Role of Calbindin-D28k in Diabetes-Associated Advanced Glycation End-Products-Induced Renal Proximal Tubule Cell Injury

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Immunohistochemistry
2.3. Double Immunofluorescence Staining
2.4. Cell Culture
2.5. Preparation of AGEs
2.6. Protein Extraction
2.7. Western Blot Analysis
2.8. RNA Interference
2.9. Delivering siRNA in Vivo
2.10. Statistical Analysis
3. Results
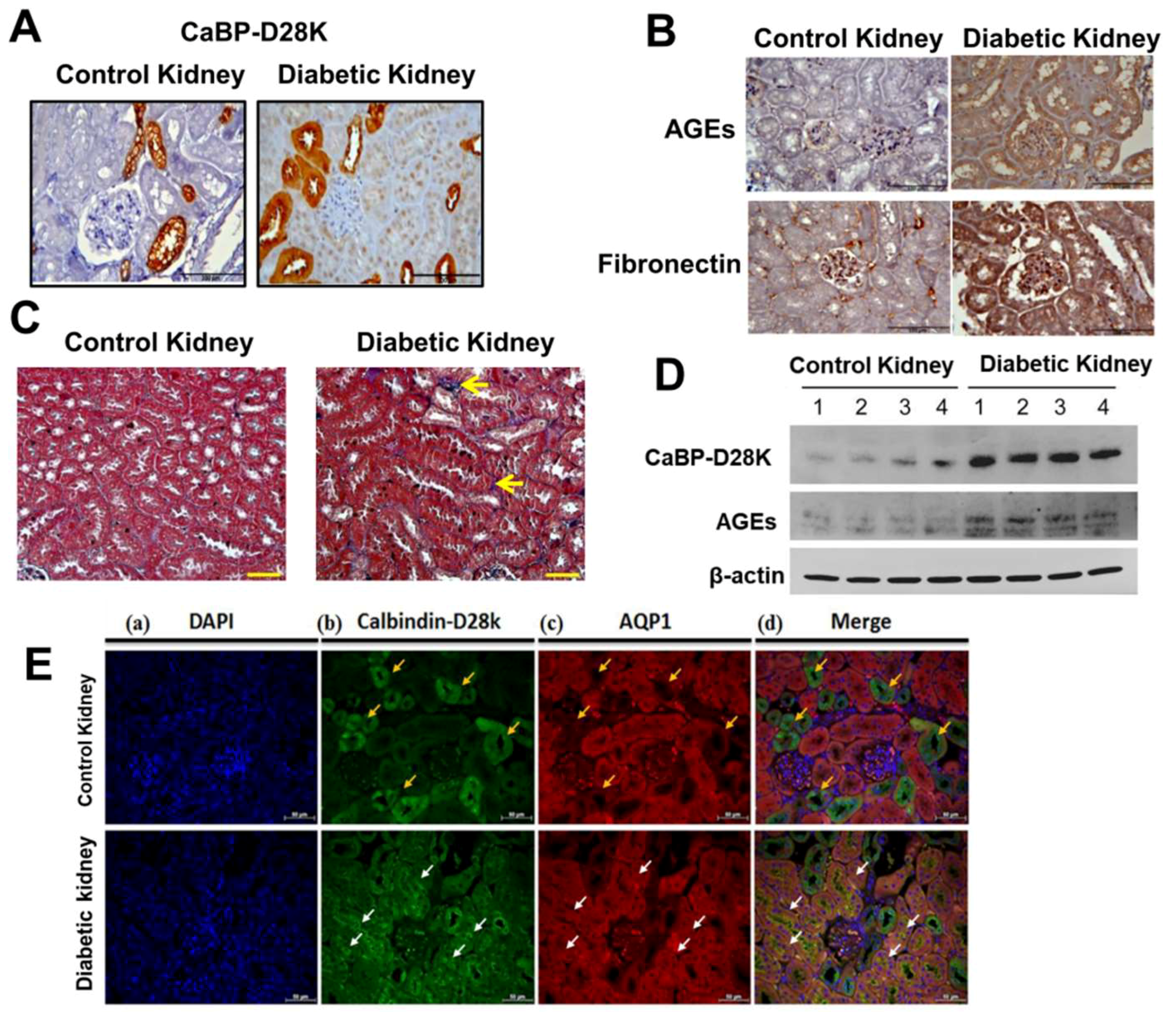
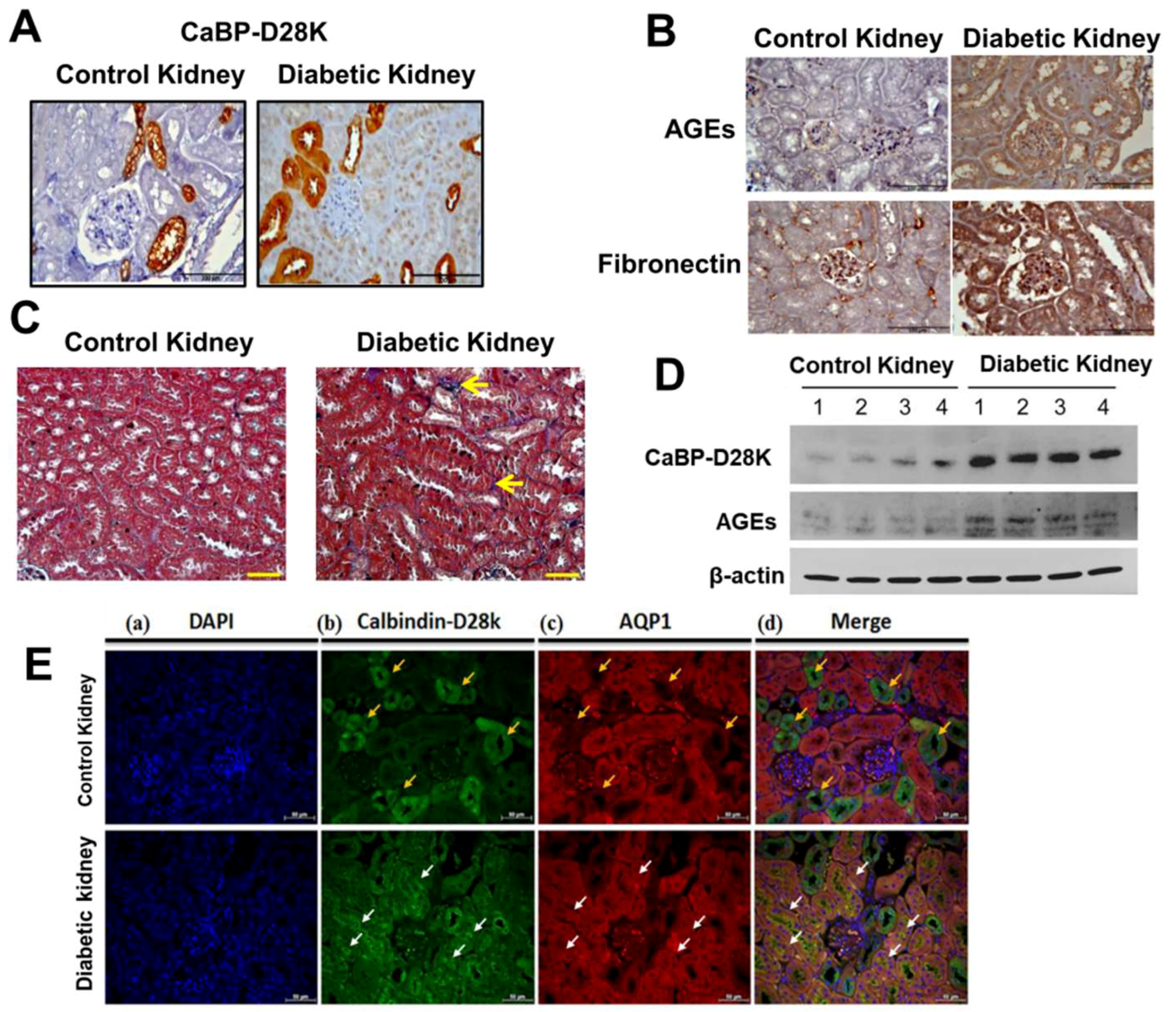
3.1. Induction of Calbindin-D28k and AGEs Accumulation in the Kidney of Diabetic Mice
3.2. AGEs Induce Calbindin-D28k Expression in Renal Proximal Tubular Cells
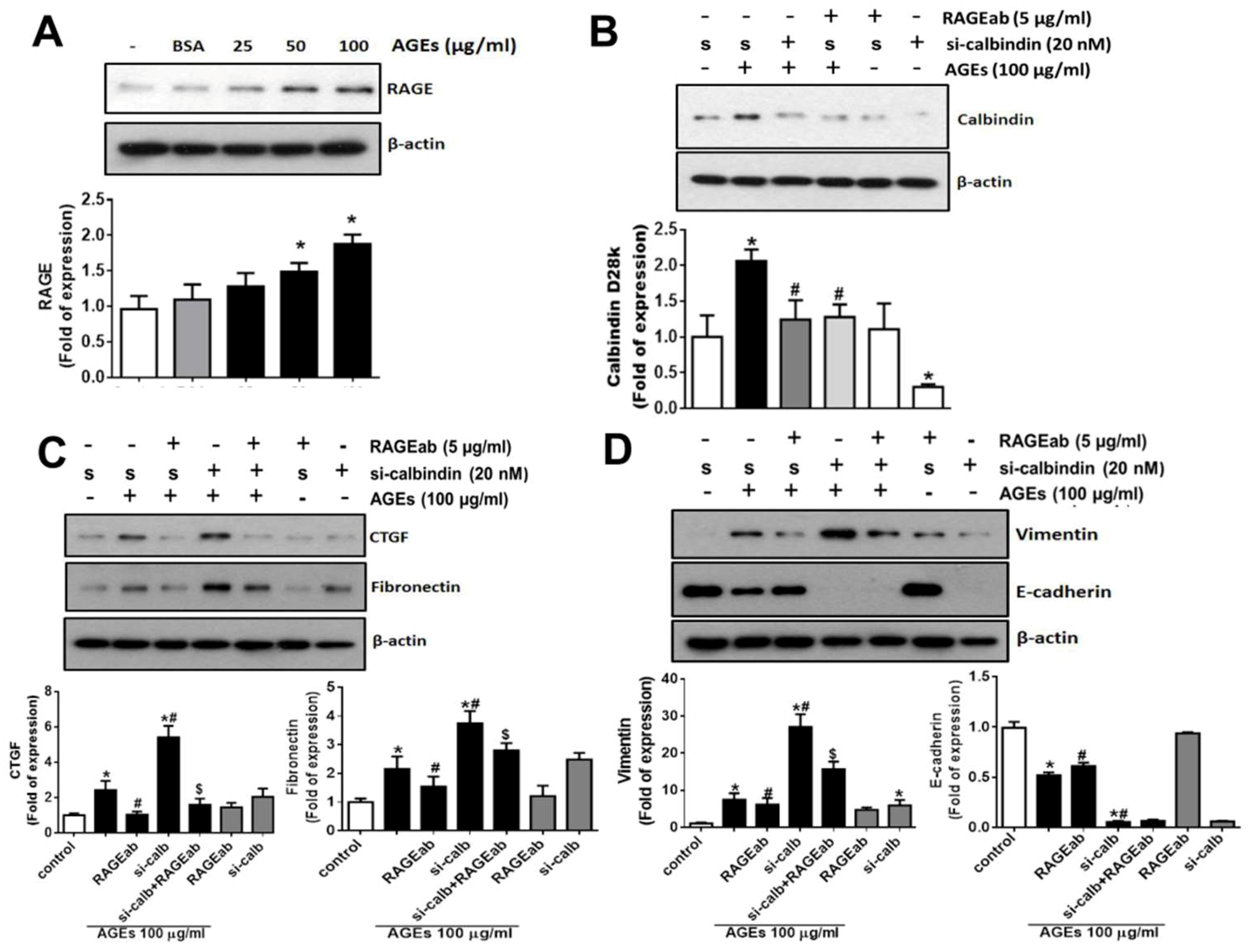
3.3. Interference of Calbindin-D28k Decreases Cell Viability and Enhances AGEs-Induced Fibrotic Responses
3.4. Role of RAGE in the AGEs-Induced Expressions of Calbindin-D28k and Fibrotic Markers in HK2 Cells
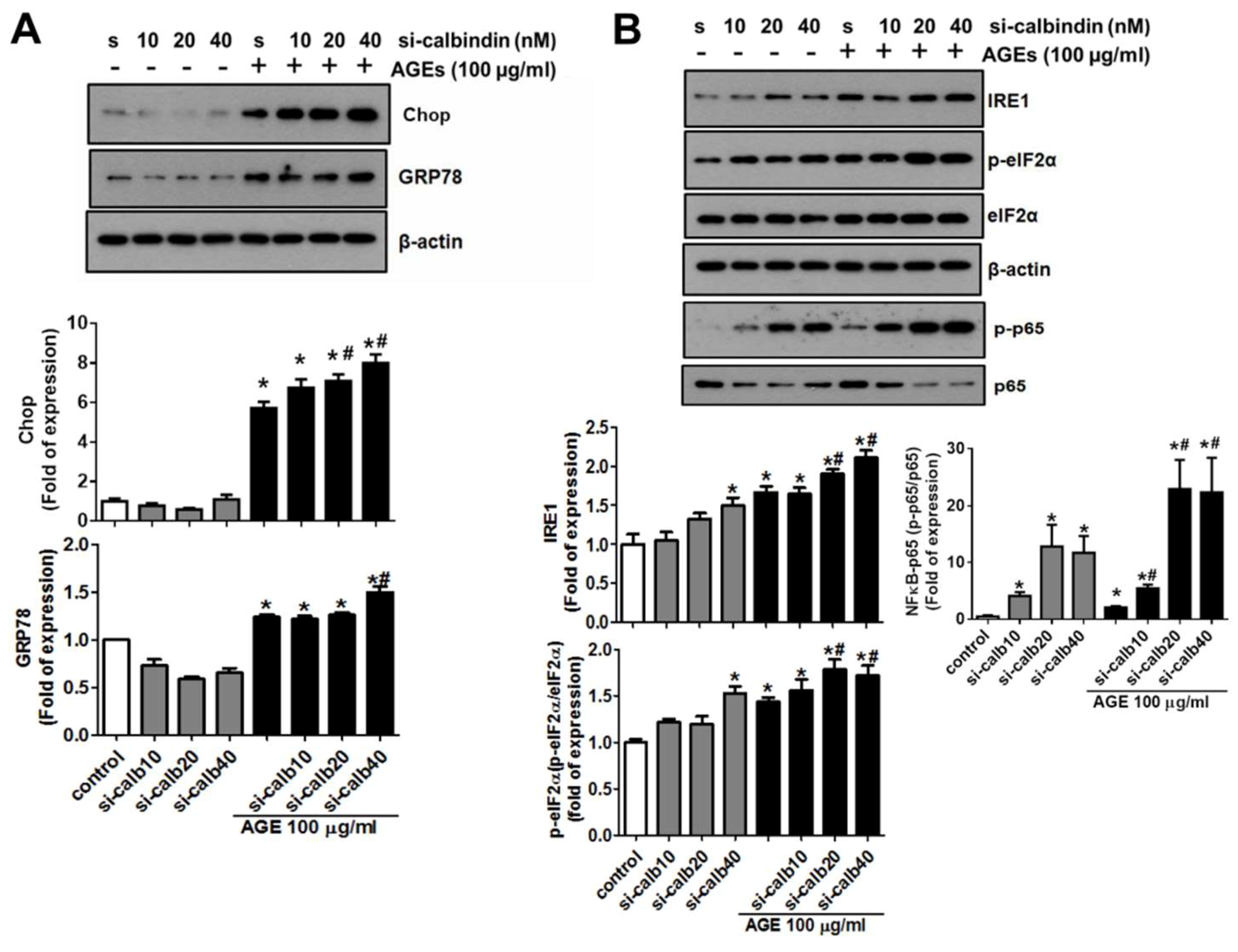
3.5. Calbindin-D28k Deficiency Enhances AGEs-Induced ER Stress Signaling in HK2 Cells
3.6. 4-Phenylbutyric Acid (4-PBA) Counteracts the Effects of Calbindin-D28k Deficiency on ER Stress and EMT Signals in HK2 Cells
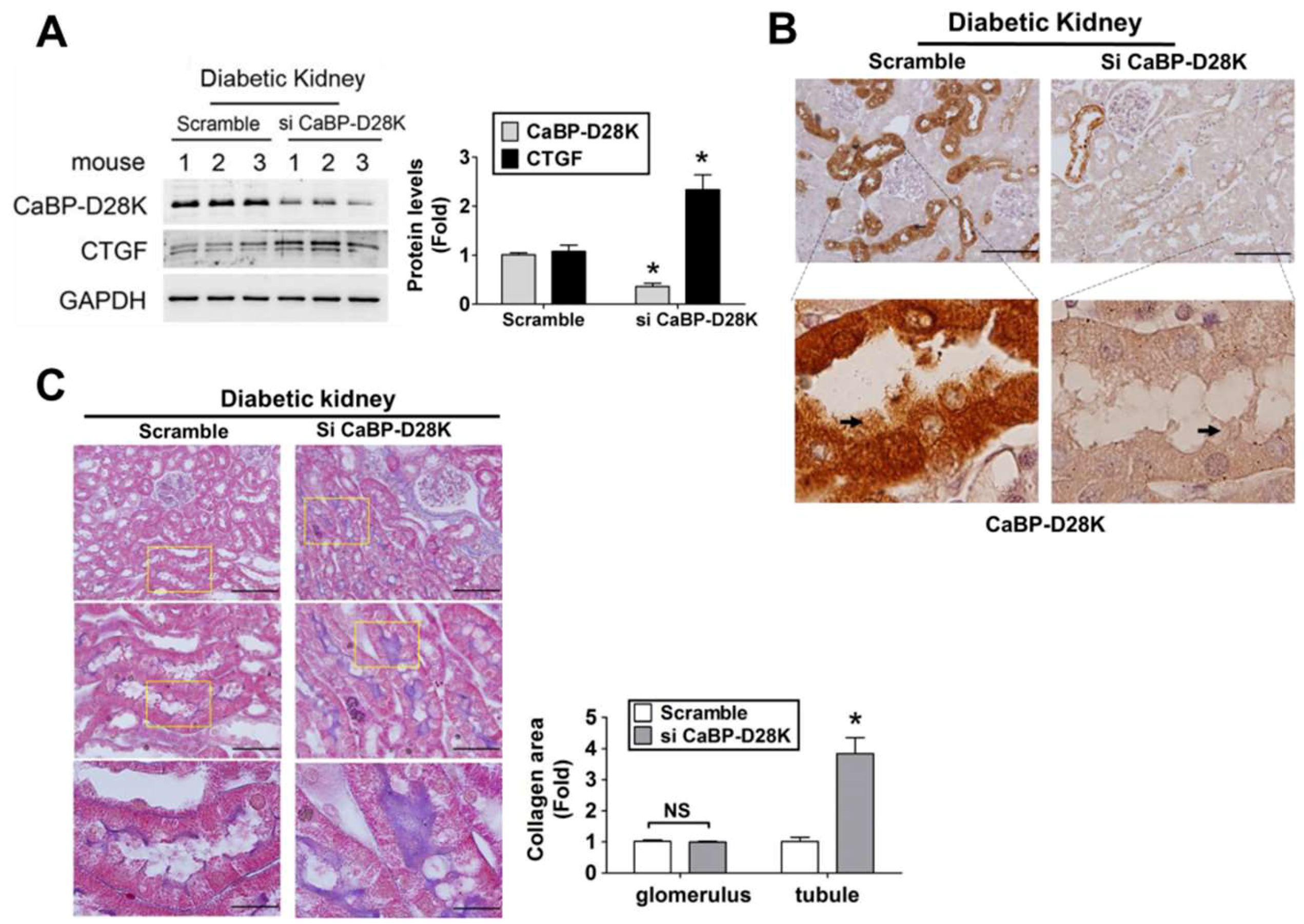
3.7. Calbindin-D28k siRNA in Vivo Delivery Enhanced Renal Fibrosis in db/db Diabetic Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- American Diabetes Association. Classification and diagnosis of diabetes. Diabetes Care 2015, 38, S8–S16. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Randive, R.; Stewart, J.A. Molecular mechanisms of AGE/RAGE-mediated fibrosis in the diabetic heart. World J. Diabetes 2014, 5, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.K. Diabetic nephropathy—Complications and treatment. Int. J. Nephrol. Renovasc. Dis. 2014, 7, 361–381. [Google Scholar]
- Sun, Y.M.; Su, Y.; Li, J.; Wang, L.F. Recent advances in understanding the biochemical and molecular mechanism of diabetic nephropathy. Biochem. Biophys. Res. Commun. 2013, 433, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Busch, M.; Franke, S.; Ruster, C.; Wolf, G. Advanced glycation end-products and the kidney. Eur. J. Clin. Investig. 2010, 40, 742–755. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Higashimoto, Y.; Nishino, Y.; Nakamura, N.; Fukami, K.; Yamagishi, S.I. RAGE-aptamer blocks the development and progression of experimental diabetic nephropathy. Diabetes 2017, 66, 1683–1695. [Google Scholar] [CrossRef] [PubMed]
- Sooy, K.; Kohut, J.; Christakos, S. The role of calbindin and 1,25dihydroxyvitamin D3 in the kidney. Curr. Opin. Nephrol. Hypertens. 2000, 9, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Jena, G.; Tikoo, K.; Kumar, V. Valproate attenuates the proteinuria, podocyte and renal injury by facilitating autophagy and inactivation of NF-κB/iNOS signaling in diabetic rat. Biochimie 2015, 110, 1–16. [Google Scholar] [CrossRef]
- Thongboonkerd, V.; Zheng, S.; McLeish, K.R.; Epstein, P.N.; Klein, J.B. Proteomic identification and immunolocalization of increased renal calbindin-D28k expression in OVE26 diabetic mice. Rev. Diabet. Stud. 2005, 2, 19–26. [Google Scholar] [CrossRef]
- Wu, M.J.; Lai, L.W.; Lien, Y.H. Effect of calbindin-D28K on cyclosporine toxicity in cultured renal proximal tubular cells. J. Cell. Physiol. 2004, 200, 395–399. [Google Scholar] [CrossRef]
- Rabinovitch, A.; Suarez-Pinzon, W.L.; Sooy, K.; Strynadka, K.; Christakos, S. Expression of calbindin-D(28k) in a pancreatic islet beta-cell line protects against cytokine-induced apoptosis and necrosis. Endocrinology 2001, 142, 3649–3655. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.K.; Wang, C.C.; Lu, T.F.; Huang, K.H.; Sheu, M.L.; Liu, S.H.; Hung, K.Y. Involvement of endoplasmic reticulum stress, autophagy, and apoptosis in advanced glycation end products-induced glomerular mesangial cell injury. Sci. Rep. 2016, 6, 34167. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.; Brown, D.; Norman, A.W.; Orci, L. Localization of the vitamin D-dependent calcium-binding protein in mammalian kidney. Am. J. Physiol. 1982, 243, F243–F252. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.P.; Bali, A.; Singh, N.; Jaggi, A.S. Advanced glycation end products and diabetic complications. Korean J. Physiol. Pharmacol. 2014, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, M.T.; Thorburn, D.R.; Penfold, S.A.; Laskowski, A.; Harcourt, B.E.; Sourris, K.C.; Tan, A.L.; Fukami, K.; Thallas-Bonke, V.; Nawroth, P.P.; et al. RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J. Am. Soc. Nephrol. 2009, 20, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, M.K. Diabetic nephropathy: Recent advances in pathophysiology and challenges in dietary management. Diabetol. Metab. Syndr. 2019, 11, 7. [Google Scholar] [CrossRef]
- Tanji, N.; Markowitz, G.S.; Fu, C.; Kislinger, T.; Taguchi, A.; Pischetsrieder, M.; Stern, D.; Schmidt, A.M.; D’Agati, V.D. Expression of advanced glycation end products and their cellular receptor RAGE in diabetic nephropathy and nondiabetic renal disease. J. Am. Soc. Nephrol. 2000, 11, 1656–1666. [Google Scholar]
- Sanajou, D.; Ghorbani Haghjo, A.; Argani, H.; Aslani, S. AGE-RAGE axis blockade in diabetic nephropathy: Current status and future directions. Eur. J. Pharmacol. 2018, 833, 158–164. [Google Scholar] [CrossRef]
- Lee, C.T.; Lien, Y.H.; Lai, L.W.; Chen, J.B.; Lin, C.R.; Chen, H.C. Increased renal calcium and magnesium transporter abundance in streptozotocin-induced diabetes mellitus. Kidney. Int. 2006, 69, 1786–1791. [Google Scholar] [CrossRef] [Green Version]
- Vig, P.J.; Wei, J.; Shao, Q.; Lopez, M.E.; Halperin, R.; Gerber, J. Suppression of calbindin-D28k expression exacerbates SCA1 phenotype in a disease mouse model. Cerebellum 2012, 11, 718–732. [Google Scholar] [CrossRef]
- Yi, S.S. Time-dependent changes of calbindin D-28K and parvalbumin immunoreactivity in the hippocampus of rats with streptozotocin-induced type 1 diabetes. J. Vet. Sci. 2013, 14, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Jiang, C.; Yuan, H.; Xiao, C.; Gao, D. Role of calbindin-D28K in estrogen treatment for Parkinson’s disease. Neural. Regen. Res. 2013, 8, 702–707. [Google Scholar]
- Liu, G.; Sun, Y.; Li, Z.; Song, T.; Wang, H.; Zhang, Y.; Ge, Z. Apoptosis induced by endoplasmic reticulum stress involved in diabetic kidney disease. Biochem. Biophys. Res. Commun. 2008, 370, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Erguler, K.; Pieri, M.; Deltas, C. A mathematical model of the unfolded protein stress response reveals the decision mechanism for recovery, adaptation and apoptosis. BMC. Syst. Biol. 2013, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.K.; Hsu, S.P.; Wu, C.T.; Huang, J.W.; Cheng, H.T.; Chang, Y.W.; Hung, K.Y.; Wu, K.D.; Liu, S.H. Endoplasmic reticulum stress implicated in the development of renal fibrosis. Mol. Med. 2011, 17, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Yang, Y.; Liu, Y.; Jiang, S.; Di, S.; Hu, W.; Ma, Z.; Li, T.; Zhu, Y.; Xin, Z.; et al. Icariin displays anticancer activity against human esophageal cancer cells via regulating endoplasmic reticulum stress-mediated apoptotic signaling. Sci. Rep. 2016, 6, 21145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Guo, Y.; Fu, H.; Hu, S.; Pan, J.; Wang, Y.; Cheng, J.; Song, J.; Yu, Q.; Zhang, S.; et al. Chop deficiency prevents UUO-induced renal fibrosis by attenuating fibrotic signals originated from Hmgb1/TLR4/NFkappaB/IL-1beta signaling. Cell Death Dis. 2015, 6, e1847. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- Mekahli, D.; Bultynck, G.; Parys, J.B.; De Smedt, H.; Missiaen, L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a004317. [Google Scholar] [CrossRef]
- Kanekura, K.; Ma, X.; Murphy, J.T.; Zhu, L.J.; Diwan, A.; Urano, F. IRE1 prevents endoplasmic reticulum membrane permeabilization and cell death under pathological conditions. Sci. Signal. 2015, 8, ra62. [Google Scholar] [CrossRef]
- Cui, W.; Ma, J.; Wang, X.; Yang, W.; Zhang, J.; Ji, Q. Free fatty acid induces endoplasmic reticulum stress and apoptosis of beta-cells by Ca2+/calpain-2 pathways. PLoS ONE 2013, 8, e59921. [Google Scholar]
- Shin, H.S.; Ryu, E.S.; Oh, E.S.; Kang, D.H. Endoplasmic reticulum stress as a novel target to ameliorate epithelial-to-mesenchymal transition and apoptosis of human peritoneal mesothelial cells. Lab. Investig. 2015, 95, 1157–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanjore, H.; Lawson, W.E.; Blackwell, T.S. Endoplasmic reticulum stress as a pro-fibrotic stimulus. Biochim. Biophys. Acta 2013, 1832, 940–947. [Google Scholar] [CrossRef] [PubMed]
- Chiappisi, E.; Ringseis, R.; Eder, K.; Gessner, D.K. Effect of endoplasmic reticulum stress on metabolic and stress signaling and kidney-specific functions in Madin-Darby bovine kidney cells. J. Dairy Sci. 2017, 100, 6689–6706. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Mu, J.; Luo, Z.F.; Zeng, W.; Guo, Y.H.; Pang, Q.; Ye, Z.L.; Liu, L.; Yuan, F.H.; Feng, B. Attenuation of diabetic nephropathy in diabetes rats induced by streptozotocin by regulating the endoplasmic reticulum stress inflammatory response. Metabolism 2011, 60, 594–603. [Google Scholar] [CrossRef]
- Grande, M.T.; Sanchez-Laorden, B.; Lopez-Blau, C.; De Frutos, C.A.; Boutet, A.; Arevalo, M.; Rowe, R.G.; Weiss, S.J.; Lopez-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef] [Green Version]
- Pallet, N.; Thervet, E.; Anglicheau, D. c-Jun-N-terminal kinase signaling is involved in cyclosporine-induced epithelial phenotypic changes. J. Transplant. 2012, 2012, 348604. [Google Scholar] [CrossRef]
- Lan, H.Y. Diverse roles of TGF-beta/Smads in renal fibrosis and inflammation. Int. J. Biol. Sci. 2011, 7, 1056–1067. [Google Scholar] [CrossRef]
- Lee, E.J.; Kang, M.K.; Kim, D.Y.; Kim, Y.H.; Oh, H.; Kang, Y.H. Chrysin inhibits advanced glycation end products-induced kidney fibrosis in renal mesangial cells and diabetic kidneys. Nutrients 2018, 10, 882. [Google Scholar] [CrossRef]
- Guan, S.S.; Sheu, M.L.; Wu, C.T.; Chiang, C.K.; Liu, S.H. ATP synthase subunit-beta down-regulation aggravates diabetic nephropathy. Sci. Rep. 2015, 5, 14561. [Google Scholar] [CrossRef]
- Sun, S.; Li, F.; Gao, X.; Zhu, Y.; Chen, J.; Zhu, X.; Yuan, H.; Gao, D. Calbindin-D28K inhibits apoptosis in dopaminergic neurons by activation of the PI3-kinase-Akt signaling pathway. Neuroscience 2011, 199, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Yoo, Y.M.; Jeung, E.B. Calbindin-D28k in the brain influences the expression of cellular prion protein. Oxid. Med. Cell Longev. 2018, 2018, 4670210. [Google Scholar] [CrossRef] [PubMed]
- Pujol, J.B.; Heikkila, E.; Savoia, C.; Hajibeigi, A.; De Marchi, U.; Battiprolu, P.K.; Oz, O.K.; Dioum, E.H.M. Isx9 regulates calbindin D28K expression in pancreatic beta cells and promotes beta cell survival and function. Int. J. Mol. Sci. 2018, 19, 2542. [Google Scholar] [CrossRef] [PubMed]
- Borke, J.L.; Caride, A.; Verma, A.K.; Penniston, J.T.; Kumar, R. Plasma membrane calcium pump and 28-kDa calcium binding protein in cells of rat kidney distal tubules. Am. J. Physiol. 1989, 257, F842–F849. [Google Scholar] [CrossRef] [PubMed]
- Biner, H.L.; Arpin-Bott, M.P.; Loffing, J.; Wang, X.; Knepper, M.; Hebert, S.C.; Kaissling, B. Human cortical distal nephron: Distribution of electrolyte and water transport pathways. J. Am. Soc. Nephrol. 2002, 13, 836–847. [Google Scholar] [PubMed]
- Karan, M.; Timurkaan, S.; Atalar, Ö. An immunohistochemical study of distribution of calbindin-D28k in the kidney of Martes foina. Vet. Arhiv. 2007, 77, 195–201. [Google Scholar]
- Yamagishi, S.; Amano, S.; Inagaki, Y.; Okamoto, T.; Koga, K.; Sasaki, N.; Yamamoto, H.; Takeuchi, M.; Makita, Z. Advanced glycation end products-induced apoptosis and overexpression of vascular endothelial growth factor in bovine retinal pericytes. Biochem. Biophys. Res. Commun. 2002, 290, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.C.; Chiu, C.Y.; Kao, C.W.; Huang, K.H.; Wang, C.C.; Huang, K.T.; Tsai, K.S.; Sheu, M.L.; Liu, S.H. Advanced glycation end-products induce apoptosis in pancreatic islet endothelial cells via NF-κB-activated cyclooxygenase-2/prostaglandin E2 up-regulation. PLoS ONE 2015, 10, e0124418. [Google Scholar] [CrossRef]
- Mahali, S.; Raviprakash, N.; Raghavendra, P.B.; Manna, S.K. Advanced glycation end products (AGEs) induce apoptosis via a novel pathway: Involvement of Ca2+ mediated by interleukin-8 protein. J. Biol. Chem. 2011, 286, 34903–34913. [Google Scholar] [CrossRef]
- Ahangarpour, A.; Oroojan, A.A.; Khorsandi, L.; Shabani, R.; Mojaddami, S. Preventive effects of betulinic acid on streptozotocinnicotinamide induced diabetic nephropathy in male mouse. J. Nephropathol. 2016, 5, 128–133. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; He, J.; Wang, L.; Su, L.; Lei, L.; Huang, W.; Geng, X.; Zhang, S.; Meng, X.; Zhou, H.; et al. Dapagliflozin aggravates renal injury via promoting gluconeogenesis in db/db mice. Cell Physiol. Biochem. 2018, 45, 1747–1758. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, K.-H.; Guan, S.-S.; Lin, W.-H.; Wu, C.-T.; Sheu, M.-L.; Chiang, C.-K.; Liu, S.-H. Role of Calbindin-D28k in Diabetes-Associated Advanced Glycation End-Products-Induced Renal Proximal Tubule Cell Injury. Cells 2019, 8, 660. https://doi.org/10.3390/cells8070660
Huang K-H, Guan S-S, Lin W-H, Wu C-T, Sheu M-L, Chiang C-K, Liu S-H. Role of Calbindin-D28k in Diabetes-Associated Advanced Glycation End-Products-Induced Renal Proximal Tubule Cell Injury. Cells. 2019; 8(7):660. https://doi.org/10.3390/cells8070660
Chicago/Turabian StyleHuang, Kuo-How, Siao-Syun Guan, Wei-Han Lin, Cheng-Tien Wu, Meei-Ling Sheu, Chih-Kang Chiang, and Shing-Hwa Liu. 2019. "Role of Calbindin-D28k in Diabetes-Associated Advanced Glycation End-Products-Induced Renal Proximal Tubule Cell Injury" Cells 8, no. 7: 660. https://doi.org/10.3390/cells8070660