Autophagy in Synucleinopathy: The Overwhelmed and Defective Machinery


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Clinical Evidence of Autophagy Implication in Synucleinopathies
2.1. Genetic Evidence of Autophagic Involvement in Familial PD Cases
2.2. Post-Mortem Evidence of Autophagic Involvement in Synucleinopathies
3. Dual-loop Between Autophagy and α-Syn Pathogenesis
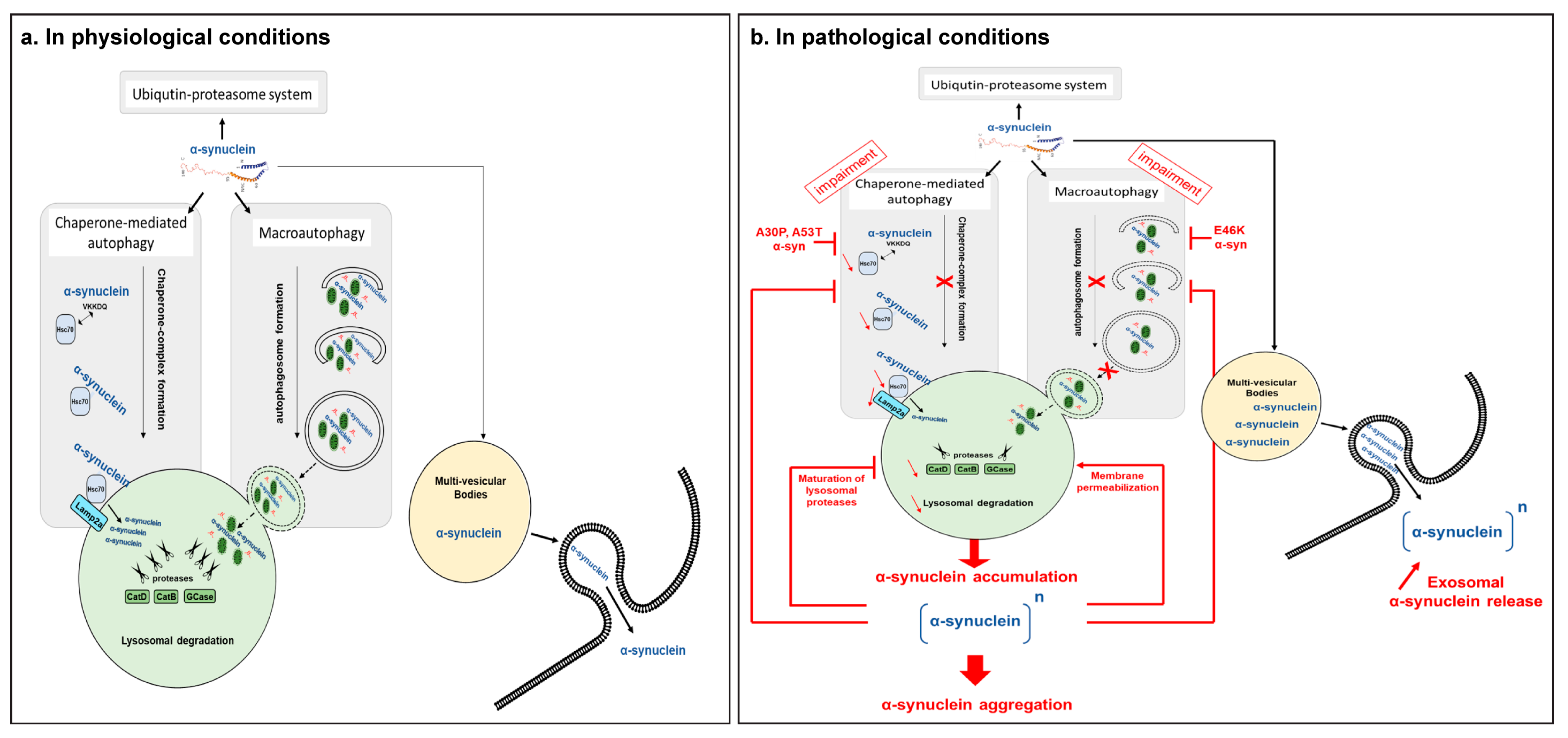
3.1. ALP Processes Participate in α-Syn Accumulation and Aggregation
3.2. ALP Pathways Participate in α-Syn Propagation
3.3. α-Syn Induces Alterations on ALP Processes
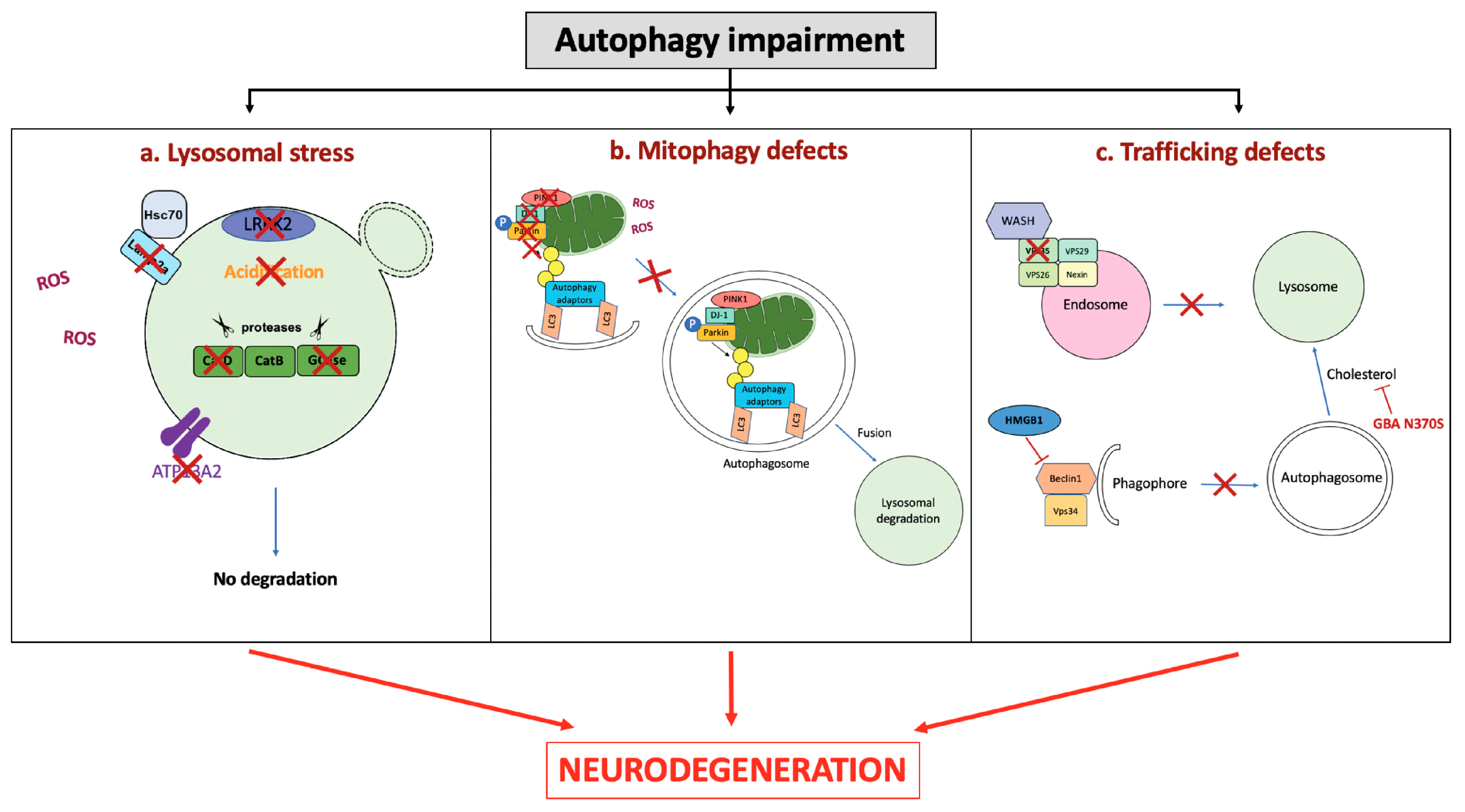
4. Role of Autophagy Impairment in Neurodegeneration
4.1. Lysosomal Dysfunction
4.2. Mitophagy Defects
4.3. Vesicular Trafficking Defects
5. Autophagy Recovery as a Therapeutic Target
5.1. mTOR-Dependent Targeting
5.2. mTOR-Independent Targeting
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| α-syn | α-synuclein |
| ALP | Autophagy-lysosomal pathway |
| Baf | BafilomycinA1 |
| CathD | Cathepsin D |
| CMA | Chaperone-mediated autophagy |
| CSF | Cerebrospinal fluid |
| DLB | Dementia with Lewy bodies |
| ER | Endoplasmic reticulum |
| GABARAP | Gamma-aminobutyric acid receptor-associated protein |
| GATE-16 | Golgi-associated ATPase enhancer of 16kDa |
| GCase | β-glucocerebrosidase |
| GCI | Glial cytoplasmic inclusions |
| GD | Gaucher’s Disease |
| Hsc70 | Heat-shock cognate 70kDa |
| Hsp | Heat shock protein |
| KO | Knock-out |
| LAMP-1 | Lysosomal-associated membrane protein 1 |
| LAMP-2 | Lysosomal-associated membrane protein 2 |
| LB | Lewy Bodies |
| LC3 | Microtubule-associated protein 1A/1B-light chain 3 |
| LMX1B | LIM homeobox transcription factor 1 beta |
| LN | Lewy neurites |
| MA | Macroautophagy |
| MEF2D | Myocyte enhancer factor 2 |
| miRNA | micro RNA |
| MSA | Multiple system atrophy |
| P-α-syn | Phosphorylated α-synuclein |
| PD | Parkinson’s Disease |
| PINK1 | Phosphate and tensin homolog-induced putative kinase 1 |
| ROS | Reactive oxygen species |
| SN | Substantia nigra |
| TFEB | Transcription factor EB |
| TLR | Toll-like receptor |
| WT | Wild-type |
| 3-MA | 3-methyladenine |
References
- Pringsheim, T.; Jette, N.; Frolkis, A.; Steeves, T.D.L. The prevalence of Parkinson’s disease: A systematic review and meta-analysis. Mov. Disord. 2014, 29, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- McCann, H.; Stevens, C.H.; Cartwright, H.; Halliday, G.M. α-Synucleinopathy phenotypes. Parkinsonism Relat. Disord. 2014, 20, S62–S67. [Google Scholar] [CrossRef]
- Emamzadeh, F.N.; Surguchov, A. Parkinson’s Disease: Biomarkers, Treatment, and Risk Factors. Front. Neurosci. 2018, 12, 612. [Google Scholar] [CrossRef] [PubMed]
- Snyder, H.; Mensah, K.; Hsu, C.; Hashimoto, M.; Surgucheva, I.G.; Festoff, B.; Surguchov, A.; Masliah, E.; Matouschek, A.; Wolozin, B. beta-Synuclein reduces proteasomal inhibition by alpha-synuclein but not gamma-synuclein. J. Biol. Chem. 2005, 280, 7562–7569. [Google Scholar] [CrossRef] [PubMed]
- Rivero-Rios, P.; Madero-Perez, J.; Fernandez, B.; Hilfiker, S. Targeting the Autophagy/Lysosomal Degradation Pathway in Parkinson’s Disease. Curr. Neuropharmacol. 2016, 14, 238–249. [Google Scholar] [CrossRef]
- Scrivo, A.; Bourdenx, M.; Pampliega, O.; Cuervo, A.M. Selective autophagy as a potential therapeutic target for neurodegenerative disorders. Lancet Neurol. 2018, 17, 802–815. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Kruger, R.; Kuhn, W.; Muller, T.; Woitalla, D.; Graeber, M.; Kosel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Zarranz, J.J.; Alegre, J.; Gomez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atares, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef]
- 1Yan, J.Q.; Yuan, Y.H.; Chu, S.F.; Li, G.H.; Chen, N.H. E46K Mutant alpha-Synuclein Is Degraded by Both Proteasome and Macroautophagy Pathway. Molecules 2018, 23. [Google Scholar] [CrossRef]
- 1Lei, Z.; Cao, G.; Wei, G. A30P mutant alpha-synuclein impairs autophagic flux by inactivating JNK signaling to enhance ZKSCAN3 activity in midbrain dopaminergic neurons. Cell Death Dis. 2019, 10, 133. [Google Scholar] [CrossRef]
- Choubey, V.; Safiulina, D.; Vaarmann, A.; Cagalinec, M.; Wareski, P.; Kuum, M.; Zharkovsky, A.; Kaasik, A. Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J. Biol. Chem. 2011, 286, 10814–10824. [Google Scholar] [CrossRef] [PubMed]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 Cause Autosomal-Dominant Parkinsonism with Pleomorphic Pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, I.; Kim, J.W.; Dawson, V.L.; Dawson, T.M. LRRK2 pathobiology in Parkinson’s disease. J. Neurochem. 2014, 131, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Dürr, A.; Tazir, M.; Lohmann, E.; Leutenegger, A.-L.; Janin, S.; Pollak, P.; Brice, A. LRRK2 G2019S as a Cause of Parkinson’s Disease in North African Arabs. N. Engl. J. Med. 2006, 354, 422–423. [Google Scholar] [CrossRef]
- Ramirez, A.; Heimbach, A.; Grundemann, J.; Stiller, B.; Hampshire, D.; Cid, L.P.; Goebel, I.; Mubaidin, A.F.; Wriekat, A.L.; Roeper, J.; et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 2006, 38, 1184–1191. [Google Scholar] [CrossRef]
- Edvardson, S.; Cinnamon, Y.; Ta-Shma, A.; Shaag, A.; Yim, Y.I.; Zenvirt, S.; Jalas, C.; Lesage, S.; Brice, A.; Taraboulos, A.; et al. A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS ONE 2012, 7, e36458. [Google Scholar] [CrossRef]
- Deng, H.; Wang, P.; Jankovic, J. The genetics of Parkinson disease. Ageing Res. Rev. 2018, 42, 72–85. [Google Scholar] [CrossRef]
- Gustavsson, E.K.; Trinh, J.; Guella, I.; Vilarino-Guell, C.; Appel-Cresswell, S.; Stoessl, A.J.; Tsui, J.K.; McKeown, M.; Rajput, A.; Rajput, A.H.; et al. DNAJC13 genetic variants in parkinsonism. Mov. Disord. 2015, 30, 273–278. [Google Scholar] [CrossRef]
- Hattori, N.; Kitada, T.; Matsumine, H.; Asakawa, S.; Yamamura, Y.; Yoshino, H.; Kobayashi, T.; Yokochi, M.; Wang, M.; Yoritaka, A.; et al. Molecular genetic analysis of a novel Parkin gene in Japanese families with autosomal recessive juvenile parkinsonism: Evidence for variable homozygous deletions in the Parkin gene in affected individuals. Ann. Neurol. 1998, 44, 935–941. [Google Scholar] [CrossRef]
- Hattori, N.; Matsumine, H.; Asakawa, S.; Kitada, T.; Yoshino, H.; Elibol, B.; Brookes, A.J.; Yamamura, Y.; Kobayashi, T.; Wang, M.; et al. Point Mutations (Thr240Arg and Ala311Stop) in theParkinGene. Biochem. Biophys. Res. Commun. 1998, 249, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Leroy, E.; Anastasopoulos, D.; Konitsiotis, S.; Lavedan, C.; Polymeropoulos, M.H. Deletions in the Parkin gene and genetic heterogeneity in a Greek family with early onset Parkinson’s disease. Hum. Genet. 1998, 103, 424–427. [Google Scholar] [CrossRef] [PubMed]
- Lücking, C.B.; Abbas, N.; Dürr, A.; Bonifati, V.; Bonnet, A.M.; de Broucker, T.; De Michele, G.; Wood, N.W.; Agid, Y.; Brice, A. Homozygous deletions in parkin gene in European and North African families with autosomal recessive juvenile parkinsonism. Lancet 1998, 352, 1355–1356. [Google Scholar] [CrossRef]
- Matsumine, H.; Saito, M.; Shimoda-Matsubayashi, S.; Tanaka, H.; Ishikawa, A.; Nakagawa-Hattori, Y.; Yokochi, M.; Kobayashi, T.; Igarashi, S.; Takano, H.; et al. Localization of a gene for an autosomal recessive form of juvenile Parkinsonism to chromosome 6q25.2-27. Am. J. Hum. Genet. 1997, 60, 588–596. [Google Scholar] [PubMed]
- Valente, E.M.; Bentivoglio, A.R.; Dixon, P.H.; Ferraris, A.; Ialongo, T.; Frontali, M.; Albanese, A.; Wood, N.W. Localization of a Novel Locus for Autosomal Recessive Early-Onset Parkinsonism, PARK6, on Human Chromosome 1p35-p36. Am. J. Hum. Genet. 2001, 68, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Brancati, F.; Caputo, V.; Graham, E.A.; Davis, M.B.; Ferraris, A.; Breteler, M.M.B.; Gasser, T.; Bonifati, V.; Bentivoglio, A.R.; et al. PARK6 is a common cause of familial parkinsonism. Neurol. Sci. 2002, 23, s117–s118. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Brancati, F.; Ferraris, A.; Graham, E.A.; Davis, M.B.; Breteler, M.M.B.; Gasser, T.; Bonifati, V.; Bentivoglio, A.R.; De Michele, G.; et al. Park6-linked parkinsonism occurs in several european families. Ann. Neurol. 2002, 51, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.J.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 Gene Associated with Autosomal Recessive Early-Onset Parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef]
- Van Duijn, C.M.; Dekker, M.C.; Bonifati, V.; Galjaard, R.J.; Houwing-Duistermaat, J.J.; Snijders, P.J.; Testers, L.; Breedveld, G.J.; Horstink, M.; Sandkuijl, L.A.; et al. Park7, a novel locus for autosomal recessive early-onset parkinsonism, on chromosome 1p36. Am. J. Hum. Genet. 2001, 69, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Hruska, K.S.; LaMarca, M.E.; Scott, C.R.; Sidransky, E. Gaucher disease: Mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum. Mutat. 2008, 29, 567–583. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.; Bras, J.; Deas, E.; O’Sullivan, S.S.; Parkkinen, L.; Lachmann, R.H.; Li, A.; Holton, J.; Guerreiro, R.; Paudel, R.; et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 2009, 132, 1783–1794. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter Analysis of Glucocerebrosidase Mutations in Parkinson’s Disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [PubMed]
- Robak, L.A.; Jansen, I.E.; van Rooij, J.; Uitterlinden, A.G.; Kraaij, R.; Jankovic, J.; International Parkinson’s Disease Genomics, C.; Heutink, P.; Shulman, J.M. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 2017, 140, 3191–3203. [Google Scholar] [CrossRef] [PubMed]
- Anglade, P.; Vyas, S.; Javoy-Agid, F.; Herrero, M.; Michel, P.; Marquez, J.; Prigent, A.; Ruberg, M.; Hirsch, E.C.; Agid, Y. Apoptosis and Autophagy in Nigral Neurons of Patients with Parkinson’s Disease. Histol. Histopathol. 1997, 12, 25–31. [Google Scholar] [PubMed]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Tanji, K.; Mori, F.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Alteration of autophagosomal proteins (LC3, GABARAP and GATE-16) in Lewy body disease. Neurobiol. Dis. 2011, 43, 690–697. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Bove, J.; Rodriguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 2010, 30, 12535–12544. [Google Scholar] [CrossRef] [PubMed]
- Higashi, S.; Moore, D.J.; Minegishi, M.; Kasanuki, K.; Fujishiro, H.; Kabuta, T.; Togo, T.; Katsuse, O.; Uchikado, H.; Furukawa, Y.; et al. Localization of MAP1-LC3 in Vulnerable Neurons and Lewy Bodies in Brains of Patients With Dementia With Lewy Bodies. J. Neuropathol. Exp. Neurol. 2011, 70, 264–280. [Google Scholar] [CrossRef] [Green Version]
- Crews, L.; Spencer, B.; Desplats, P.; Patrick, C.; Paulino, A.; Rockenstein, E.; Hansen, L.; Adame, A.; Galasko, D.; Masliah, E. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS ONE 2010, 5, e9313. [Google Scholar] [CrossRef]
- Tanji, K.; Odagiri, S.; Maruyama, A.; Mori, F.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Alteration of autophagosomal proteins in the brain of multiple system atrophy. Neurobiol. Dis. 2013, 49, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, L.; Goldbaum, O.; Bergmann, M.; Probst-Cousin, S.; Richter-Landsberg, C. Involvement of macroautophagy in multiple system atrophy and protein aggregate formation in oligodendrocytes. J. Mol. Neurosci. 2012, 47, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Ramirez, A.; Martinez-Vicente, M.; Perier, C.; Canron, M.H.; Doudnikoff, E.; Vital, A.; Vila, M.; Klein, C.; Bezard, E. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 9611–9616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohn, T.T.; Catlin, L.W. Immunolocalization of influenza A virus and markers of inflammation in the human Parkinson’s disease brain. PLoS ONE 2011, 6, e20495. [Google Scholar] [CrossRef] [PubMed]
- Papagiannakis, N.; Xilouri, M.; Koros, C.; Simitsi, A.M.; Stamelou, M.; Maniati, M.; Stefanis, L. Autophagy dysfunction in peripheral blood mononuclear cells of Parkinson’s disease patients. Neurosci. Lett. 2019, 704, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Laguna, A.; Schintu, N.; Nobre, A.; Alvarsson, A.; Volakakis, N.; Jacobsen, J.K.; Gomez-Galan, M.; Sopova, E.; Joodmardi, E.; Yoshitake, T.; et al. Dopaminergic control of autophagic-lysosomal function implicates Lmx1b in Parkinson’s disease. Nat. Neurosci. 2015, 18, 826–835. [Google Scholar] [CrossRef]
- Chu, Y.; Dodiya, H.; Aebischer, P.; Olanow, C.W.; Kordower, J.H. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: Relationship to alpha-synuclein inclusions. Neurobiol. Dis. 2009, 35, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.E.; Cottle, L.; Gysbers, A.M.; Cooper, A.A.; Halliday, G.M. ATP13A2 (PARK9) protein levels are reduced in brain tissue of cases with Lewy bodies. Acta Neuropathol. Commun. 2013, 1, 11. [Google Scholar] [CrossRef] [Green Version]
- Makioka, K.; Yamazaki, T.; Takatama, M.; Nakazato, Y.; Okamoto, K. Activation and alteration of lysosomes in multiple system atrophy. Neuroreport 2012, 23, 270–276. [Google Scholar] [CrossRef]
- Monzio Compagnoni, G.; Kleiner, G.; Samarani, M.; Aureli, M.; Faustini, G.; Bellucci, A.; Ronchi, D.; Bordoni, A.; Garbellini, M.; Salani, S.; et al. Mitochondrial Dysregulation and Impaired Autophagy in iPSC-Derived Dopaminergic Neurons of Multiple System Atrophy. Stem Cell Rep. 2018, 11, 1185–1198. [Google Scholar] [CrossRef] [Green Version]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Spiro, A.S.; Furuta, A.; Cooper, A.; Garner, B.; Kabuta, T.; Halliday, G.M. Lysosomal-associated membrane protein 2 isoforms are differentially affected in early Parkinson’s disease. Mov. Disord. 2015, 30, 1639–1647. [Google Scholar] [CrossRef] [PubMed]
- Chiba, Y.; Takei, S.; Kawamura, N.; Kawaguchi, Y.; Sasaki, K.; Hasegawa-Ishii, S.; Furukawa, A.; Hosokawa, M.; Shimada, A. Immunohistochemical localization of aggresomal proteins in glial cytoplasmic inclusions in multiple system atrophy. Neuropathol. Appl. Neurobiol. 2012, 38, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Gegg, M.E.; Burke, D.; Heales, S.J.; Cooper, J.M.; Hardy, J.; Wood, N.W.; Schapira, A.H. Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann. Neurol. 2012, 72, 455–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiasserini, D.; Paciotti, S.; Eusebi, P.; Persichetti, E.; Tasegian, A.; Kurzawa-Akanbi, M.; Chinnery, P.F.; Morris, C.M.; Calabresi, P.; Parnetti, L.; et al. Selective loss of glucocerebrosidase activity in sporadic Parkinson’s disease and dementia with Lewy bodies. Mol. Neurodegener. 2015, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Tayebi, N.; Kim, W.S.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G.M. Reduced glucocerebrosidase is associated with increased alpha-synuclein in sporadic Parkinson’s disease. Brain 2014, 137, 834–848. [Google Scholar] [CrossRef]
- Balducci, C.; Pierguidi, L.; Persichetti, E.; Parnetti, L.; Sbaragli, M.; Tassi, C.; Orlacchio, A.; Calabresi, P.; Beccari, T.; Rossi, A. Lysosomal hydrolases in cerebrospinal fluid from subjects with Parkinson’s disease. Mov. Disord. 2007, 22, 1481–1484. [Google Scholar] [CrossRef] [PubMed]
- Parnetti, L.; Chiasserini, D.; Persichetti, E.; Eusebi, P.; Varghese, S.; Qureshi, M.M.; Dardis, A.; Deganuto, M.; De Carlo, C.; Castrioto, A.; et al. Cerebrospinal fluid lysosomal enzymes and alpha-synuclein in Parkinson’s disease. Mov. Disord. 2014, 29, 1019–1027. [Google Scholar] [CrossRef]
- van Dijk, K.D.; Persichetti, E.; Chiasserini, D.; Eusebi, P.; Beccari, T.; Calabresi, P.; Berendse, H.W.; Parnetti, L.; van de Berg, W.D. Changes in endolysosomal enzyme activities in cerebrospinal fluid of patients with Parkinson’s disease. Mov. Disord. 2013, 28, 747–754. [Google Scholar] [CrossRef]
- Klucken, J.; Poehler, A.M.; Ebrahimi-Fakhari, D.; Schneider, J.; Nuber, S.; Rockenstein, E.; Schlotzer-Schrehardt, U.; Hyman, B.T.; McLean, P.J.; Masliah, E.; et al. Alpha-synuclein aggregation involves a bafilomycin A 1-sensitive autophagy pathway. Autophagy 2012, 8, 754–766. [Google Scholar] [CrossRef]
- Usenovic, M.; Tresse, E.; Mazzulli, J.R.; Taylor, J.P.; Krainc, D. Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha-synuclein accumulation, and neurotoxicity. J. Neurosci. 2012, 32, 4240–4246. [Google Scholar] [CrossRef]
- Tsunemi, T.; Perez-Rosello, T.; Ishiguro, Y.; Yoroisaka, A.; Jeon, S.; Hamada, K.; Krishna Vangipuram Suresh, M.; Wong, Y.C.; Xie, Z.; Akamatsu, W.; et al. Increased lysosomal exocytosis induced by lysosomal Ca(2+) channel agonists protects human dopaminergic neurons from alpha-synuclein toxicity. J. Neurosci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Uchihara, T.; Fukuda, T.; Noda, S.; Kondo, H.; Saiki, S.; Komatsu, M.; Uchiyama, Y.; Tanaka, K.; Hattori, N. Loss of autophagy in dopaminergic neurons causes Lewy pathology and motor dysfunction in aged mice. Sci. Rep. 2018, 8, 2813. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. α-Synuclein Is Degraded by Both Autophagy and the Proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J.; Khoshaghideh, F.; Patel, S.; Lee, S.-J. Clearance of α-Synuclein Oligomeric Intermediates via the Lysosomal Degradation Pathway. J. Neurosci. 2004, 24, 1888. [Google Scholar] [CrossRef] [PubMed]
- Spencer, B.; Potkar, R.; Trejo, M.; Rockenstein, E.; Patrick, C.; Gindi, R.; Adame, A.; Wyss-Coray, T.; Masliah, E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J. Neurosci. 2009, 29, 13578–13588. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.H.; Dorado, B.; Figueroa, H.Y.; Wang, L.; Planel, E.; Cookson, M.R.; Clark, L.N.; Duff, K.E. Metabolic activity determines efficacy of macroautophagic clearance of pathological oligomeric alpha-synuclein. Am. J. Pathol. 2009, 175, 736–747. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired Degradation of Mutant α-Synuclein by Chaperone-Mediated Autophagy. Science 2004, 305, 1292. [Google Scholar] [CrossRef] [PubMed]
- Mak, S.K.; McCormack, A.L.; Manning-Bog, A.B.; Cuervo, A.M.; Di Monte, D.A. Lysosomal degradation of alpha-synuclein in vivo. J. Biol. Chem. 2010, 285, 13621–13629. [Google Scholar] [CrossRef]
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008, 283, 23542–23556. [Google Scholar] [CrossRef]
- Xilouri, M.; Brekk, O.R.; Polissidis, A.; Chrysanthou-Piterou, M.; Kloukina, I.; Stefanis, L. Impairment of chaperone-mediated autophagy induces dopaminergic neurodegeneration in rats. Autophagy 2016, 12, 2230–2247. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Rodriguez-Oroz, M.C.; Obeso, J.A.; Cooper, J.M. Influence of microRNA deregulation on chaperone-mediated autophagy and alpha-synuclein pathology in Parkinson’s disease. Cell Death Dis. 2013, 4, e545. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yang, H.; Zhu, D.; Huang, H.; Liu, G.; Lun, P. Targeted suppression of chaperone-mediated autophagy by miR-320a promotes alpha-synuclein aggregation. Int. J. Mol. Sci 2014, 15, 15845–15857. [Google Scholar] [CrossRef] [PubMed]
- Shendelman, S.; Jonason, A.; Martinat, C.; Leete, T.; Abeliovich, A. DJ-1 is a redox-dependent molecular chaperone that inhibits alpha-synuclein aggregate formation. PLoS Biol. 2004, 2, e362. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.Y.; Kang, W.Y.; Chen, Y.M.; Jiang, T.F.; Zhang, J.; Zhang, L.N.; Ding, J.Q.; Liu, J.; Chen, S.D. DJ-1 Inhibits alpha-Synuclein Aggregation by Regulating Chaperone-Mediated Autophagy. Front. Aging Neurosci. 2017, 9, 308. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.J.; Yang, N.Y.; Lee, C.; Lee, H.J.; Kim, S.; Sardi, S.P.; Lee, S.J. Loss of glucocerebrosidase 1 activity causes lysosomal dysfunction and alpha-synuclein aggregation. Exp. Mol. Med. 2015, 47, e153. [Google Scholar] [CrossRef] [PubMed]
- Cullen, V.; Sardi, S.P.; Ng, J.; Xu, Y.H.; Sun, Y.; Tomlinson, J.J.; Kolodziej, P.; Kahn, I.; Saftig, P.; Woulfe, J.; et al. Acid beta-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter alpha-synuclein processing. Ann. Neurol. 2011, 69, 940–953. [Google Scholar] [CrossRef] [PubMed]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Cleeter, M.W.; Chau, K.Y.; Gluck, C.; Mehta, A.; Hughes, D.A.; Duchen, M.; Wood, N.W.; Hardy, J.; Mark Cooper, J.; Schapira, A.H. Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem. Int. 2013, 62, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.H.; Sun, Y.; Ran, H.; Quinn, B.; Witte, D.; Grabowski, G.A. Accumulation and distribution of alpha-synuclein and ubiquitin in the CNS of Gaucher disease mouse models. Mol. Genet. Metab. 2011, 102, 436–447. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2002, 24, 197–211. [Google Scholar] [CrossRef]
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Olanow, C.W.; Freeman, T.B. Transplanted dopaminergic neurons develop PD pathologic changes: A second case report. Mov. Disord. 2008, 23, 2303–2306. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Bjorklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Mendez, I.; Viñuela, A.; Astradsson, A.; Mukhida, K.; Hallett, P.; Robertson, H.; Tierney, T.; Holness, R.; Dagher, A.; Trojanowski, J.Q.; et al. Dopamine neurons implanted into people with Parkinson’s disease survive without pathology for 14 years. Nat. Med. 2008, 14, 507–509. [Google Scholar] [CrossRef] [PubMed]
- Desplats, P.; Lee, H.J.; Bae, E.J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl Acad Sci USA 2009, 106, 13010–13015. [Google Scholar] [CrossRef] [PubMed]
- Recasens, A.; Dehay, B.; Bove, J.; Carballo-Carbajal, I.; Dovero, S.; Perez-Villalba, A.; Fernagut, P.O.; Blesa, J.; Parent, A.; Perier, C.; et al. Lewy body extracts from Parkinson disease brains trigger alpha-synuclein pathology and neurodegeneration in mice and monkeys. Ann. Neurol. 2014, 75, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Soria, F.N.; Pampliega, O.; Bourdenx, M.; Meissner, W.G.; Bezard, E.; Dehay, B. Exosomes, an Unmasked Culprit in Neurodegenerative Diseases. Front. Neurosci. 2017, 11, 26. [Google Scholar] [CrossRef] [Green Version]
- Minakaki, G.; Menges, S.; Kittel, A.; Emmanouilidou, E.; Schaeffner, I.; Barkovits, K.; Bergmann, A.; Rockenstein, E.; Adame, A.; Marxreiter, F.; et al. Autophagy inhibition promotes SNCA/alpha-synuclein release and transfer via extracellular vesicles with a hybrid autophagosome-exosome-like phenotype. Autophagy 2018, 14, 98–119. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Gardiner, C.; Sargent, I.L.; Wood, M.J.; Cooper, J.M. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 2011, 42, 360–367. [Google Scholar] [CrossRef] [Green Version]
- Danzer, K.M.; Kranich, L.R.; Ruf, W.P.; Cagsal-Getkin, O.; Winslow, A.R.; Zhu, L.; Vanderburg, C.R.; McLean, P.J. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 2012, 7, 42. [Google Scholar] [CrossRef]
- Lee, H.J.; Cho, E.D.; Lee, K.W.; Kim, J.H.; Cho, S.G.; Lee, S.J. Autophagic failure promotes the exocytosis and intercellular transfer of alpha-synuclein. Exp. Mol. Med. 2013, 45, e22. [Google Scholar] [CrossRef]
- Poehler, A.M.; Xiang, W.; Spitzer, P.; May, V.E.; Meixner, H.; Rockenstein, E.; Chutna, O.; Outeiro, T.F.; Winkler, J.; Masliah, E.; et al. Autophagy modulates SNCA/alpha-synuclein release, thereby generating a hostile microenvironment. Autophagy 2014, 10, 2171–2192. [Google Scholar] [CrossRef] [PubMed]
- Tsunemi, T.; Hamada, K.; Krainc, D. ATP13A2/PARK9 regulates secretion of exosomes and alpha-synuclein. J. Neurosci. 2014, 34, 15281–15287. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.M.; Chan, B.K.; Park, J.S.; Hill, K.J.; Aitken, J.B.; Cottle, L.; Farghaian, H.; Cole, A.R.; Lay, P.A.; Sue, C.M.; et al. Parkinson’s disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes alpha-Synuclein externalization via exosomes. Hum. Mol. Genet. 2014, 23, 2816–2833. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.J.; Yang, N.Y.; Song, M.; Lee, C.S.; Lee, J.S.; Jung, B.C.; Lee, H.J.; Kim, S.; Masliah, E.; Sardi, S.P.; et al. Glucocerebrosidase depletion enhances cell-to-cell transmission of alpha-synuclein. Nat. Commun. 2014, 5, 4755. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L.; Larsen, K.E.; Rideout, H.J.; Sulzer, D.; Greene, L.A. Expression of A53T Mutant But Not Wild-Type α-Synuclein in PC12 Cells Induces Alterations of the Ubiquitin-Dependent Degradation System, Loss of Dopamine Release, and Autophagic Cell Death. J. Neurosci. 2001, 21, 9549–9560. [Google Scholar] [CrossRef] [PubMed]
- Song, J.X.; Lu, J.H.; Liu, L.F.; Chen, L.L.; Durairajan, S.S.; Yue, Z.; Zhang, H.Q.; Li, M. HMGB1 is involved in autophagy inhibition caused by SNCA/alpha-synuclein overexpression: A process modulated by the natural autophagy inducer corynoxine B. Autophagy 2014, 10, 144–154. [Google Scholar] [CrossRef]
- Winslow, A.R.; Chen, C.W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. alpha-Synuclein impairs macroautophagy: Implications for Parkinson’s disease. J. Cell Biol. 2010, 190, 1023–1037. [Google Scholar] [CrossRef]
- Xilouri, M.; Vogiatzi, T.; Vekrellis, K.; Park, D.; Stefanis, L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS ONE 2009, 4, e5515. [Google Scholar] [CrossRef]
- Martinez-Vicente, M.; Talloczy, Z.; Kaushik, S.; Massey, A.C.; Mazzulli, J.; Mosharov, E.V.; Hodara, R.; Fredenburg, R.; Wu, D.C.; Follenzi, A.; et al. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J. Clin. Invest. 2008, 118, 777–788. [Google Scholar] [CrossRef]
- Hoffmann, A.C.; Minakaki, G.; Menges, S.; Salvi, R.; Savitskiy, S.; Kazman, A.; Vicente Miranda, H.; Mielenz, D.; Klucken, J.; Winkler, J.; et al. Extracellular aggregated alpha synuclein primarily triggers lysosomal dysfunction in neural cells prevented by trehalose. Sci Rep. 2019, 9, 544. [Google Scholar] [CrossRef]
- Yang, Q.; She, H.; Gearing, M.; Colla, E.; Lee, M.; Shacka, J.J.; Mao, Z. Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy. Science 2009, 323, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.-Q.; Yuan, Y.-H.; Gao, Y.-N.; Huang, J.-Y.; Ma, K.-L.; Gao, Y.; Zhang, W.-Q.; Guo, X.-F.; Chen, N.-H. Overexpression of Human E46K Mutant α-Synuclein Impairs Macroautophagy via Inactivation of JNK1-Bcl-2 Pathway. Mol. Neurobiol. 2014, 50, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Tanik, S.A.; Schultheiss, C.E.; Volpicelli-Daley, L.A.; Brunden, K.R.; Lee, V.M. Lewy body-like alpha-synuclein aggregates resist degradation and impair macroautophagy. J. Biol. Chem. 2013, 288, 15194–15210. [Google Scholar] [CrossRef] [PubMed]
- Freeman, D.; Cedillos, R.; Choyke, S.; Lukic, Z.; McGuire, K.; Marvin, S.; Burrage, A.M.; Sudholt, S.; Rana, A.; O’Connor, C.; et al. Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS ONE 2013, 8, e62143. [Google Scholar] [CrossRef] [PubMed]
- Stefanovic, A.N.; Stockl, M.T.; Claessens, M.M.; Subramaniam, V. alpha-Synuclein oligomers distinctively permeabilize complex model membranes. FEBS J. 2014, 281, 2838–2850. [Google Scholar] [CrossRef] [PubMed]
- Mazzulli, J.R.; Zunke, F.; Isacson, O.; Studer, L.; Krainc, D. alpha-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc. Natl. Acad. Sci. USA 2016, 113, 1931–1936. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.L.; Gruschus, J.M.; Velayati, A.; Westbroek, W.; Goldin, E.; Moaven, N.; Sidransky, E.; Lee, J.C. Alpha-synuclein interacts with Glucocerebrosidase providing a molecular link between Parkinson and Gaucher diseases. J. Biol. Chem. 2011, 286, 28080–28088. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Komatsu, M.; Wang, Q.J.; Holstein, G.R.; Friedrich, V.L., Jr.; Iwata, J.; Kominami, E.; Chait, B.T.; Tanaka, K.; Yue, Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc. Natl. Acad. Sci. USA 2007, 104, 14489–14494. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, S.M. Neuronal lysosomes. Neurosci. Lett. 2019, 697, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jäger, S.; Bucci, C.; Tanida, I.; Ueno, T.; Kominami, E.; Saftig, P.; Eskelinen, E.L. Role for Rab7 in maturation of late autophagic vacuoles. J. Cell. Sci. 2004, 117, 4837–4848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourdenx, M.; Daniel, J.; Genin, E.; Soria, F.N.; Blanchard-Desce, M.; Bezard, E.; Dehay, B. Nanoparticles restore lysosomal acidification defects: Implications for Parkinson and other lysosomal-related diseases. Autophagy 2016, 12, 472–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kett, L.R.; Stiller, B.; Bernath, M.M.; Tasset, I.; Blesa, J.; Jackson-Lewis, V.; Chan, R.B.; Zhou, B.; Di Paolo, G.; Przedborski, S.; et al. alpha-Synuclein-independent histopathological and motor deficits in mice lacking the endolysosomal Parkinsonism protein Atp13a2. J. Neurosci. 2015, 35, 5724–5742. [Google Scholar] [CrossRef] [PubMed]
- Henry, A.G.; Aghamohammadzadeh, S.; Samaroo, H.; Chen, Y.; Mou, K.; Needle, E.; Hirst, W.D. Pathogenic LRRK2 mutations, through increased kinase activity, produce enlarged lysosomes with reduced degradative capacity and increase ATP13A2 expression. Hum. Mol. Genet. 2015, 24, 6013–6028. [Google Scholar] [CrossRef] [Green Version]
- Tong, Y.; Yamaguchi, H.; Giaime, E.; Boyle, S.; Kopan, R.; Kelleher, R.J., 3rd; Shen, J. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc. Natl. Acad. Sci. USA 2010, 107, 9879–9884. [Google Scholar] [CrossRef]
- Giaime, E.; Tong, Y.; Wagner, L.K.; Yuan, Y.; Huang, G.; Shen, J. Age-Dependent Dopaminergic Neurodegeneration and Impairment of the Autophagy-Lysosomal Pathway in LRRK-Deficient Mice. Neuron 2017, 96, 796–807. [Google Scholar] [CrossRef]
- Qiao, L.; Hamamichi, S.; Caldwell, K.A.; Caldwell, G.A.; Yacoubian, T.A.; Wilson, S.; Xie, Z.L.; Speake, L.D.; Parks, R.; Crabtree, D.; et al. Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol. Brain 2008, 1, 17. [Google Scholar] [CrossRef]
- Sardi, S.P.; Clarke, J.; Kinnecom, C.; Tamsett, T.J.; Li, L.; Stanek, L.M.; Passini, M.A.; Grabowski, G.A.; Schlossmacher, M.G.; Sidman, R.L.; et al. CNS expression of glucocerebrosidase corrects alpha-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc. Natl. Acad. Sci. USA 2011, 108, 12101–12106. [Google Scholar] [CrossRef]
- Lewis, M.R.; Lewis, W.H. Mitochondria in tissue culture. Science 1914, 39, 330–333. [Google Scholar] [CrossRef]
- Greene, J.C.; Whitworth, A.J.; Kuo, I.; Andrews, L.A.; Feany, M.B.; Pallanck, L.J. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. USA 2003, 100, 4078–4083. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, S.B.; Lee, S.; Kim, Y.; Song, S.; Kim, S.; Bae, E.; Kim, J.; Shong, M.; Kim, J.M.; et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 2006, 441, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Poole, A.C.; Thomas, R.E.; Andrews, L.A.; McBride, H.M.; Whitworth, A.J.; Pallanck, L.J. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2008, 105, 1638–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, J.; Ding, J.; Simon-Sanchez, J.; Paisan-Ruiz, C.; Singleton, A.B.; Scholz, S.W. Parkin and PINK1 mutations in early-onset Parkinson’s disease: Comprehensive screening in publicly available cases and control. J. Med. Genet. 2009, 46, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D.A.; Lee, Y.; Kang, H.C.; Lee, B.D.; Lee, Y.I.; Bower, A.; Jiang, H.; Kang, S.U.; Andrabi, S.A.; Dawson, V.L.; et al. Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proc. Natl. Acad. Sci. USA 2015, 112, 11696–11701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Corti, O. Neuronal Mitophagy: Lessons from a Pathway Linked to Parkinson’s Disease. Neurotox. Res. 2019. [Google Scholar] [CrossRef]
- van der Merwe, C.; Jalali Sefid Dashti, Z.; Christoffels, A.; Loos, B.; Bardien, S. Evidence for a common biological pathway linking three Parkinson’s disease-causing genes: Parkin, PINK1 and DJ-1. Eur. J. Neurosci. 2015, 41, 1113–1125. [Google Scholar] [CrossRef]
- Zondler, L.; Miller-Fleming, L.; Repici, M.; Goncalves, S.; Tenreiro, S.; Rosado-Ramos, R.; Betzer, C.; Straatman, K.R.; Jensen, P.H.; Giorgini, F.; et al. DJ-1 interactions with alpha-synuclein attenuate aggregation and cellular toxicity in models of Parkinson’s disease. Cell Death Dis. 2014, 5, e1350. [Google Scholar] [CrossRef]
- Thomas, K.J.; McCoy, M.K.; Blackinton, J.; Beilina, A.; van der Brug, M.; Sandebring, A.; Miller, D.; Maric, D.; Cedazo-Minguez, A.; Cookson, M.R. DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum. Mol. Genet. 2011, 20, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Cai, Z.; Tao, K.; Zeng, W.; Lu, F.; Yang, R.; Feng, D.; Gao, G.; Yang, Q. Essential control of mitochondrial morphology and function by chaperone-mediated autophagy through degradation of PARK7. Autophagy 2016, 12, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ham, A.; Ma, T.C.; Kuo, S.H.; Kanter, E.; Kim, D.; Ko, H.S.; Quan, Y.; Sardi, S.P.; Li, A.; et al. Mitochondrial dysfunction and mitophagy defect triggered by heterozygous GBA mutations. Autophagy 2019, 15, 113–130. [Google Scholar] [CrossRef] [PubMed]
- Zimprich, A.; Benet-Pages, A.; Struhal, W.; Graf, E.; Eck, S.H.; Offman, M.N.; Haubenberger, D.; Spielberger, S.; Schulte, E.C.; Lichtner, P.; et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet. 2011, 89, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Zavodszky, E.; Seaman, M.N.; Moreau, K.; Jimenez-Sanchez, M.; Breusegem, S.Y.; Harbour, M.E.; Rubinsztein, D.C. Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat. Commun. 2014, 5, 3828. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.T.; Glauser, L.; Tsika, E.; Jiang, H.; Islam, S.; Moore, D.J. Parkin mediates the ubiquitination of VPS35 and modulates retromer-dependent endosomal sorting. Hum. Mol. Genet. 2018, 27, 3189–3205. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Yang, J.; Shen, Y.; Jiang, H.; Han, C.; Zhang, G.; Liu, L.; Xu, X.; Li, J.; Lin, Z.; et al. HMGB1 Mediates Autophagy Dysfunction via Perturbing Beclin1-Vps34 Complex in Dopaminergic Cell Model. Front. Mol. Neurosci. 2017, 10, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Sanz, P.; Orgaz, L.; Fuentes, J.M.; Vicario, C.; Moratalla, R. Cholesterol and multilamellar bodies: Lysosomal dysfunction in GBA-Parkinson disease. Autophagy 2018, 14, 717–718. [Google Scholar] [CrossRef]
- Kim, M.J.; Jeon, S.; Burbulla, L.F.; Krainc, D. Acid ceramidase inhibition ameliorates alpha-synuclein accumulation upon loss of GBA1 function. Hum. Mol. Genet. 2018, 27, 1972–1988. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Schuchman, E.H. Acid ceramidase and human disease. Biochim. Biophys. Acta 2006, 1758, 2133–2138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lie, P.P.Y.; Nixon, R.A. Lysosome trafficking and signaling in health and neurodegenerative diseases. Neurobiol. Dis. 2019, 122, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Rawal, P.; Wu, Y.; Xie, W.; Jankovic, J.; Le, W. Rapamycin protects against rotenone-induced apoptosis through autophagy induction. Neuroscience 2009, 164, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Wey, M.C.; Fernandez, E.; Hart, M.J.; Gelfond, J.; Bokov, A.F.; Rani, S.; Strong, R. Rapamycin improves motor function, reduces 4-hydroxynonenal adducted protein in brain, and attenuates synaptic injury in a mouse model of synucleinopathy. Pathobiol. Aging Age Relat. Dis. 2015, 5, 28743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malagelada, C.; Jin, Z.H.; Jackson-Lewis, V.; Przedborski, S.; Greene, L.A. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J. Neurosci. 2010, 30, 1166–1175. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef] [Green Version]
- Kilpatrick, K.; Zeng, Y.; Hancock, T.; Segatori, L. Genetic and chemical activation of TFEB mediates clearance of aggregated alpha-synuclein. PLoS ONE 2015, 10, e0120819. [Google Scholar] [CrossRef]
- Decressac, M.; Mattsson, B.; Weikop, P.; Lundblad, M.; Jakobsson, J.; Bjorklund, A. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc. Natl. Acad. Sci. USA 2013, 110, E1817–E1826. [Google Scholar] [CrossRef]
- Tan, S.; Yu, C.Y.; Sim, Z.W.; Low, Z.S.; Lee, B.; See, F.; Min, N.; Gautam, A.; Chu, J.J.H.; Ng, K.W.; et al. Pomegranate activates TFEB to promote autophagy-lysosomal fitness and mitophagy. Sci. Rep. 2019, 9, 727. [Google Scholar] [CrossRef]
- Kim, C.; Rockenstein, E.; Spencer, B.; Kim, H.K.; Adame, A.; Trejo, M.; Stafa, K.; Lee, H.J.; Lee, S.J.; Masliah, E. Antagonizing Neuronal Toll-like Receptor 2 Prevents Synucleinopathy by Activating Autophagy. Cell Rep. 2015, 13, 771–782. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Ho, D.H.; Suk, J.E.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.J.; et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef]
- Rodríguez-Navarro, J.A.; Rodríguez, L.; Casarejos, M.J.; Solano, R.M.; Gómez, A.; Perucho, J.; Cuervo, A.M.; García de Yébenes, J.; Mena, M.A. Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiol. Dis. 2010, 39, 423–438. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J. Biol. Chem. 2007, 282, 5641–5652. [Google Scholar] [CrossRef] [PubMed]
- Luan, Y.; Ren, X.; Zheng, W.; Zeng, Z.; Guo, Y.; Hou, Z.; Guo, W.; Chen, X.; Li, F.; Chen, J.F. Chronic Caffeine Treatment Protects Against alpha-Synucleinopathy by Reestablishing Autophagy Activity in the Mouse Striatum. Front. Neurosci. 2018, 12, 301. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Xiong, N.; Liu, L.; Huang, J.; Han, C.; Zhang, G.; Li, J.; Xu, X.; Lin, Z.; Wang, T. Lithium protects dopaminergic cells from rotenone toxicity via autophagy enhancement. BMC Neurosci. 2015, 16, 82. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Z.; Chen, X.P.; Zhao, K.; Bai, L.M.; Zhang, H.; Zhou, X.P. Therapeutic effects of valproate combined with lithium carbonate on MPTP-induced parkinsonism in mice: Possible mediation through enhanced autophagy. Int. J. Neurosci. 2013, 123, 73–79. [Google Scholar] [CrossRef]
- Williams, A.; Sarkar, S.; Cuddon, P.; Ttofi, E.K.; Saiki, S.; Siddiqi, F.H.; Jahreiss, L.; Fleming, A.; Pask, D.; Goldsmith, P.; et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol. 2008, 4, 295–305. [Google Scholar] [CrossRef]
- Pagan, F.; Hebron, M.; Valadez, E.H.; Torres-Yaghi, Y.; Huang, X.; Mills, R.R.; Wilmarth, B.M.; Howard, H.; Dunn, C.; Carlson, A.; et al. Nilotinib Effects in Parkinson’s disease and Dementia with Lewy bodies. J. Parkinsons Dis. 2016, 6, 503–517. [Google Scholar] [CrossRef]
- Pagan, F.L.; Hebron, M.L.; Wilmarth, B.; Torres-Yaghi, Y.; Lawler, A.; Mundel, E.E.; Yusuf, N.; Starr, N.J.; Arellano, J.; Howard, H.H.; et al. Pharmacokinetics and pharmacodynamics of a single dose Nilotinib in individuals with Parkinson’s disease. Pharm. Res. Perspect. 2019, 7, e00470. [Google Scholar] [CrossRef]
- Lee, J.-H.; McBrayer, M.K.; Wolfe, D.M.; Haslett, L.J.; Kumar, A.; Sato, Y.; Lie, P.P.Y.; Mohan, P.; Coffey, E.E.; Kompella, U.; et al. Presenilin 1 Maintains Lysosomal Ca(2+) Homeostasis via TRPML1 by Regulating vATPase-Mediated Lysosome Acidification. Cell Rep. 2015, 12, 1430–1444. [Google Scholar] [CrossRef]
- Prévot, G.; Soria, F.N.; Thiolat, M.-L.; Daniel, J.; Verlhac, J.B.; Blanchard-Desce, M.; Bezard, E.; Barthélémy, P.; Crauste-Manciet, S.; Dehay, B. Harnessing Lysosomal pH through PLGA Nanoemulsion as a Treatment of Lysosomal-Related Neurodegenerative Diseases. Bioconjugate Chem. 2018, 29, 4083–4089. [Google Scholar] [CrossRef]
- Koh, J.-Y.; Kim, H.N.; Hwang, J.J.; Kim, Y.-H.; Park, S.E. Lysosomal dysfunction in proteinopathic neurodegenerative disorders: Possible therapeutic roles of cAMP and zinc. Mol. Brain 2019, 12, 18. [Google Scholar] [CrossRef] [PubMed]
- Ambrosi, G.; Ghezzi, C.; Zangaglia, R.; Levandis, G.; Pacchetti, C.; Blandini, F. Ambroxol-induced rescue of defective glucocerebrosidase is associated with increased LIMP-2 and saposin C levels in GBA1 mutant Parkinson’s disease cells. Neurobiol. Dis. 2015, 82, 235–242. [Google Scholar] [CrossRef] [PubMed]
- McNeill, A.; Magalhaes, J.; Shen, C.; Chau, K.Y.; Hughes, D.; Mehta, A.; Foltynie, T.; Cooper, J.M.; Abramov, A.Y.; Gegg, M.; et al. Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain 2014, 137, 1481–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silveira, C.R.A.; MacKinley, J.; Coleman, K.; Li, Z.; Finger, E.; Bartha, R.; Morrow, S.A.; Wells, J.; Borrie, M.; Tirona, R.G.; et al. Ambroxol as a novel disease-modifying treatment for Parkinson’s disease dementia: Protocol for a single-centre, randomized, double-blind, placebo-controlled trial. BMC Neurol. 2019, 19, 20. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.H.; Tan, J.Q.; Durairajan, S.S.; Liu, L.F.; Zhang, Z.H.; Ma, L.; Shen, H.M.; Chan, H.Y.; Li, M. Isorhynchophylline, a natural alkaloid, promotes the degradation of alpha-synuclein in neuronal cells via inducing autophagy. Autophagy 2012, 8, 98–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Huang, J.; Xie, W.; Huang, L.; Zhong, C.; Chen, Z. Beclin1 and HMGB1 ameliorate the alpha-synuclein-mediated autophagy inhibition in PC12 cells. Diagn. Pathol. 2016, 11, 15. [Google Scholar] [CrossRef] [PubMed]
- Xilouri, M.; Brekk, O.R.; Landeck, N.; Pitychoutis, P.M.; Papasilekas, T.; Papadopoulou-Daifoti, Z.; Kirik, D.; Stefanis, L. Boosting chaperone-mediated autophagy in vivo mitigates alpha-synuclein-induced neurodegeneration. Brain 2013, 136, 2130–2146. [Google Scholar] [CrossRef] [PubMed]
- Anguiano, J.; Garner, T.P.; Mahalingam, M.; Das, B.C.; Gavathiotis, E.; Cuervo, A.M. Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat. Chem. Biol. 2013, 9, 374–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullett, S.J.; Di Maio, R.; Greenamyre, J.T.; Hinkle, D.A. DJ-1 expression modulates astrocyte-mediated protection against neuronal oxidative stress. J. Mol. Neurosci. 2013, 49, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Mullett, S.J.; Hinkle, D.A. DJ-1 knock-down in astrocytes impairs astrocyte-mediated neuroprotection against rotenone. Neurobiol. Dis. 2009, 33, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Mullett, S.J.; Hinkle, D.A. DJ-1 deficiency in astrocytes selectively enhances mitochondrial Complex I inhibitor-induced neurotoxicity. J. Neurochem. 2011, 117, 375–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Miranda, B.R.; Rocha, E.M.; Bai, Q.; El Ayadi, A.; Hinkle, D.; Burton, E.A.; Timothy Greenamyre, J. Astrocyte-specific DJ-1 overexpression protects against rotenone-induced neurotoxicity in a rat model of Parkinson’s disease. Neurobiol. Dis. 2018, 115, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Pupyshev, A.B.; Tikhonova, M.A.; Akopyan, A.A.; Tenditnik, M.V.; Dubrovina, N.I.; Korolenko, T.A. Therapeutic activation of autophagy by combined treatment with rapamycin and trehalose in a mouse MPTP-induced model of Parkinson’s disease. Pharm. Biochem. Behav. 2019, 177, 1–11. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arotcarena, M.-L.; Teil, M.; Dehay, B. Autophagy in Synucleinopathy: The Overwhelmed and Defective Machinery. Cells 2019, 8, 565. https://doi.org/10.3390/cells8060565
Arotcarena M-L, Teil M, Dehay B. Autophagy in Synucleinopathy: The Overwhelmed and Defective Machinery. Cells. 2019; 8(6):565. https://doi.org/10.3390/cells8060565
Chicago/Turabian StyleArotcarena, Marie-Laure, Margaux Teil, and Benjamin Dehay. 2019. "Autophagy in Synucleinopathy: The Overwhelmed and Defective Machinery" Cells 8, no. 6: 565. https://doi.org/10.3390/cells8060565
APA StyleArotcarena, M.-L., Teil, M., & Dehay, B. (2019). Autophagy in Synucleinopathy: The Overwhelmed and Defective Machinery. Cells, 8(6), 565. https://doi.org/10.3390/cells8060565