X-rays Activate Telomeric Homologous Recombination Mediated Repair in Primary Cells

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Irradiation Procedure
2.3. Long-Term Proliferation Assessment
2.4. Growth Curves
2.5. Flow Cytometry
2.6. Collection of Chromosome Spreads
2.7. Telomeric Quantitative FISH (Q-FISH)
2.8. Intracellular Reactive Oxygen Species (ROS) Determination
2.9. N-acetylcysteine (NAC) Administration
2.10. Telomere Dysfunction-Induced Foci (TIFs) Co-Immuno Staining
2.11. Real Time Quantitative–Telomerase Repeat Amplification Protocol Assay (RTQ-TRAP)
2.12. PML/RPA2-Telomere Immunofluorescence-FISH Staining
2.13. Chromosome Orientation-FISH (CO-FISH) Analysis
2.14. Whole Cell Extracts and Western Blotting
2.15. Chromatin Immunoprecipitation (ChIP) Assay and Telomere Dot-Blot
2.16. Statistical Analysis
3. Results
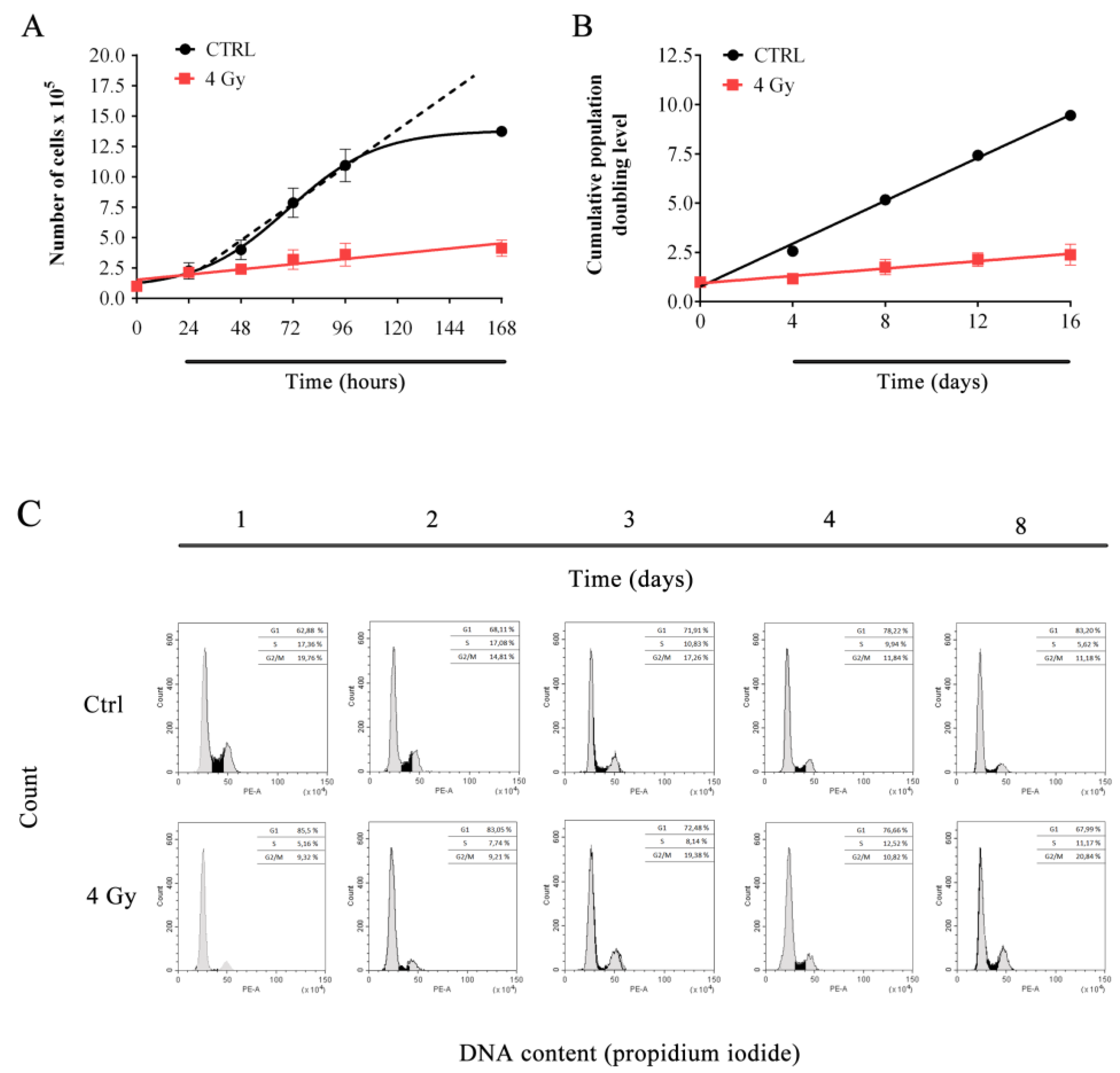
3.1. X-ray Irradiation Affects HFFF2 Proliferation.
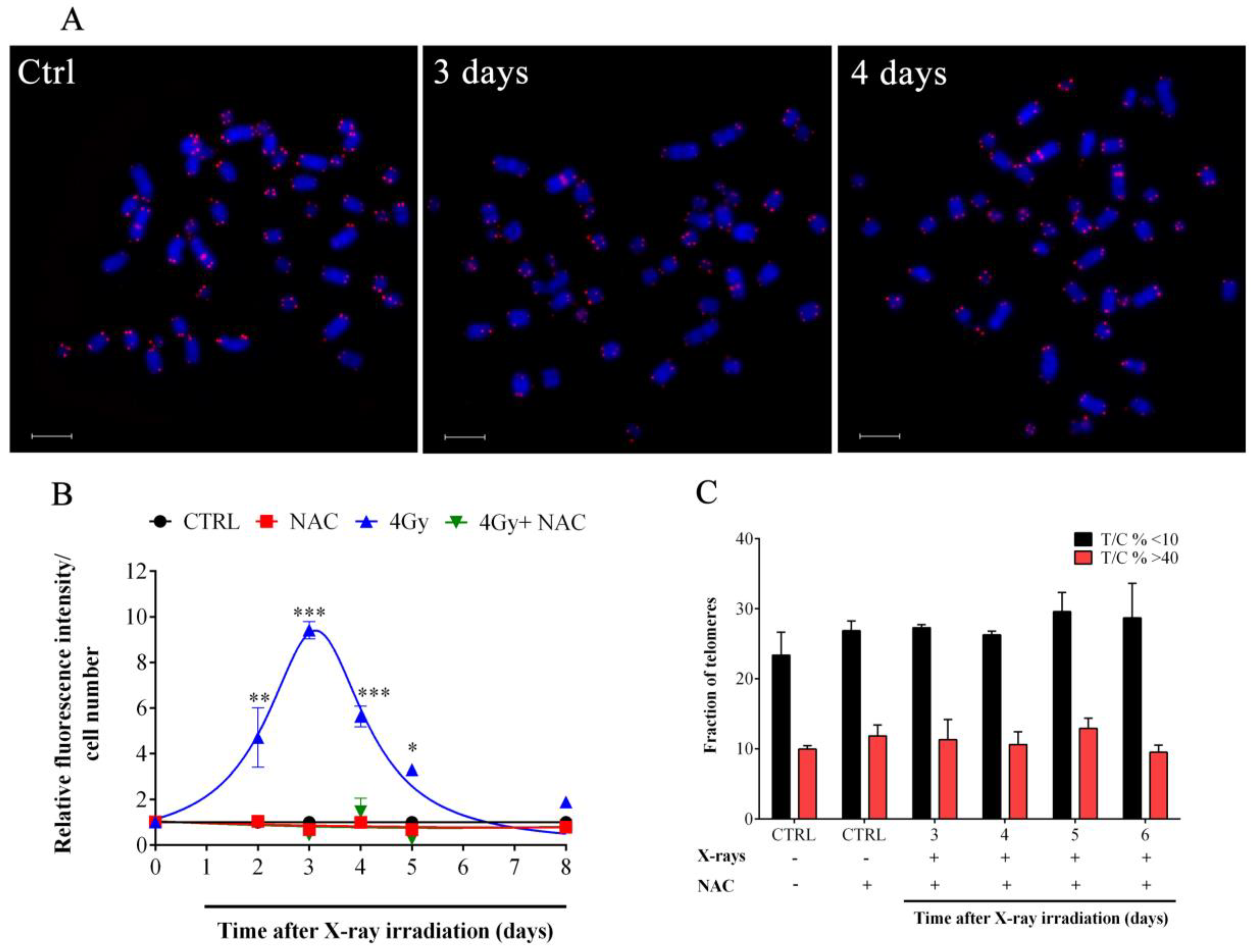
3.2. X-rays Induced Telomere Length Modulation Depends on the Level of IR-Induced OS
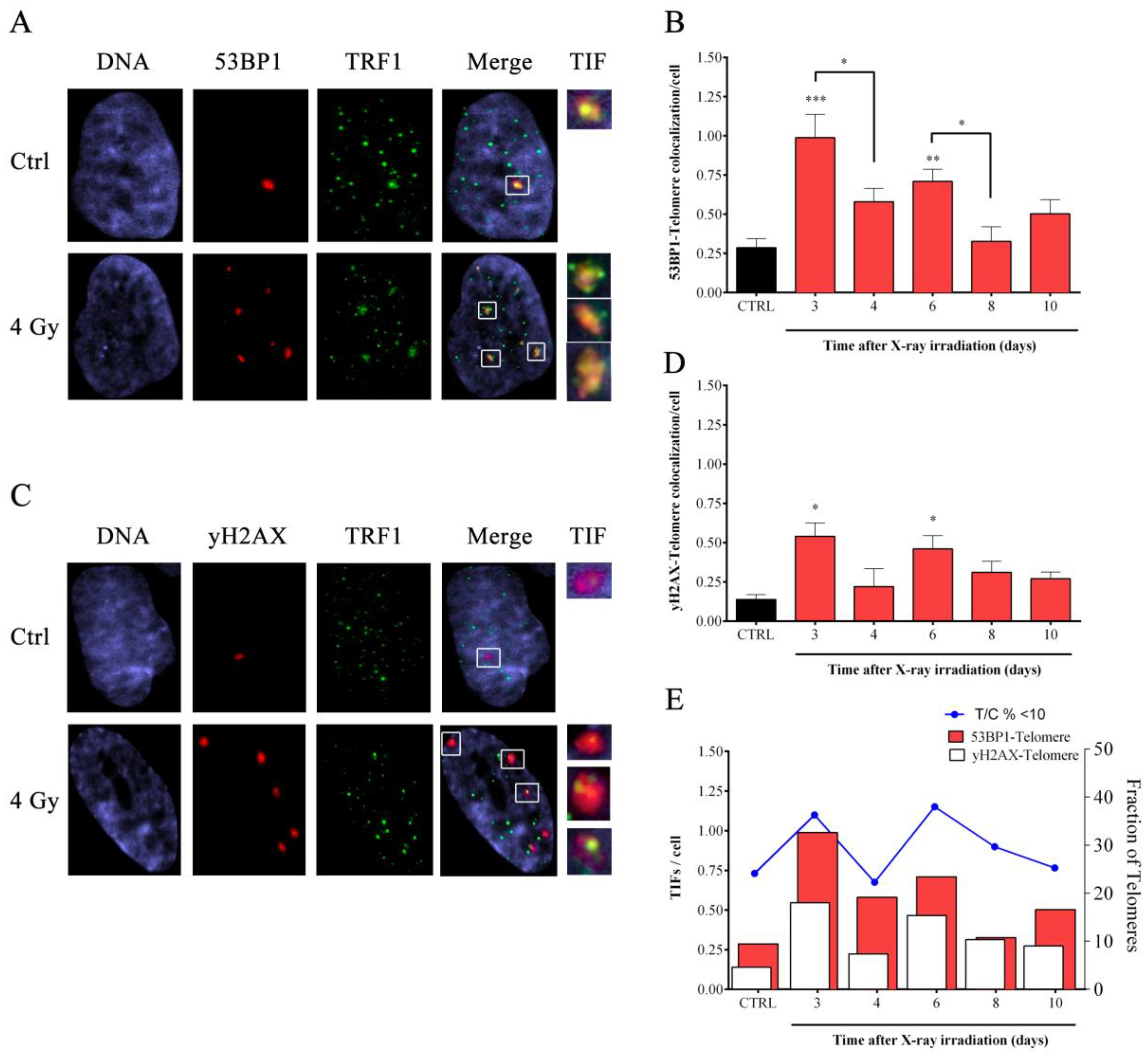
3.3. X-ray-Induced Telomere Damage Is Strictly Correlated to Telomere Shortening
3.4. IR-Induced Telomere Length Modulation Is Not Dependent on Telomerase Reactivation in Fibroblasts
3.5. Induction of APBs and TSCE in Response to X-rays Irradiation
3.6. IR-Dependent Induction of Homologous Recombination Proteins RAD51 and RPA2 Was Coupled to the Suppression of the Chromatin-Remodeling Factor ATRX
3.7. Reduction of Telomeric Histone Modification Mark in Correspondence to Telomeric HR Activation
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALT | Alternative lengthening of telomeres |
| IR | Ionizing radiations |
| HR | Homologous recombination |
| DSB | Double strand break |
| NHEJ | Non-homologous end-joining |
| T-SCE | Telomere-sister chromatid exchange |
| ECTR | Extrachromosomal telomeric repeats |
| PML | Promyelocytic leukemia protein |
| APB | ALT-associated PML (promyelocytic leukemia protein) body |
| DDR | DNA damage response |
| OS | Oxidative stress |
| CPDL | Cumulative population doubling level |
| TIF | Telomere dysfunction-induced foci |
| PCC | Prematurely condensed chromosomes |
References
- Pandita, T.K.; Pathak, S.; Geard, C.R. Chromosome end associations, telomeres and telomerase activity in ataxia telangiectasia cells. Cytogenet. Cell Genet. 1995, 71, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E.H. Switching and signaling at the telomere. Cell 2001, 106, 661–673. [Google Scholar] [CrossRef]
- de Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- Tommerup, H.; Dousmanis, A.; de Lange, T. Unusual chromatin in human telomeres. Mol. Cell. Biol. 1994, 14, 5777–5785. [Google Scholar] [CrossRef] [PubMed]
- Lejnine, S.; Makarov, V.L.; Langmore, J.P. Conserved nucleoprotein structure at the ends of vertebrate and invertebrate chromosomes. Proc. Natl. Acad. Sci. USA 1995, 92, 2393–2397. [Google Scholar] [CrossRef] [PubMed]
- Nikitina, T.; Woodcock, C.L. Closed chromatin loops at the ends of chromosomes. J. Cell Biol. 2004, 166, 161–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broccoli, D.; Smogorzewska, A.; Chong, L.; de Lange, T. Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2. Nat. Genet. 1997, 17, 231–235. [Google Scholar] [CrossRef] [PubMed]
- de Lange, T. Protection of mammalian telomeres. Oncogene 2002, 21, 532–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smogorzewska, A.; de Lange, T. Regulation of telomerase by telomeric proteins. Annu. Rev. Biochem. 2004, 73, 177–208. [Google Scholar] [CrossRef] [PubMed]
- Blasco, M.A. The epigenetic regulation of mammalian telomeres. Nat. Rev. Genet. 2007, 8, 299–309. [Google Scholar] [CrossRef]
- van Steensel, B.; Smogorzewska, A.; de Lange, T. TRF2 protects human telomeres from end-to-end fusions. Cell 1998, 92, 401–413. [Google Scholar] [CrossRef]
- Karlseder, J.; Broccoli, D.; Dai, Y.; Hardy, S.; de Lange, T. p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science 1999, 283, 1321–1325. [Google Scholar] [CrossRef]
- Sfeir, A.; de Lange, T. Removal of shelterin reveals the telomere end-protection problem. Science 2012, 336, 593–597. [Google Scholar] [CrossRef] [PubMed]
- Kruk, P.A.; Rampino, N.J.; Bohr, V.A. DNA damage and repair in telomeres: Relation to aging. Proc. Natl. Acad. Sci. USA 1995, 92, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Petersen, S.; Saretzki, G.; von Zglinicki, T. Preferential accumulation of single-stranded regions in telomeres of human fibroblasts. Exp. Cell Res. 1998, 239, 152–160. [Google Scholar] [CrossRef]
- Mao, P.; Liu, J.; Zhang, Z.; Zhang, H.; Liu, H.; Gao, S.; Rong, Y.S.; Zhao, Y. Homologous recombination-dependent repair of telomeric DSBs in proliferating human cells. Nat. Commun. 2016, 7, 12154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oikawa, S.; Tada-Oikawa, S.; Kawanishi, S. Site-specific DNA damage at the GGG sequence by UVA involves acceleration of telomere shortening. Biochemistry 2001, 40, 4763–4768. [Google Scholar] [CrossRef]
- Londono-Vallejo, J.A.; Der-Sarkissian, H.; Cazes, L.; Bacchetti, S.; Reddel, R.R. Alternative lengthening of telomeres is characterized by high rates of telomeric exchange. Cancer Res. 2004, 64, 2324–2327. [Google Scholar] [CrossRef]
- Cesare, A.J.; Reddel, R.R. Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 2010, 11, 319–330. [Google Scholar] [CrossRef]
- Murnane, J.P.; Sabatier, L.; Marder, B.A.; Morgan, W.F. Telomere dynamics in an immortal human cell line. EMBO J. 1994, 13, 4953–4962. [Google Scholar] [CrossRef]
- Bryan, T.M.; Englezou, A.; Gupta, J.; Bacchetti, S.; Reddel, R.R. Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J. 1995, 14, 4240–4248. [Google Scholar] [CrossRef] [PubMed]
- Cesare, A.J.; Griffith, J.D. Telomeric DNA in ALT cells is characterized by free telomeric circles and heterogeneous t-loops. Mol. Cell. Biol. 2004, 24, 9948–9957. [Google Scholar] [CrossRef] [PubMed]
- Nabetani, A.; Ishikawa, F. Unusual telomeric DNAs in human telomerase-negative immortalized cells. Mol. Cell. Biol. 2009, 29, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Yeager, T.R.; Neumann, A.A.; Englezou, A.; Huschtscha, L.I.; Noble, J.R.; Reddel, R.R. Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res. 1999, 59, 4175–4179. [Google Scholar] [PubMed]
- Tang, J.; Wu, S.; Liu, H.; Stratt, R.; Barak, O.G.; Shiekhattar, R.; Picketts, D.J.; Yang, X. A novel transcription regulatory complex containing death domain-associated protein and the ATR-X syndrome protein. J. Biol. Chem. 2004, 279, 20369–20377. [Google Scholar] [CrossRef] [PubMed]
- Heaphy, C.M.; de Wilde, R.F.; Jiao, Y.; Klein, A.P.; Edil, B.H.; Shi, C.; Bettegowda, C.; Rodriguez, F.J.; Eberhart, C.G.; Hebbar, S.; et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science 2011, 333, 425. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, C.A.; Li, W.; Reisenweber, S.; Thongthip, S.; Bruno, J.; de Lange, T.; De, S.; Petrini, J.H.; Sung, P.A.; Jasin, M.; et al. Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PloS Genet. 2012, 8, e1002772. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tonjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Napier, C.E.; Huschtscha, L.I.; Harvey, A.; Bower, K.; Noble, J.R.; Hendrickson, E.A.; Reddel, R.R. ATRX represses alternative lengthening of telomeres. Oncotarget 2015, 6, 16543–16558. [Google Scholar] [CrossRef] [Green Version]
- Tagami, H.; Ray-Gallet, D.; Almouzni, G.; Nakatani, Y. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 2004, 116, 51–61. [Google Scholar] [CrossRef]
- O’Sullivan, R.J.; Almouzni, G. Assembly of telomeric chromatin to create ALTernative endings. Trends Cell Biol. 2014, 24, 675–685. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, R.J.; Kubicek, S.; Schreiber, S.L.; Karlseder, J. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat. Struct. Mol. Biol. 2010, 17, 1218–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dilley, R.L.; Verma, P.; Cho, N.W.; Winters, H.D.; Wondisford, A.R.; Greenberg, R.A. Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 2016, 539, 54–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobinoff, A.P.; Pickett, H.A. Alternative Lengthening of Telomeres: DNA Repair Pathways Converge. Trends Genet. 2017, 33, 921–932. [Google Scholar] [CrossRef] [PubMed]
- Doksani, Y.; de Lange, T. Telomere-Internal Double-Strand Breaks Are Repaired by Homologous Recombination and PARP1/Lig3-Dependent End-Joining. Cell Rep. 2016, 17, 1646–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, N.W.; Dilley, R.L.; Lampson, M.A.; Greenberg, R.A. Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell 2014, 159, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Berardinelli, F.; Antoccia, A.; Cherubini, R.; De Nadal, V.; Gerardi, S.; Cirrone, G.A.; Tanzarella, C.; Sgura, A. Transient activation of the ALT pathway in human primary fibroblasts exposed to high-LET radiation. Radiat. Res. 2010, 174, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Gnocchi, D.; Leoni, S.; Incerpi, S.; Bruscalupi, G. 3,5,3′-triiodothyronine (T3) stimulates cell proliferation through the activation of the PI3K/Akt pathway and reactive oxygen species (ROS) production in chick embryo hepatocytes. Steroids 2012, 77, 589–595. [Google Scholar] [CrossRef]
- Wege, H.; Chui, M.S.; Le, H.T.; Tran, J.M.; Zern, M.A. SYBR Green real-time telomeric repeat amplification protocol for the rapid quantification of telomerase activity. Nucleic Acids Res. 2003, 31, E3. [Google Scholar] [CrossRef]
- Bailey, S.M.; Goodwin, E.H.; Cornforth, M.N. Strand-specific fluorescence in situ hybridization: The CO-FISH family. Cytogenet. Genome Res. 2004, 107, 14–17. [Google Scholar] [CrossRef]
- Coluzzi, E.; Leone, S.; Sgura, A. Oxidative Stress Induces Telomere Dysfunction and Senescence by Replication Fork Arrest. Cells 2019, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Benetti, R.; Garcia-Cao, M.; Blasco, M.A. Telomere length regulates the epigenetic status of mammalian telomeres and subtelomeres. Nat. Genet. 2007, 39, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Sgura, A.; Antoccia, A.; Berardinelli, F.; Cherubini, R.; Gerardi, S.; Zilio, C.; Tanzarella, C. Telomere length in mammalian cells exposed to low- and high-LET radiations. Radiat. Prot. Dosim. 2006, 122, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Berardinelli, F.; Nieri, D.; Sgura, A.; Tanzarella, C.; Antoccia, A. Telomere loss, not average telomere length, confers radiosensitivity to TK6-irradiated cells. Mutat. Res. 2012, 740, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Berardinelli, F.; Antoccia, A.; Buonsante, R.; Gerardi, S.; Cherubini, R.; De Nadal, V.; Tanzarella, C.; Sgura, A. The role of telomere length modulation in delayed chromosome instability induced by ionizing radiation in human primary fibroblasts. Environ. Mol. Mutagenesis 2013, 54, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Opresko, P.L.; Fan, J.; Danzy, S.; Wilson, D.M., 3rd; Bohr, V.A. Oxidative damage in telomeric DNA disrupts recognition by TRF1 and TRF2. Nucleic Acids Res. 2005, 33, 1230–1239. [Google Scholar] [CrossRef]
- von Zglinicki, T. Role of oxidative stress in telomere length regulation and replicative senescence. Ann. N. Y. Acad. Sci. 2000, 908, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Conomos, D.; Pickett, H.A.; Reddel, R.R. Alternative lengthening of telomeres: Remodeling the telomere architecture. Front. Oncol. 2013, 3, 27. [Google Scholar] [CrossRef]
- Morrish, T.A.; Greider, C.W. Short telomeres initiate telomere recombination in primary and tumor cells. PloS Genet. 2009, 5, e1000357. [Google Scholar] [CrossRef]
- Neumann, A.A.; Watson, C.M.; Noble, J.R.; Pickett, H.A.; Tam, P.P.; Reddel, R.R. Alternative lengthening of telomeres in normal mammalian somatic cells. Genes Dev. 2013, 27, 18–23. [Google Scholar] [CrossRef] [Green Version]
- McEachern, M.J.; Haber, J.E. Break-induced replication and recombinational telomere elongation in yeast. Annu. Rev. Biochem. 2006, 75, 111–135. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beausejour, C.M.; et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 2012, 14, 355–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, J.F. Nature of Lesions Formed by Ionizing Radiation; Humana Press: Totowa, NJ, USA, 1998; pp. 65–84. [Google Scholar] [CrossRef]
- Coluzzi, E.; Colamartino, M.; Cozzi, R.; Leone, S.; Meneghini, C.; O’Callaghan, N.; Sgura, A. Oxidative stress induces persistent telomeric DNA damage responsible for nuclear morphology change in mammalian cells. PloS ONE 2014, 9, e110963. [Google Scholar] [CrossRef] [PubMed]
- von Zglinicki, T.; Pilger, R.; Sitte, N. Accumulation of single-strand breaks is the major cause of telomere shortening in human fibroblasts. Free Radic. Biol. Med. 2000, 28, 64–74. [Google Scholar] [CrossRef]
- Rugo, R.E.; Secretan, M.B.; Schiestl, R.H. X radiation causes a persistent induction of reactive oxygen species and a delayed reinduction of TP53 in normal human diploid fibroblasts. Radiat. Res. 2002, 158, 210–219. [Google Scholar] [CrossRef]
- Kobashigawa, S.; Suzuki, K.; Yamashita, S. Ionizing radiation accelerates Drp1-dependent mitochondrial fission, which involves delayed mitochondrial reactive oxygen species production in normal human fibroblast-like cells. Biochem. Biophys. Res. Commun. 2011, 414, 795–800. [Google Scholar] [CrossRef]
- Dettmering, T.; Zahnreich, S.; Colindres-Rojas, M.; Durante, M.; Taucher-Scholz, G.; Fournier, C. Increased effectiveness of carbon ions in the production of reactive oxygen species in normal human fibroblasts. J. Radiat. Res. 2015, 56, 67–76. [Google Scholar] [CrossRef]
- Hu, Y.; Shi, G.; Zhang, L.; Li, F.; Jiang, Y.; Jiang, S.; Ma, W.; Zhao, Y.; Songyang, Z.; Huang, J. Switch telomerase to ALT mechanism by inducing telomeric DNA damages and dysfunction of ATRX and DAXX. Sci. Rep. 2016, 6, 32280. [Google Scholar] [CrossRef]
- Liu, H.; Xie, Y.; Zhang, Z.; Mao, P.; Liu, J.; Ma, W.; Zhao, Y. Telomeric Recombination Induced by DNA Damage Results in Telomere Extension and Length Heterogeneity. Neoplasia 2018, 20, 905–916. [Google Scholar] [CrossRef]
- Tanaka, H.; Mendonca, M.S.; Bradshaw, P.S.; Hoelz, D.J.; Malkas, L.H.; Meyn, M.S.; Gilley, D. DNA damage-induced phosphorylation of the human telomere-associated protein TRF2. Proc. Natl. Acad. Sci. USA 2005, 102, 15539–15544. [Google Scholar] [CrossRef] [Green Version]
- Huda, N.; Tanaka, H.; Mendonca, M.S.; Gilley, D. DNA damage-induced phosphorylation of TRF2 is required for the fast pathway of DNA double-strand break repair. Mol. Cell. Biol. 2009, 29, 3597–3604. [Google Scholar] [CrossRef] [PubMed]
- Karlseder, J.; Hoke, K.; Mirzoeva, O.K.; Bakkenist, C.; Kastan, M.B.; Petrini, J.H.; de Lange, T. The telomeric protein TRF2 binds the ATM kinase and can inhibit the ATM-dependent DNA damage response. PloS Biol. 2004, 2, E240. [Google Scholar] [CrossRef] [PubMed]
- Denchi, E.L.; de Lange, T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 2007, 448, 1068–1071. [Google Scholar] [CrossRef] [PubMed]
- Sfeir, A.; Denchi, E.L. Stressed telomeres without POT1 enhance tumorigenesis. Oncotarget 2016, 7, 46833–46834. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.L.; Cox, K.E.; Jeitany, M.; Wakimoto, H.; Bryll, A.R.; Ganem, N.J.; Bersani, F.; Pineda, J.R.; Suva, M.L.; Benes, C.H.; et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 2015, 347, 273–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clynes, D.; Jelinska, C.; Xella, B.; Ayyub, H.; Scott, C.; Mitson, M.; Taylor, S.; Higgs, D.R.; Gibbons, R.J. Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat. Commun. 2015, 6, 7538. [Google Scholar] [CrossRef]
- Coluzzi, E.; Buonsante, R.; Leone, S.; Asmar, A.J.; Miller, K.L.; Cimini, D.; Sgura, A. Transient ALT activation protects human primary cells from chromosome instability induced by low chronic oxidative stress. Sci. Rep. 2017, 7, 43309. [Google Scholar] [CrossRef]
- Russell, J.S.; Brady, K.; Burgan, W.E.; Cerra, M.A.; Oswald, K.A.; Camphausen, K.; Tofilon, P.J. Gleevec-mediated inhibition of Rad51 expression and enhancement of tumor cell radiosensitivity. Cancer Res. 2003, 63, 7377–7383. [Google Scholar]
- Lee, E.S.; Won, Y.J.; Kim, B.C.; Park, D.; Bae, J.H.; Park, S.J.; Noh, S.J.; Kang, Y.R.; Choi, S.H.; Yoon, J.H.; et al. Low-dose irradiation promotes Rad51 expression by down-regulating miR-193b-3p in hepatocytes. Sci. Rep. 2016, 6, 25723. [Google Scholar] [CrossRef]
- Bo, H.; Ghazizadeh, M.; Shimizu, H.; Kurihara, Y.; Egawa, S.; Moriyama, Y.; Tajiri, T.; Kawanami, O. Effect of ionizing irradiation on human esophageal cancer cell lines by cDNA microarray gene expression analysis. J. Nippon Med. Sch. = Nippon Ika Daigaku Zasshi 2004, 71, 172–180. [Google Scholar] [CrossRef]
- Caslini, C.; Connelly, J.A.; Serna, A.; Broccoli, D.; Hess, J.L. MLL associates with telomeres and regulates telomeric repeat-containing RNA transcription. Mol. Cell. Biol. 2009, 29, 4519–4526. [Google Scholar] [CrossRef] [PubMed]
- Gottschling, D.E. Telomere-proximal DNA in Saccharomyces cerevisiae is refractory to methyltransferase activity in vivo. Proc. Natl. Acad. Sci. USA 1992, 89, 4062–4065. [Google Scholar] [CrossRef] [PubMed]
- Boivin, A.; Dura, J.M. In vivo chromatin accessibility correlates with gene silencing in Drosophila. Genetics 1998, 150, 1539–1549. [Google Scholar] [PubMed]
- Murr, R.; Loizou, J.I.; Yang, Y.G.; Cuenin, C.; Li, H.; Wang, Z.Q.; Herceg, Z. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Shen, X. Chromatin remodeling in DNA double-strand break repair. Curr. Opin. Genet. Dev. 2007, 17, 126–131. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Vitis, M.; Berardinelli, F.; Coluzzi, E.; Marinaccio, J.; O’Sullivan, R.J.; Sgura, A. X-rays Activate Telomeric Homologous Recombination Mediated Repair in Primary Cells. Cells 2019, 8, 708. https://doi.org/10.3390/cells8070708
De Vitis M, Berardinelli F, Coluzzi E, Marinaccio J, O’Sullivan RJ, Sgura A. X-rays Activate Telomeric Homologous Recombination Mediated Repair in Primary Cells. Cells. 2019; 8(7):708. https://doi.org/10.3390/cells8070708
Chicago/Turabian StyleDe Vitis, Marco, Francesco Berardinelli, Elisa Coluzzi, Jessica Marinaccio, Roderick J. O’Sullivan, and Antonella Sgura. 2019. "X-rays Activate Telomeric Homologous Recombination Mediated Repair in Primary Cells" Cells 8, no. 7: 708. https://doi.org/10.3390/cells8070708