC Comparative Sequencing Program; Belkaid, Y.; Segre, J.A.; Kong, H.H. Staphylococcus aureus and Staphylococcus epidermidis strain diversity underlying pediatric atopic dermatitis. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Thyssen, J.P.; Godoy-Gijon, E.; Elias, P.M. Ichthyosis vulgaris: the filaggrin mutation disease. Br. J. Dermatol. 2013, 168, 1155–1166. [Google Scholar] [CrossRef]
- Patel, N.; Spencer, L.A.; English, J.C., 3rd; Zirwas, M.J. Acquired ichthyosis. J. Am. Acad. Dermatol. 2006, 55, 647–656. [Google Scholar] [CrossRef]
- Schmuth, M.; Gruber, R.; Elias, P.M.; Williams, M.L. Ichthyosis update: towards a function-driven model of pathogenesis of the disorders of cornification and the role of corneocyte proteins in these disorders. Adv. Dermatol. 2007, 23, 231–256. [Google Scholar] [CrossRef]
- Gruber, R.; Elias, P.M.; Crumrine, D.; Lin, T.K.; Brandner, J.M.; Hachem, J.P.; Presland, R.B.; Fleckman, P.; Janecke, A.R.; Sandilands, A.; et al. Filaggrin genotype in ichthyosis vulgaris predicts abnormalities in epidermal structure and function. Am. J. Pathol. 2011, 178, 2252–2263. [Google Scholar] [CrossRef]
- Sybert, V.P.; Dale, B.A.; Holbrook, K.A. Ichthyosis vulgaris: identification of a defect in synthesis of filaggrin correlated with an absence of keratohyaline granules. J. Investig. Dermatol. 1985, 84, 191–194. [Google Scholar] [CrossRef]
- Fleckman, P.; Holbrook, K.A.; Dale, B.A.; Sybert, V.P. Keratinocytes cultured from subjects with ichthyosis vulgaris are phenotypically abnormal. J. Investig. Dermatol. 1987, 88, 640–645. [Google Scholar] [CrossRef]
- Hoetzenecker, W.; Schanz, S.; Schaller, M.; Fierlbeck, G. Generalized tinea corporis due to Trichophyton rubrum in ichthyosis vulgaris. J. Eur. Acad. Dermatol. Venereol. 2007, 21, 1129–1131. [Google Scholar] [CrossRef]
- Agostini, G.; Geti, V.; Difonzo, E.M.; Giannotti, B. Dermatophyte infection in ichthyosis vulgaris. Mycoses 1992, 35, 197–199. [Google Scholar] [CrossRef]
- Blunder, S.; Kõks, S.; Kõks, G.; Reimann, E.; Hackl, H.; Gruber, R.; Moosbrugger-Martinz, V.; Schmuth, M.; Dubrac, S. Enhanced Expression of Genes Related to Xenobiotic Metabolism in the Skin of Patients with Atopic Dermatitis but Not with Ichthyosis Vulgaris. J. Investig. Dermatol. 2018, 138, 98–108. [Google Scholar] [CrossRef] [Green Version]
- Ransohoff, R.M. All (animal) models (of neurodegeneration) are wrong. Are they also useful? J. Exp. Med. 2018, 215, 2955–2958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the Protection of Animals Used for Scientific Purposes Text with EEA Relevance. Available online: https://eur-lex.europa.eu/eli/dir/2010/63/oj (accessed on 21 May 2019).
- Engelhart, K.; El Hindi, T.; Biesalski, H.K.; Pfitzner, I. In vitro reproduction of clinical hallmarks of eczematous dermatitis in organotypic skin models. Arch. Dermatol. Res. 2005, 297, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bechetoille, N.; Vachon, H.; Gaydon, A.; Boher, A.; Fontaine, T.; Schaeffer, E.; Decossas, M.; André-Frei, V.; Mueller, C.G. A new organotypic model containing dermal-type macrophages. Exp. Dermatol. 2011, 20, 1035–1037. [Google Scholar] [CrossRef] [PubMed]
- van den Bogaard, E.H.; Tjabringa, G.S.; Joosten, I.; Vonk-Bergers, M.; van Rijssen, E.; Tijssen, H.J.; Erkens, M.; Schalkwijk, J.; Koenen, H.J.P.M. Crosstalk between keratinocytes and T cells in a 3D microenvironment: A model to study inflammatory skin diseases. J. Investig. Dermatol. 2014, 134, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Bechetoille, N.; Dezutter-Dambuyant, C.; Damour, O.; André, V.; Orly, I.; Perrier, E. Effects of solar ultraviolet radiation on engineered human skin equivalent containing both Langerhans cells and dermal dendritic cells. Tissue Eng. 2007, 13, 2667–2679. [Google Scholar] [CrossRef]
- Schuster, C.; Mildner, M.; Mairhofer, M.; Bauer, W.; Fiala, C.; Prior, M.; Eppel, W.; Kolbus, A.; Tschachler, E.; Stingl, G.; et al. Human embryonic epidermis contains a diverse Langerhans cell precursor pool. Development 2014, 141, 807–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, N.; Hosseini, M.; Vainio, S.; Taïeb, A.; Cario-André, M.; Rezvani, H.R. Skin equivalents: Skin from reconstructions as models to study skin development and diseases. Br. J. Dermatol. 2015, 173, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Niehues, H.; Bouwstra, J.A.; El Ghalbzouri, A.; Brandner, J.M.; Zeeuwen, P.L.J.M.; van den Bogaard, E.H. 3D skin models for 3R research: The potential of 3D reconstructed skin models to study skin barrier function. Exp. Dermatol. 2018, 27, 501–511. [Google Scholar] [CrossRef] [Green Version]
- Smits, J.P.H.; Niehues, H.; Rikken, G.; van Vlijmen-Willems, I.M.J.J.; van de Zande, G.W.H.J.F.; Zeeuwen, P.L.J.M.; Schalkwijk, J.; van den Bogaard, E.H. Immortalized N/TERT keratinocytes as an alternative cell source in 3D human epidermal models. Sci. Rep. 2017, 7, 11838. [Google Scholar] [CrossRef]
- Mathes, S.H.; Ruffner, H.; Graf-Hausner, U. The use of skin models in drug development. Adv. Drug Deliv. Rev. 2014, 69–70, 81–102. [Google Scholar] [CrossRef] [PubMed]
- Laco, F.; Kun, M.; Weber, H.J.; Ramakrishna, S.; Chan, C.K. The dose effect of human bone marrow-derived mesenchymal stem cells on epidermal development in organotypic co-culture. J. Dermatol. Sci. 2009, 55, 150–160. [Google Scholar] [CrossRef]
- van de Kamp, J.; Kramann, R.; Anraths, J.; Schöler, H.R.; Ko, K.; Knüchel, R.; Zenke, M.; Neuss, S.; Schneider, R.K. Epithelial morphogenesis of germline-derived pluripotent stem cells on organotypic skin equivalents in vitro. Differentiation 2012, 83, 138–147. [Google Scholar] [CrossRef]
- Petrova, A.; Capalbo, A.; Jacquet, L.; Hazelwood-Smith, S.; Dafou, D.; Hobbs, C.; Arno, M.; Farcomeni, A.; Devito, L.; Badraiq, H.; et al. Induced Pluripotent Stem Cell Differentiation and Three-Dimensional Tissue Formation Attenuate Clonal Epigenetic Differences in Trichohyalin. Stem Cells Dev. 2016, 25, 1366–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrova, A.; Celli, A.; Jacquet, L.; Dafou, D.; Crumrine, D.; Hupe, M.; Arno, M.; Hobbs, C.; Cvoro, A.; Karagiannis, P.; et al. 3D In vitro model of a functional epidermal permeability barrier from human embryonic stem cells and induced pluripotent stem cells. Stem Cell Rep. 2014, 2, 675–689. [Google Scholar] [CrossRef]
- Hoeller, D.; Huppertz, B.; Roos, T.C.; Poblete Gutiérrez, P.; Merk, H.F.; Frank, J.; Jugert, F.K. An improved and rapid method to construct skin equivalents from human hair follicles and fibroblasts. Exp. Dermatol. 2001, 10, 264–271. [Google Scholar] [CrossRef]
- Wagner, T.; Gschwandtner, M.; Strajeriu, A.; Elbe-Bürger, A.; Grillari, J.; Grillari-Voglauer, R.; Greiner, G.; Golabi, B.; Tschachler, E.; Mildner, M. Establishment of keratinocyte cell lines from human hair follicles. Sci. Rep. 2018, 8, 13434. [Google Scholar] [CrossRef]
- Muller, Q.; Beaudet, M.J.; De Serres-Bérard, T.; Bellenfant, S.; Flacher, V.; Berthod, F. Development of an innervated tissue-engineered skin with human sensory neurons and Schwann cells differentiated from iPS cells. Acta Biomater. 2018, 82, 93–101. [Google Scholar] [CrossRef] [Green Version]
- McAleer, M.A.; Irvine, A.D. The multifunctional role of filaggrin in allergic skin disease. J. Allergy Clin. Immunol. 2013, 131, 280–291. [Google Scholar] [CrossRef]
- Sandilands, A.; Sutherland, C.; Irvine, A.D.; McLean, W.H. Filaggrin in the frontline: role in skin barrier function and disease. J. Cell Sci. 2009, 122, 1285–1294. [Google Scholar] [CrossRef] [Green Version]
- Miajlovic, H.; Fallon, P.G.; Irvine, A.D.; Foster, T.J. Effect of filaggrin breakdown products on growth of and protein expression by Staphylococcus aureus. J. Allergy Clin. Immunol. 2010, 126, 1184–1190.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mildner, M.; Jin, J.; Eckhart, L.; Kezic, S.; Gruber, F.; Barresi, C.; Stremnitzer, C.; Buchberger, M.; Mlitz, V.; Ballaun, C.; et al. Knockdown of filaggrin impairs diffusion barrier function and increases UV sensitivity in a human skin model. J. Investig. Dermatol. 2010, 130, 2286–2294. [Google Scholar] [CrossRef]
- Pendaries, V.; Malaisse, J.; Pellerin, L.; Le Lamer, M.; Nachat, R.; Kezic, S.; Schmitt, A.M.; Paul, C.; Poumay, Y.; Serre, G.; et al. Knockdown of filaggrin in a three-dimensional reconstructed human epidermis impairs keratinocyte differentiation. J. Investig. Dermatol. 2014, 134, 2938–2946. [Google Scholar] [CrossRef]
- Niehues, H.; Schalkwijk, J.; van Vlijmen-Willems, I.M.J.J.; Rodijk-Olthuis, D.; van Rossum, M.M.; Wladykowski, E.; Brandner, J.M.; van den Bogaard, E.H.J.; Zeeuwen, P.L.J.M. Epidermal equivalents of filaggrin null keratinocytes do not show impaired skin barrier function. J. Allergy Clin. Immunol. 2017, 139, 1979–1981.e13. [Google Scholar] [CrossRef] [Green Version]
- Blunder, S.; Rühl, R.; Moosbrugger-Martinz, V.; Krimmel, C.; Geisler, A.; Zhu, H.; Crumrine, D.; Elias, P.M.; Gruber, R.; Schmuth, M.; et al. Alterations in Epidermal Eicosanoid Metabolism Contribute to Inflammation and Impaired Late Differentiation in FLG-Mutated Atopic Dermatitis. J. Investig. Dermatol. 2017, 137, 706–715. [Google Scholar] [CrossRef]
- Vávrová, K.; Henkes, D.; Strüver, K.; Sochorová, M.; Školová, B.; Witting, M.Y.; Friess, W.; Schreml, S.; Meier, R.J.; Schäfer-Korting, M.; et al. Filaggrin deficiency leads to impaired lipid profile and altered acidification pathways in a 3D skin construct. J. Investig. Dermatol. 2014, 134, 746–753. [Google Scholar] [CrossRef]
- Wallmeyer, L.; Dietert, K.; Sochorová, M.; Gruber, A.D.; Kleuser, B.; Vávrová, K.; Hedtrich, S. TSLP is a direct trigger for T cell migration in filaggrin-deficient skin equivalents. Sci. Rep. 2017, 7, 774. [Google Scholar] [CrossRef] [PubMed]
- van Drongelen, V.; Alloul-Ramdhani, M.; Danso, M.O.; Mieremet, A.; Mulder, A.; van Smeden, J.; Bouwstra, J.A.; El Ghalbzouri, A. Knock-down of filaggrin does not affect lipid organization and composition in stratum corneum of reconstructed human skin equivalents. Exp. Dermatol. 2013, 22, 807–812. [Google Scholar] [CrossRef]
- Reynier, M.; Allart, S.; Goudounèche, D.; Moga, A.; Serre, G.; Simon, M.; Leprince, C. The Actin-Based Motor Myosin Vb is Crucial to Maintain the Epidermal Barrier Integrity. J. Investig. Dermatol. 2019, in press. [Google Scholar] [CrossRef]
- Küchler, S.; Henkes, D.; Eckl, K.M.; Ackermann, K.; Plendl, J.; Korting, H.C.; Hennies, H.C.; Schäfer-Korting, M. Hallmarks of atopic skin mimicked in vitro by means of a skin disease model based on FLG knock-down. Altern. Lab. Anim. 2011, 39, 471–480. [Google Scholar] [CrossRef]
- de Jager, M.; Groenink, W.; Bielsa i Guivernau, R.; Andersson, E.; Angelova, N.; Ponec, M.; Bouwstra, J. A novel in vitro percutaneous penetration model: evaluation of barrier properties with p-aminobenzoic acid and two of its derivatives. Pharm. Res. 2006, 23, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Borrás-Blasco, J.; Díez-Sales, O.; López, A.; Herráez-Domínguez, M. A mathematical approach to predicting the percutaneous absorption enhancing effect of sodium lauryl sulphate. Int. J. Pharm. 2004, 269, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.B. Percutaneous penetration through slightly damaged skin. Arch. Dermatol. Res. 2005, 296, 560–567. [Google Scholar] [CrossRef]
- Mansbridge, J.N.; Knapp, A.M. Penetration of lucifer yellow into human skin: A lateral diffusion channel in the stratum corneum. J. Histochem. Cytochem. 1993, 41, 909–914. [Google Scholar] [CrossRef] [PubMed]
- Hönzke, S.; Wallmeyer, L.; Ostrowski, A.; Radbruch, M.; Mundhenk, L.; Schäfer-Korting, M.; Hedtrich, S. Influence of Th2 Cytokines on the Cornified Envelope, Tight Junction Proteins, and ß-Defensins in Filaggrin-Deficient Skin Equivalents. J. Investig. Dermatol. 2016, 136, 631–639. [Google Scholar] [CrossRef]
- Wang, X.W.; Wang, J.J.; Gutowska-Owsiak, D.; Salimi, M.; Selvakumar, T.A.; Gwela, A.; Chen, L.Y.; Wang, Y.J.; Giannoulatou, E.; Ogg, G. Deficiency of filaggrin regulates endogenous cysteine protease activity, leading to impaired skin barrier function. Clin. Exp. Dermatol. 2017, 42, 622–631. [Google Scholar] [CrossRef]
- Agren, J.; Sjörs, G.; Sedin, G. Ambient humidity influences the rate of skin barrier maturation in extremely preterm infants. J. Pediatr. 2006, 148, 613–617. [Google Scholar] [CrossRef]
- Sun, R.; Celli, A.; Crumrine, D.; Hupe, M.; Adame, L.C.; Pennypacker, S.D.; Park, K.; Uchida, Y.; Feingold, K.R.; Elias, P.M.; et al. Lowered humidity produces human epidermal equivalents with enhanced barrier properties. Tissue Eng. Part. C Methods 2015, 21, 15–22. [Google Scholar] [CrossRef]
- Mieremet, A.; van Dijk, R.; Boiten, W.; Gooris, G.; Bouwstra, J.A.; El Ghalbzouri, A. Characterization of human skin equivalents developed at body’s core and surface temperatures. J. Tissue Eng. Regen. Med. 2019, in press. [Google Scholar] [CrossRef]
- Kennedy, K.; Heimall, J.; Spergel, J.M. Advances in atopic dermatitis in 2017. J. Allergy Clin. Immunol. 2018, 142, 1740–1747. [Google Scholar] [CrossRef] [Green Version]
- Meylan, P.; Lang, C.; Mermoud, S.; Johannsen, A.; Norrenberg, S.; Hohl, D.; Vial, Y.; Prod’hom, G.; Greub, G.; Kypriotou, M.; et al. Skin Colonization by Staphylococcus aureus Precedes the Clinical Diagnosis of Atopic Dermatitis in Infancy. J. Investig. Dermatol. 2017, 137, 2497–2504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paller, A.S.; Kong, H.H.; Seed, P.; Naik, S.; Scharschmidt, T.C.; Gallo, R.L.; Luger, T.; Irvine, A.D. The microbiome in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2019, 143, 26–35. [Google Scholar] [CrossRef]
- Nakatsuji, T.; Chen, T.H.; Two, A.M.; Chun, K.A.; Narala, S.; Geha, R.S.; Hata, T.R.; Gallo, R.L. Staphylococcus aureus Exploits Epidermal Barrier Defects in Atopic Dermatitis to Trigger Cytokine Expression. J. Investig. Dermatol. 2016, 136, 2192–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Drongelen, V.; Haisma, E.M.; Out-Luiting, J.J.; Nibbering, P.H.; El Ghalbzouri, A. Reduced filaggrin expression is accompanied by increased Staphylococcus aureus colonization of epidermal skin models. Clin. Exp. Allergy 2014, 44, 1515–1524. [Google Scholar] [CrossRef]
- Zeeuwen, P.L.; Ederveen, T.H.; van der Krieken, D.A.; Niehues, H.; Boekhorst, J.; Kezic, S.; Hanssen, D.A.; Otero, M.E.; van Vlijmen-Willems, I.M.; Rodijk-Olthuis, D.; et al. Gram-positive anaerobe cocci are underrepresented in the microbiome of filaggrin-deficient human skin. J. Allergy Clin. Immunol. 2017, 139, 1368–1371. [Google Scholar] [CrossRef] [Green Version]
- Brandner, J.M. Tight junctions and tight junction proteins in mammalian epidermis. Eur. J. Pharm. Biopharm. 2009, 72, 289–294. [Google Scholar] [CrossRef]
- Moriwaki, K.; Tsukita, S.; Furuse, M. Tight junctions containing claudin 4 and 6 are essential for blastocyst formation in preimplantation mouse embryos. Dev. Biol. 2007, 312, 509–522. [Google Scholar] [CrossRef] [Green Version]
- Yuki, T.; Komiya, A.; Kusaka, A.; Kuze, T.; Sugiyama, Y.; Inoue, S. Impaired tight junctions obstruct stratum corneum formation by altering polar lipid and profilaggrin processing. J. Dermatol. Sci. 2013, 69, 148–158. [Google Scholar] [CrossRef]
- Aho, S.; Harding, C.R.; Lee, J.M.; Meldrum, H.; Bosko, C.A. Regulatory role for the profilaggrin N-terminal domain in epidermal homeostasis. J. Investig. Dermatol. 2012, 132, 2376–2385. [Google Scholar] [CrossRef] [PubMed]
- Henry, J.; Hsu, C.Y.; Haftek, M.; Nachat, R.; de Koning, H.D.; Gardinal-Galera, I.; Hitomi, K.; Balica, S.; Jean-Decoster, C.; Schmitt, A.M.; et al. Hornerin is a component of the epidermal cornified cell envelopes. FASEB J. 2011, 25, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Bouameur, J.E.; Bär, J.; Rice, R.H.; Hornig-Do, H.T.; Roop, D.R.; Schwarz, N.; Brodesser, S.; Thiering, S.; Leube, R.E.; et al. A keratin scaffold regulates epidermal barrier formation, mitochondrial lipid composition, and activity. J. Cell Biol. 2015, 211, 1057–1075. [Google Scholar] [CrossRef] [PubMed]
- Molin, S.; Merl, J.; Dietrich, K.A.; Regauer, M.; Flaig, M.; Letulé, V.; Saucke, T.; Herzinger, T.; Ruzicka, T.; Hauck, S.M. The hand eczema proteome: imbalance of epidermal barrier proteins. Br. J. Dermatol. 2015, 172, 994–1001. [Google Scholar] [CrossRef]
- Pellerin, L.; Henry, J.; Hsu, C.Y.; Balica, S.; Jean-Decoster, C.; Méchin, M.C.; Hansmann, B.; Rodriguez, E.; Weindinger, S.; Schmitt, A.M.; et al. Defects of filaggrin-like proteins in both lesional and nonlesional atopic skin. J. Allergy Clin. Immunol. 2013, 131, 1094–1102. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.Y.; Gasc, G.; Raymond, A.A.; Burlet-Schiltz, O.; Takahara, H.; Serre, G.; Méchin, M.C.; Simon, M. Deimination of Human Hornerin Enhances its Processing by Calpain-1 and its Cross-Linking by Transglutaminases. J. Investig. Dermatol. 2017, 137, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Garach-Jehoshua, O.; Ravid, A.; Liberman, U.A.; Reichrath, J.; Glaser, T.; Koren, R. Upregulation of the calcium-dependent protease, calpain, during keratinocyte differentiation. Br. J. Dermatol. 1998, 139, 950–957. [Google Scholar] [CrossRef]
- Leloup, L.; Wells, A. Calpains as potential anti-cancer targets. Expert Opin. Ther. Targets 2011, 15, 309–323. [Google Scholar] [CrossRef] [Green Version]
- Rice, R.H.; Bradshaw, K.M.; Durbin-Johnson, B.P.; Rocke, D.M.; Eigenheer, R.A.; Phinney, B.S.; Schmuth, M.; Gruber, R. Distinguishing ichthyoses by protein profiling. PLoS ONE 2013, 8, e75355. [Google Scholar] [CrossRef]
- O’Regan, G.M.; Sandilands, A.; McLean, W.H.I.; Irvine, A.D. Filaggrin in atopic dermatitis. J. Allergy Clin. Immunol. 2008, 122, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Behne, M.J.; Meyer, J.W.; Hanson, K.M.; Barry, N.P.; Murata, S.; Crumrine, D.; Clegg, R.W.; Gratton, E.; Holleran, W.M.; Elias, P.M.; et al. NHE1 regulates the stratum corneum permeability barrier homeostasis. Microenvironment acidification assessed with fluorescence lifetime imaging. J. Biol. Chem. 2002, 277, 47399–47406. [Google Scholar] [CrossRef] [PubMed]
- Fluhr, J.W.; Elias, P.M.; Man, M.Q.; Hupe, M.; Selden, C.; Sundberg, J.P.; Tschachler, E.; Eckhart, L.; Mauro, T.M.; Feingold, K.R. Is the filaggrin-histidine-urocanic acid pathway essential for stratum corneum acidification? J. Investig. Dermatol. 2010, 130, 2141–2144. [Google Scholar] [CrossRef]
- Fluhr, J.W.; Behne, M.J.; Brown, B.E.; Moskowitz, D.G.; Selden, C.; Mao-Qiang, M.; Mauro, T.M.; Elias, P.M.; Feingold, K.R. Stratum corneum acidification in neonatal skin: Secretory phospholipase A2 and the sodium/hydrogen antiporter-1 acidify neonatal rat stratum corneum. J. Investig. Dermatol. 2004, 122, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Ilic, D.; Bollinger, J.M.; Gelb, M.; Mauro, T.M. sPLA2 and the epidermal barrier. Biochim. Biophys. Acta 2014, 1841, 416–421. [Google Scholar] [CrossRef]
- Gibbs, N.K.; Tye, J.; Norval, M. Recent advances in urocanic acid photochemistry, photobiology and photoimmunology. Photochem. Photobiol. Sci. 2008, 7, 655–667. [Google Scholar] [CrossRef]
- Kendall, A.C.; Nicolaou, A. Bioactive lipid mediators in skin inflammation and immunity. Prog. Lipid Res. 2013, 52, 141–164. [Google Scholar] [CrossRef]
- Kendall, A.C.; Pilkington, S.M.; Massey, K.A.; Sassano, G.; Rhodes, L.E.; Nicolaou, A. Distribution of bioactive lipid mediators in human skin. J. Investig. Dermatol. 2015, 135, 1510–1520. [Google Scholar] [CrossRef]
- Yoo, H.; Jeon, B.; Jeon, M.S.; Lee, H.; Kim, T.Y. Reciprocal regulation of 12- and 15-lipoxygenases by UV-irradiation in human keratinocytes. FEBS Lett. 2008, 582, 3249–3253. [Google Scholar] [CrossRef] [Green Version]
- Kragballe, K.; Pinnamaneni, G.; Desjarlais, L.; Duell, E.A.; Voorhees, J.J. Dermis-derived 15-hydroxy-eicosatetraenoic acid inhibits epidermal 12-lipoxygenase activity. J. Investig. Dermatol. 1986, 87, 494–498. [Google Scholar] [CrossRef]
- Nicolaou, A.; Masoodi, M.; Gledhill, K.; Haylett, A.K.; Thody, A.J.; Tobin, D.J.; Rhodes, L.E. The eicosanoid response to high dose UVR exposure of individuals prone and resistant to sunburn. Photochem. Photobiol. Sci. 2012, 11, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, L.E.; Gledhill, K.; Masoodi, M.; Haylett, A.K.; Brownrigg, M.; Thody, A.J.; Tobin, D.J.; Nicolaou, A. The sunburn response in human skin is characterized by sequential eicosanoid profiles that may mediate its early and late phases. FASEB J. 2009, 23, 3947–3956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, J.M.; Bryant, R.W.; Low, C.E.; Pupillo, M.B.; Vanderhoek, J.Y. Regulation of T-lymphocyte mitogenesis by the leukocyte product 15-hydroxy-eicosatetraenoic acid (15-HETE). Cell Immunol. 1982, 67, 112–120. [Google Scholar] [CrossRef]
- Camp, R.D.; Fincham, N.J. Inhibition of ionophore-stimulated leukotriene B4 production in human leucocytes by monohydroxy fatty acids. Br. J. Pharmacol. 1985, 85, 837–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gualde, N.; Atluru, D.; Goodwin, J.S. Effect of lipoxygenase metabolites of arachidonic acid on proliferation of human T cells and T cell subsets. J. Immunol. 1985, 134, 1125–1129. [Google Scholar]
- Waldman, J.S.; Marcus, A.J.; Soter, N.A.; Lim, H.W. Cutaneous inflammation: Effects of hydroxy acids and eicosanoid pathway inhibitors on vascular permeability. J. Investig. Dermatol. 1989, 92, 112–116. [Google Scholar] [CrossRef]
- Fogh, K.; Herlin, T.; Kragballe, K. In vitro inhibition of leukotriene B4 formation by exogeneous 5-lipoxygenase inhibitors is associated with enhanced generation of 15-hydroxy-eicosatetraenoic acid (15-HETE) by human neutrophils. Arch. Dermatol. Res. 1988, 280, 430–436. [Google Scholar] [CrossRef]
- Eckl, K.M.; Alef, T.; Torres, S.; Hennies, H.C. Full-thickness human skin models for congenital ichthyosis and related keratinization disorders. J. Investig. Dermatol. 2011, 131, 1938–1942. [Google Scholar] [CrossRef]
- Plank, R.; Yealland, G.; Miceli, E.; Lima Cunha, D.; Graff, P.; Thomforde, S.; Gruber, R.; Moosbrugger-Martinz, V.; Eckl, K.; Calderón, M.; et al. Transglutaminase 1 Replacement Therapy Successfully Mitigates the Autosomal Recessive Congenital Ichthyosis Phenotype in Full-Thickness Skin Disease Equivalents. J. Investig. Dermatol. 2018, in press. [Google Scholar] [CrossRef]
- Gerecke, C.; Edlich, A.; Giulbudagian, M.; Schumacher, F.; Zhang, N.; Said, A.; Yealland, G.; Lohan, S.B.; Neumann, F.; Meinke, M.C.; et al. Biocompatibility and characterization of polyglycerol-based thermoresponsive nanogels designed as novel drug-delivery systems and their intracellular localization in keratinocytes. Nanotoxicology 2017, 11, 267–277. [Google Scholar] [CrossRef]
- Stout, T.E.; McFarland, T.; Mitchell, J.C.; Appukuttan, B.; Timothy Stout, J. Recombinant filaggrin is internalized and processed to correct filaggrin deficiency. J. Investig. Dermatol. 2014, 134, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Clarysse, K.; Pfaff, C.M.; Marquardt, Y.; Huth, L.; Kortekaas Krohn, I.; Kluwig, D.; Lüscher, B.; Gutermuth, J.; Baron, J. JAK1/3 inhibition preserves epidermal morphology in full-thickness 3D skin models of atopic dermatitis and psoriasis. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 367–375. [Google Scholar] [CrossRef]
- Nygaard, U.; van den Bogaard, E.H.; Niehues, H.; Hvid, M.; Deleuran, M.; Johansen, C.; Vestergaard, C. The “Alarmins” HMBG1 and IL-33 Downregulate Structural Skin Barrier Proteins and Impair Epidermal Growth. Acta Derm. Venereol. 2017, 97, 305–312. [Google Scholar] [CrossRef]
- Hubaux, R.; Bastin, C.; Salmon, M. On the relevance of an in vitro reconstructed human epidermis model for drug screening in atopic dermatitis. Exp. Dermatol. 2018, 27, 1403–1407. [Google Scholar] [CrossRef]
- Jang, Y.; Jeong, S.H.; Park, Y.H.; Bae, H.C.; Lee, H.; Ryu, W.I.; Park, G.H.; Son, S.W. UVB induces HIF-1α-dependent TSLP expression via the JNK and ERK pathways. J. Investig. Dermatol. 2013, 133, 2601–2608. [Google Scholar] [CrossRef] [PubMed]
- Bernard, M.; Carrasco, C.; Laoubi, L.; Guiraud, B.; Rozières, A.; Goujon, C.; Duplan, H.; Bessou-Touya, S.; Nicolas, J.F.; Vocanson, M.; et al. IL-1β induces thymic stromal lymphopoietin and an atopic dermatitis-like phenotype in reconstructed healthy human epidermis. J. Pathol. 2017, 242, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Malaisse, J.; Bourguignon, V.; De Vuyst, E.; Lambert de Rouvroit, C.; Nikkels, A.F.; Flamion, B.; Poumay, Y. Hyaluronan metabolism in human keratinocytes and atopic dermatitis skin is driven by a balance of hyaluronan synthases 1 and 3. J. Investig. Dermatol. 2014, 134, 2174–2182. [Google Scholar] [CrossRef] [PubMed]
- Gilhar, A.; Keren, A.; Paus, R. JAK inhibitors and alopecia areata. Lancet 2019, 393, 318–319. [Google Scholar] [CrossRef]
- Yamaguchi, Y. Periostin in skin tissue and skin-related diseases. Allergol. Int. 2014, 63, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.Y.; Lecland, N.; Pendaries, V.; Viodé, C.; Redoulès, D.; Paul, C.; Merdes, A.; Simon, M.; Bierkamp, C. Stabilization of microtubules restores barrier function after cytokine-induced defects in reconstructed human epidermis. J. Dermatol. Sci. 2018, 91, 87–96. [Google Scholar] [CrossRef]
- Simons, F.E.; Simons, K.J. The pharmacology and use of H1-receptor-antagonist drugs. N. Engl. J. Med. 1994, 330, 1663–1670. [Google Scholar] [PubMed]
- Gschwandtner, M.; Mildner, M.; Mlitz, V.; Gruber, F.; Eckhart, L.; Werfel, T.; Gutzmer, R.; Elias, P.M.; Tschachler, E. Histamine suppresses epidermal keratinocyte differentiation and impairs skin barrier function in a human skin model. Allergy 2013, 68, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M. AHR: Making the keratinocytes thick skinned. Immunity 2014, 40, 863–864. [Google Scholar] [CrossRef]
- van den Bogaard, E.H.; Bergboer, J.G.; Vonk-Bergers, M.; van Vlijmen-Willems, I.M.; Hato, S.V.; van der Valk, P.G.; Schröder, J.M.; Joosten, I.; Zeeuwen, P.L.; Schalkwijk, J. Coal tar induces AHR-dependent skin barrier repair in atopic dermatitis. J. Clin. Invest. 2013, 123, 917–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidaka, T.; Ogawa, E.; Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Fujimura, T.; Aiba, S.; Nakayama, K.; Okuyama, R.; et al. The aryl hydrocarbon receptor AhR links atopic dermatitis and air pollution via induction of the neurotrophic factor artemin. Nat. Immunol. 2017, 18, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Tauchi, M.; Hida, A.; Negishi, T.; Katsuoka, F.; Noda, S.; Mimura, J.; Hosoya, T.; Yanaka, A.; Aburatani, H.; Fujii-Kuriyama, Y.; et al. Constitutive expression of aryl hydrocarbon receptor in keratinocytes causes inflammatory skin lesions. Mol. Cell Biol. 2005, 25, 9360–9368. [Google Scholar] [CrossRef] [PubMed]
- Rademacher, F.; Simanski, M.; Hesse, B.; Dombrowsky, G.; Vent, N.; Gläser, R.; Harder, J. Staphylococcus epidermidis Activates Aryl Hydrocarbon Receptor Signaling in Human Keratinocytes: Implications for Cutaneous Defense. J. Innate Immun. 2018, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Larigot, L.; Juricek, L.; Dairou, J.; Coumoul, X. AhR signaling pathways and regulatory functions. Biochim. Open 2018, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Julliard, W.; Fechner, J.H.; Mezrich, J.D. The aryl hydrocarbon receptor meets immunology: Friend or foe? A little of both. Front. Immunol. 2014, 5, 458. [Google Scholar] [CrossRef]
- Roggenkamp, D.; Köpnick, S.; Stäb, F.; Wenck, H.; Schmelz, M.; Neufang, G. Epidermal nerve fibers modulate keratinocyte growth via neuropeptide signaling in an innervated skin model. J. Investig. Dermatol. 2013, 133, 1620–1628. [Google Scholar] [CrossRef]
- Peters, E.M.; Ericson, M.E.; Hosoi, J.; Seiffert, K.; Hordinsky, M.K.; Ansel, J.C.; Paus, R.; Scholzen, T.E. Neuropeptide control mechanisms in cutaneous biology: Physiological and clinical significance. J. Investig. Dermatol. 2006, 126, 1937–1947. [Google Scholar] [CrossRef] [PubMed]
- Werner, Y.; Lindberg, M.; Forslind, B. Membrane-coating granules in “dry” non-eczematous skin of patients with atopic dermatitis. A quantitative electron microscopic study. Acta Derm. Venereol. 1987, 67, 385–390. [Google Scholar] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leman, G.; Moosbrugger-Martinz, V.; Blunder, S.; Pavel, P.; Dubrac, S. 3D-Organotypic Cultures to Unravel Molecular and Cellular Abnormalities in Atopic Dermatitis and Ichthyosis Vulgaris. Cells 2019, 8, 489. https://doi.org/10.3390/cells8050489
Leman G, Moosbrugger-Martinz V, Blunder S, Pavel P, Dubrac S. 3D-Organotypic Cultures to Unravel Molecular and Cellular Abnormalities in Atopic Dermatitis and Ichthyosis Vulgaris. Cells. 2019; 8(5):489. https://doi.org/10.3390/cells8050489
Chicago/Turabian StyleLeman, Géraldine, Verena Moosbrugger-Martinz, Stefan Blunder, Petra Pavel, and Sandrine Dubrac. 2019. "3D-Organotypic Cultures to Unravel Molecular and Cellular Abnormalities in Atopic Dermatitis and Ichthyosis Vulgaris" Cells 8, no. 5: 489. https://doi.org/10.3390/cells8050489
APA StyleLeman, G., Moosbrugger-Martinz, V., Blunder, S., Pavel, P., & Dubrac, S. (2019). 3D-Organotypic Cultures to Unravel Molecular and Cellular Abnormalities in Atopic Dermatitis and Ichthyosis Vulgaris. Cells, 8(5), 489. https://doi.org/10.3390/cells8050489