The Intrinsic GDP/GTP Exchange Activities of Cdc42 and Rac1 Are Critical Determinants for Their Specific Effects on Mobilization of the Actin Filament System

Abstract
:1. Introduction
2. Experimental
2.1. Mutant Small GTPases
2.2. Antibodies and Reagents
2.3. Cell Cultivation and Transfection
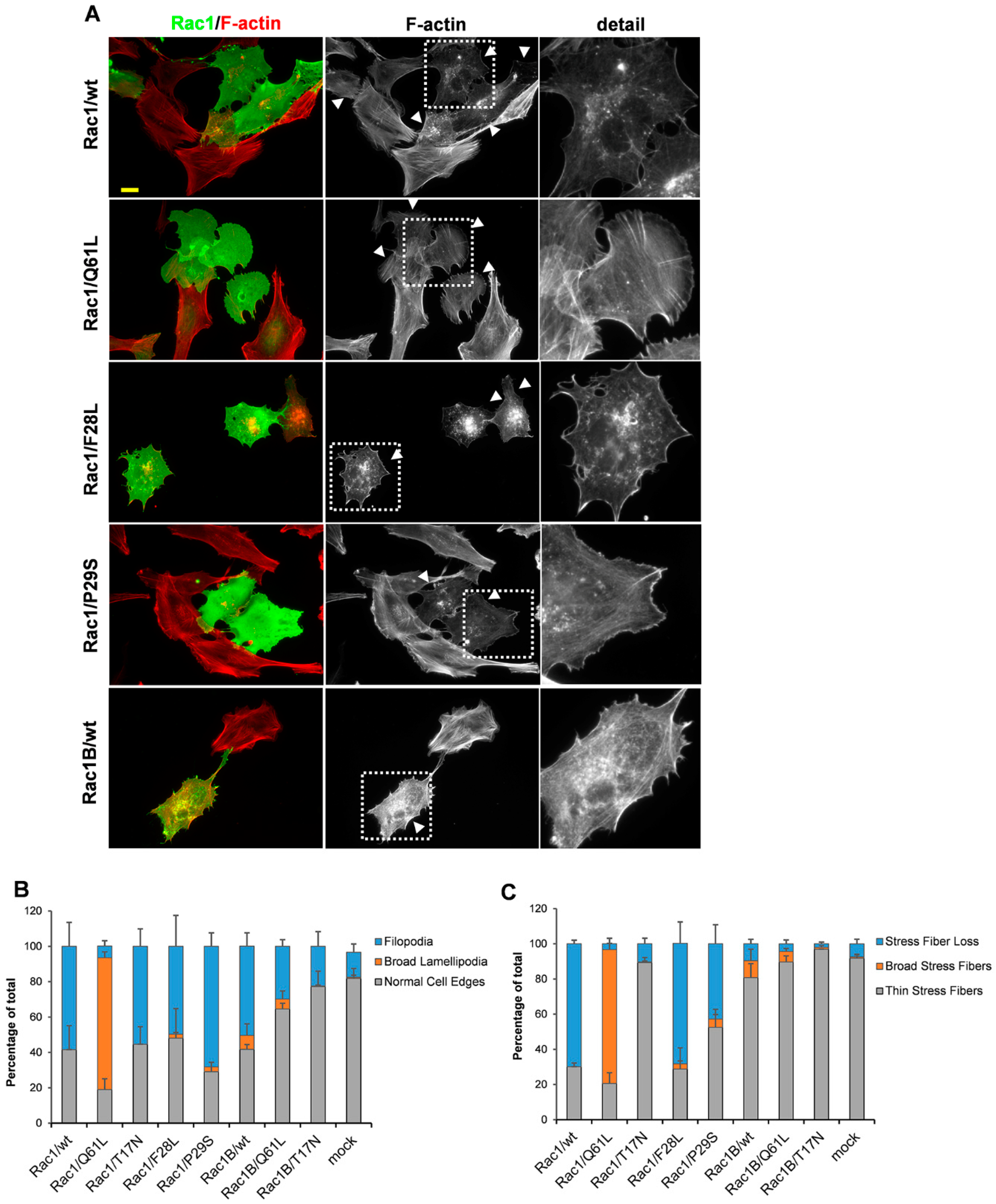
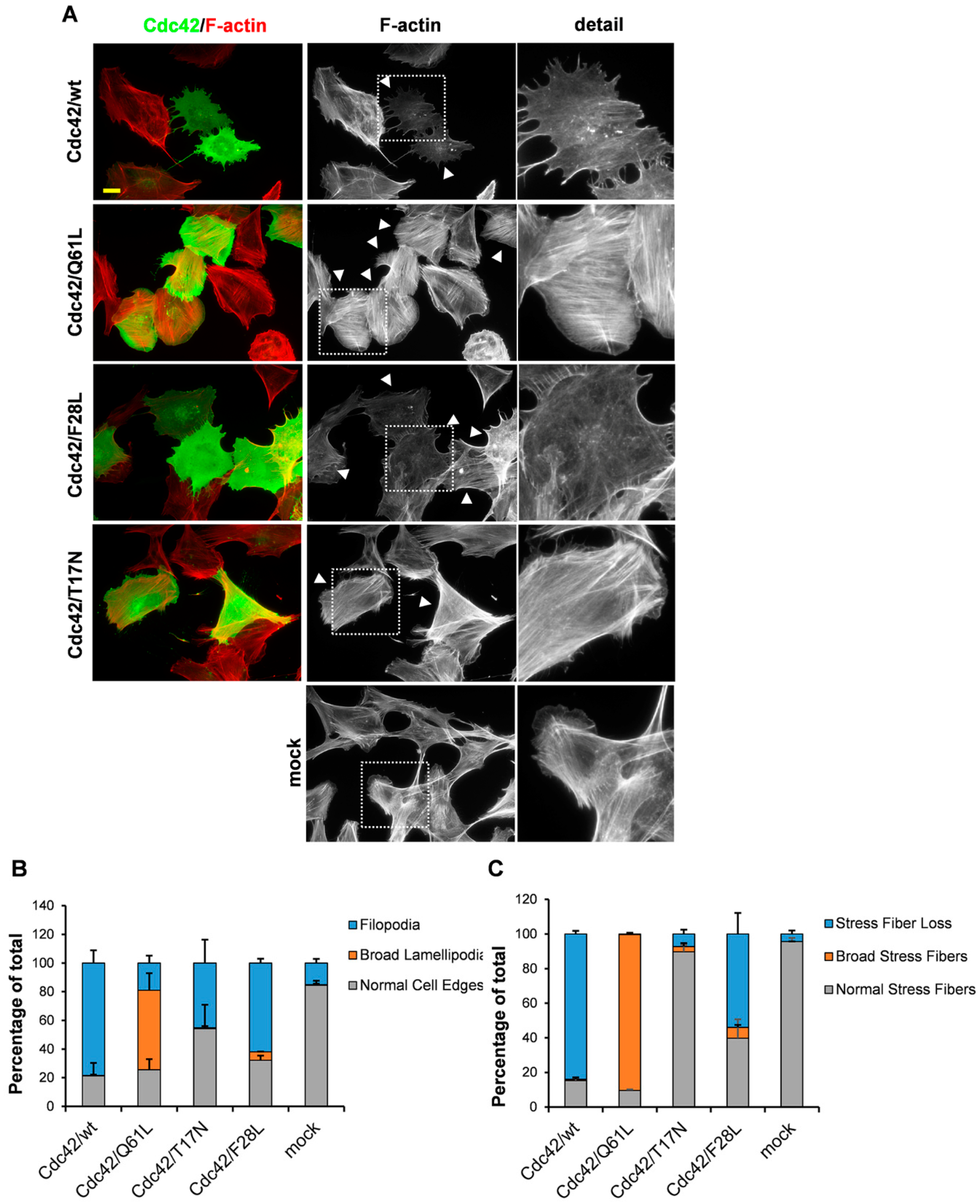
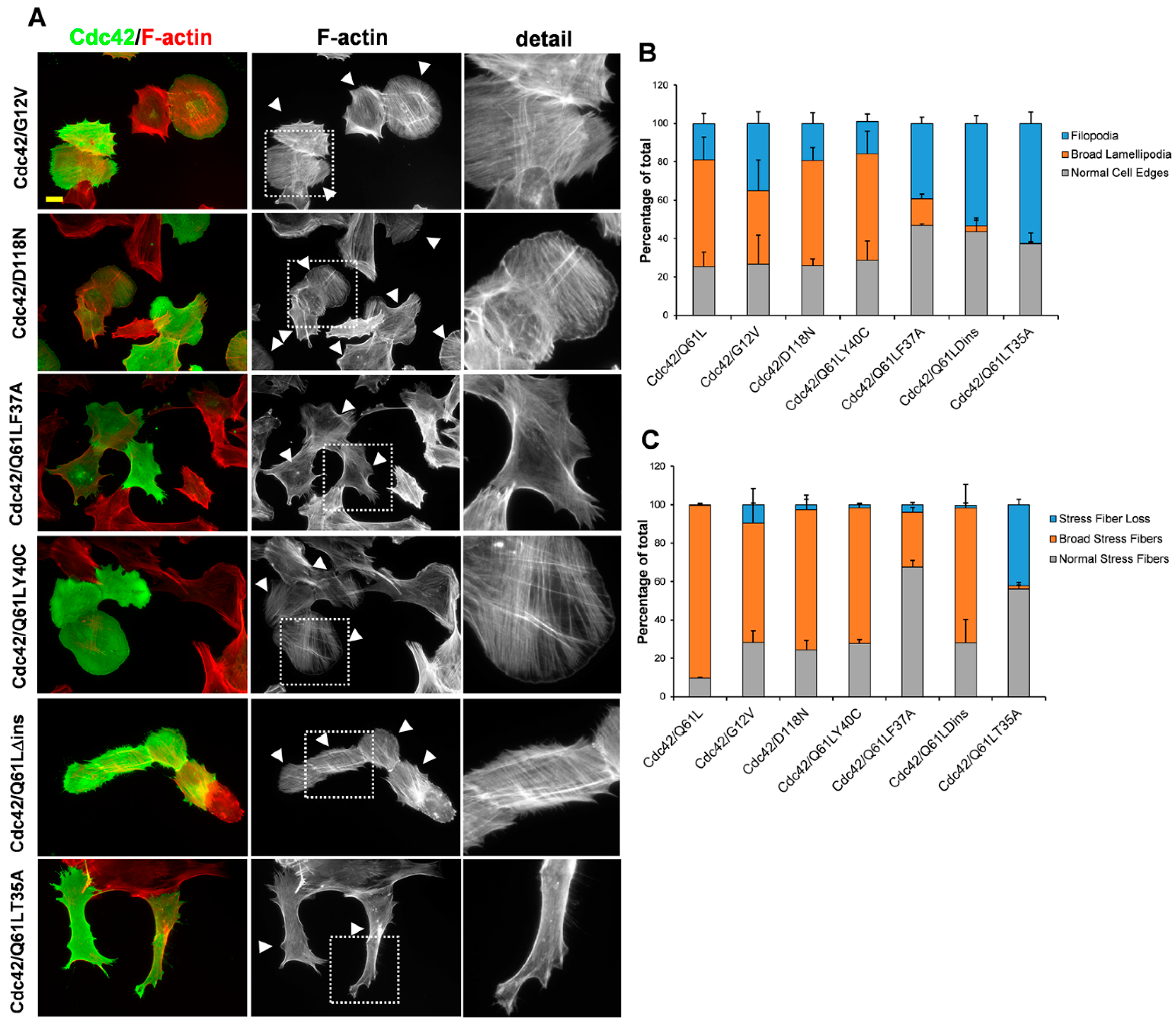
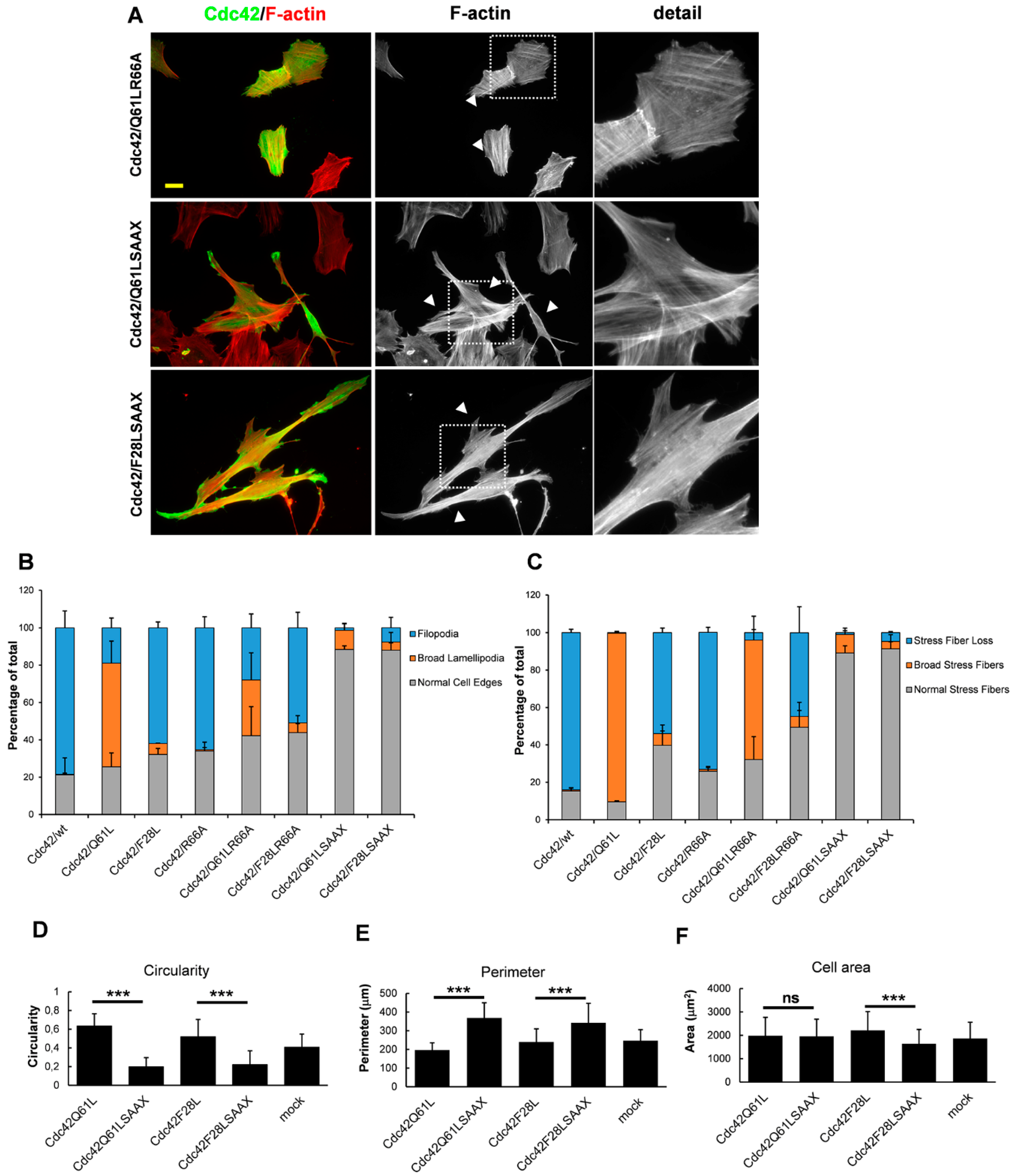
3. Results
3.1. An Intact GDP/GTP Exchange Activity is the Basis for Cdc42-Induced Filopodia Formation
3.2. The Role of the Effector Loop in Cdc42-Induced Filopodia Formation
3.3. RhoGDI Binding Is Not Necessary for Cdc42-Dependent Actin Reorganization
3.4. Membrane Targeting Is Necessary for Cdc42-Dependent Actin Reorganization
3.5. Rac1 Mutants with Elevated Intrinsic GDP/GTP Exchange Activities Induce Filopodia
3.6. The Involvement of Formins and Arp2/3 in Cdc42- and Rac1-Induced Actin Reorganization
4. Discussion
Supplementary Materials
Funding
Conflicts of Interest
References
- Ridley, A.; Hall, A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 1992, 70, 389–399. [Google Scholar] [CrossRef]
- Ridley, A.J.; Paterson, H.F.; Johnston, C.L.; Diekmann, D.; Hall, A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 1992, 70, 401–410. [Google Scholar] [CrossRef]
- Nobes, C.D.; Hall, A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 1995, 81, 53–62. [Google Scholar] [CrossRef]
- Aspenström, P.; Fransson, Å.; Saras, J.; Aspenstrm, P. Rho GTPases have diverse effects on the organization of the actin filament system. Biochem. J. 2004, 377, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Colicelli, J. Human RAS Superfamily Proteins and Related GTPases. Sci. STKE 2004, 2004, re13. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Der, C.J. Ras history: The saga continues. Small GTPases 2010, 1, 2–27. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.R.; Rossman, K.L.; Der, C.J. Rho guanine nucleotide exchange factors: Regulators of Rho GTPase activity in development and disease. Oncogene 2014, 33, 4021–4035. [Google Scholar] [CrossRef]
- Tcherkezian, J.; Lamarche-Vane, N.; Lamarche-Vane, N. Current knowledge of the large RhoGAP family of proteins. Biol. Cell 2007, 99, 67–86. [Google Scholar] [CrossRef]
- Xie, F.; Shao, S.; Aziz, A.U.R.; Zhang, B.; Wang, H.; Liu, B. Role of Rho-specific guanine nucleotide dissociation inhibitor α regulation in cell migration. Acta Histochem. 2017, 119, 183–189. [Google Scholar] [CrossRef]
- Chardin, P. Function and regulation of Rnd proteins. Nat. Rev. Mol. Cell Biol. 2006, 7, 54–62. [Google Scholar] [CrossRef]
- Aspenström, P. Fast-cycling Rho GTPases. Small GTPases 2018, 1–8. Available online: https://www.tandfonline.com/doi/full/10.1080/21541248.2017.1391365 (accessed on 17 July 2019).
- Traut, T.W. Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 1994, 140, 1–22. [Google Scholar] [CrossRef]
- Boureux, A.; Vignal, E.; Faure, S.; Fort, P. Evolution of the Rho family of Ras-like GTPases in eukaryotes. Mol. Biol. Evol. 2007, 24, 203–216. [Google Scholar] [CrossRef]
- Krengel, U.; Schlichting, I.; Scherer, A.; Schümann, R.; Frech, M.; John, J.; Kabsch, W.; Pai, E.F.; Wittinghofer, A. Three-dimensional structures of H-ras p21 mutants: Molecular basis for their inability to function as signal switch molecules. Cell 1990, 62, 539–548. [Google Scholar] [CrossRef]
- Lin, R.; Bagrodia, S.; Cerione, R.; Manor, D. A novel Cdc42Hs mutant induces cellular transformation. Curr. Biol. 1997, 7, 794–797. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Wang, Y.; Barry, D.C.; Chanock, S.J.; Bokoch, G.M. Guanine Nucleotide Binding Properties of Rac2 Mutant Proteins and Analysis of the Responsiveness to Guanine Nucleotide Dissociation Stimulator. Biochemistry 1997, 36, 626–632. [Google Scholar] [CrossRef]
- Reinstein, J.; Schlichting, I.; Frech, M.; Goody, R.S.; Wittinghofer, A. p21 with a phenylalanine 28—leucine mutation reacts normally with the GTPase activating protein GAP but nevertheless has transforming properties. J. Biol. Chem. 1991, 266, 17700–17706. [Google Scholar]
- Jordan, P.; Brazåo, R.; Boavida, M.G.; Gespach, C.; Chastre, E. Cloning of a novel human Rac1b splice variant with increased expression in colorectal tumors. Oncogene 1999, 18, 6835–6839. [Google Scholar] [CrossRef] [Green Version]
- Schnelzer, A.; Prechtel, D.; Knaus, U.; Dehne, K.; Gerhard, M.; Graeff, H.; Harbeck, N.; Schmitt, M.; Lengyel, E. Rac1 in human breast cancer: Overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene 2000, 19, 3013–3020. [Google Scholar] [CrossRef]
- Fiegen, D.; Haeusler, L.C.; Blumenstein, L.; Herbrand, U.; Dvorský, R.; Vetter, I.R.; Ahmadian, M.R. Alternative splicing of Rac1 generates Rac1b, a self-activating GTPase. J. Biol. Chem. 2004, 2004, 4743–4749. [Google Scholar] [CrossRef]
- Matos, P.; Collard, J.G.; Jordan, P. Tumor-related Alternatively Spliced Rac1b Is Not Regulated by Rho-GDP Dissociation Inhibitors and Exhibits Selective Downstream Signaling. J. Biol. Chem. 2003, 278, 50442–50448. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Karnoub, A.E.; Palmby, T.R.; Lengyel, E.; Sondek, J.; Der, C.J. Rac1b, a tumor associated, constitutively active Rac1 splice variant, promotes cellular transformation. Oncogene 2004, 23, 9369–9380. [Google Scholar] [CrossRef] [Green Version]
- Hall, A. Rho family GTPases. Biochem. Soc. Trans. 2012, 40, 1378–1382. [Google Scholar] [CrossRef] [Green Version]
- Kozma, R.; Ahmed, S.; Best, A.; Lim, L. The Ras-related protein Cdc42Hs and bradykinin promote formation of peripheral actin microspikes and filopodia in Swiss 3T3 fibroblasts. Mol. Cell. Biol. 1995, 15, 1942–1952. [Google Scholar] [CrossRef] [Green Version]
- Tu, S.S.; Wu, W.J.; Yang, W.; Nolbant, P.; Hahn, K.; Cerione, R.A. Antiapoptotic Cdc42 Mutants Are Potent Activators of Cellular Transformation. Biochemistry 2002, 41, 12350–12358. [Google Scholar] [CrossRef]
- Lamarche, N.; Tapon, N.; Stowers, L.; Burbelo, P.D.; Aspenström, P.; Bridges, T.; Chant, J.; Hall, A. Rac and Cdc42 Induce Actin Polymerization and G1 Cell Cycle Progression Independently of p65PAK and the JNK/SAPK MAP Kinase Cascade. Cell 1996, 87, 519–529. [Google Scholar] [CrossRef] [Green Version]
- Abo, A.; Freeman, J.L.; Lambeth, J.D. Rac “Insert Region” Is a Novel Effector Region That Is Implicated in the Activation of NADPH Oxidase, but Not PAK65. J. Biol. Chem. 1996, 271, 19794–19801. [Google Scholar]
- Wu, W.-J.; Leonard, D.A.; A-Cerione, R.; Manor, D. Interaction between Cdc42Hs and RhoGDI Is Mediated through the Rho Insert Region. J. Biol. Chem. 1997, 272, 26153–26158. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; Fuji, R.N.; Yang, W.; Cerione, R.A. RhoGDI Is Required for Cdc42-Mediated Cellular Transformation. Curr. Biol. 2003, 13, 1469–1479. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Liao, J.; Yang, G.Y. CAAX-box protein, prenylation process and carcinogenesis. Am. J. Transl. Res. 2009, 1, 312–325. [Google Scholar]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, S.A.; Neidt, E.M.; Cui, J.; Feiger, Z.; Skau, C.T.; Gardel, M.L.; Kozmin, S.A.; Kovar, D.R. Identification and Characterization of a Small Molecule Inhibitor of Formin-Mediated Actin Assembly. Chem. Biol. 2009, 16, 1158–1168. [Google Scholar] [CrossRef] [Green Version]
- Hetrick, B.; Han, M.S.; Helgeson, L.A.; Nolen, B.J. Small molecules CK-666 and CK-869 inhibit Arp2/3 complex by blocking an activating conformational change. Chem. Biol. 2013, 20, 701–712. [Google Scholar] [CrossRef]
- Czuchra, A.; Wu, X.; Meyer, H.; Van Hengel, J.; Schroeder, T.; Geffers, R.; Rottner, K.; Brakebusch, C.; Ginsberg, M. Cdc42 Is Not Essential for Filopodium Formation, Directed Migration, Cell Polarization, and Mitosis in Fibroblastoid Cells. Mol. Biol. Cell 2005, 16, 4473–4484. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Wang, L.; Zheng, Y. Gene Targeting of Cdc42 and Cdc42GAP Affirms the Critical Involvement of Cdc42 in Filopodia Induction, Directed Migration, and Proliferation in Primary Mouse Embryonic Fibroblasts. Mol. Biol. Cell 2006, 17, 4675–4685. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Dickerson, J.B.; Guo, F.; Zheng, J.; Zheng, Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 7618–7623. [Google Scholar] [CrossRef] [Green Version]
- Surviladze, Z.; Waller, A.; Strouse, J.J.; Bologa, C.; Ursu, O.; Salas, V.; Parkinson, J.F.; Phillips, G.K.; Romero, E.; Wandinger-Ness, A.; et al. A Potent and Selective Inhibitor of Cdc42 GTPase. In Probe Reports from the NIH Molecular Libraries Program [Internet]; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2010. [Google Scholar]
- Owen, D.; Mott, H.R.; Laue, E.D.; Lowe, P.N. Residues in Cdc42 That Specify Binding to Individual CRIB Effector Proteins. Biochemistry 2000, 39, 1243–1250. [Google Scholar] [CrossRef]
- Spoerner, M.; Herrmann, C.; Wittinghofer, A.; Vetter, I.R.; Kalbitzer, H.R. Dynamic properties of the Ras switch I region and its importance for binding to effectors. Proc. Natl. Acad. Sci. USA 2001, 98, 4944–4949. [Google Scholar] [CrossRef] [Green Version]
- McCallum, S.J.; Wu, W.J.; Cerione, R.A. Identification of a Putative Effector for Cdc42Hs with High Sequence Similarity to the RasGAP-related Protein IQGAP1 and a Cdc42Hs Binding Partner with Similarity to IQGAP2. J. Biol. Chem. 1996, 271, 18825–18830. [Google Scholar] [CrossRef]
- Derivery, E.; Gautreau, A. Generation of branched actin networks: Assembly and regulation of the N-WASP and WAVE molecular machines. Bioessays 2010, 32, 119–131. [Google Scholar] [CrossRef]
- Kuhn, S.; Geyer, M. Formins as effector proteins of Rho GTPases. Small GTPases 2014, 5, e983876. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A. Cdc42 and Rac1 variants used in the study | ||
| Small GTPase | Mutant | Phenotype |
| Cdc42 | Q61L | GTPase defective |
| G12V | GTPase defective | |
| T17N | Dominant negative (nucleotide binding-defective) | |
| F28L | Fast-cycling (increased GDP/GTP exchange) | |
| D118N | Elevated GDP/GTP exchange | |
| R66A | RhoGDI binding-defective | |
| T35A | Effector loop mutant | |
| F37A | Effector loop mutant | |
| Y40C | Effector loop mutant | |
| Δins | Insert domain mutant | |
| SAAX | CAAX box mutant | |
| Rac1 | Q61L | GTPase defective |
| T17N | Dominant negative (nucleotide binding- defective) | |
| F28L | Fast-cycling (increased GDP/GTP exchange) | |
| P29S | Fast-cycling (increased GDP/GTP exchange). Cancer mutation | |
| Rac1B | Fast-cycling (increased GDP/GTP exchange). Cancer mutation | |
| B. Inhibitors used in the study | ||
| Inhibitor | Concentration Used | Targeted Pathway |
| GGTI298 | 10 μM | Inhibitor of geranylgeranylation |
| FFT277 | 10 μM | Inhibitor of farnesylation |
| 2-bromopalmitate (2-BP) | 100 μM | Inhibitor of palmitoylation |
| SU6656 | 2 μM | Inhibitor of Src family kinases |
| LY294002 | 10 μM | Inhibitor of PI3 kinases |
| Y27632 | 10 μM | Inhibitor of Rho kinase (ROCK) |
| NSC23766 | 30 μM | Inhibitor of Rac |
| ML-141 | 10 μM | Inhibitor of Cdc42 |
| SMIFH2 | 30 μM | Inhibitor of formins |
| CK-666 | 100 μM | Inhibitor of Arp2/3 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aspenström, P. The Intrinsic GDP/GTP Exchange Activities of Cdc42 and Rac1 Are Critical Determinants for Their Specific Effects on Mobilization of the Actin Filament System. Cells 2019, 8, 759. https://doi.org/10.3390/cells8070759
Aspenström P. The Intrinsic GDP/GTP Exchange Activities of Cdc42 and Rac1 Are Critical Determinants for Their Specific Effects on Mobilization of the Actin Filament System. Cells. 2019; 8(7):759. https://doi.org/10.3390/cells8070759
Chicago/Turabian StyleAspenström, Pontus. 2019. "The Intrinsic GDP/GTP Exchange Activities of Cdc42 and Rac1 Are Critical Determinants for Their Specific Effects on Mobilization of the Actin Filament System" Cells 8, no. 7: 759. https://doi.org/10.3390/cells8070759