Loss of Protein Kinase Csnk2b/CK2β at Neuromuscular Junctions Affects Morphology and Dynamics of Aggregated Nicotinic Acetylcholine Receptors, Neuromuscular Transmission, and Synaptic Gene Expression


 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice Strains
2.2. Grid Test, Treadmill Performance
2.3. Nerve-Muscle Preparations and Extracellular Recordings
2.4. Intracellular Recordings and Data Analysis
2.5. Quantitative 3D Morphometrical Imaging
2.6. Tissue Sections, Immunohistochemistry
2.7. Tissue Culture, Culturing of Primary Muscle Cells, Transfection
2.8. DNA and RNA Preparation, Reverse Transcription, PCR
2.9. In Vivo Microscopy and Analysis of AChR Turnover and NMJ Fragmentation
2.10. Statistical Analysis
3. Results
3.1. The Ablation of CKβ in Skeletal Muscle Fibers of Mice Results in Impaired Neuromuscular Transmission and Reduced Grip Strength
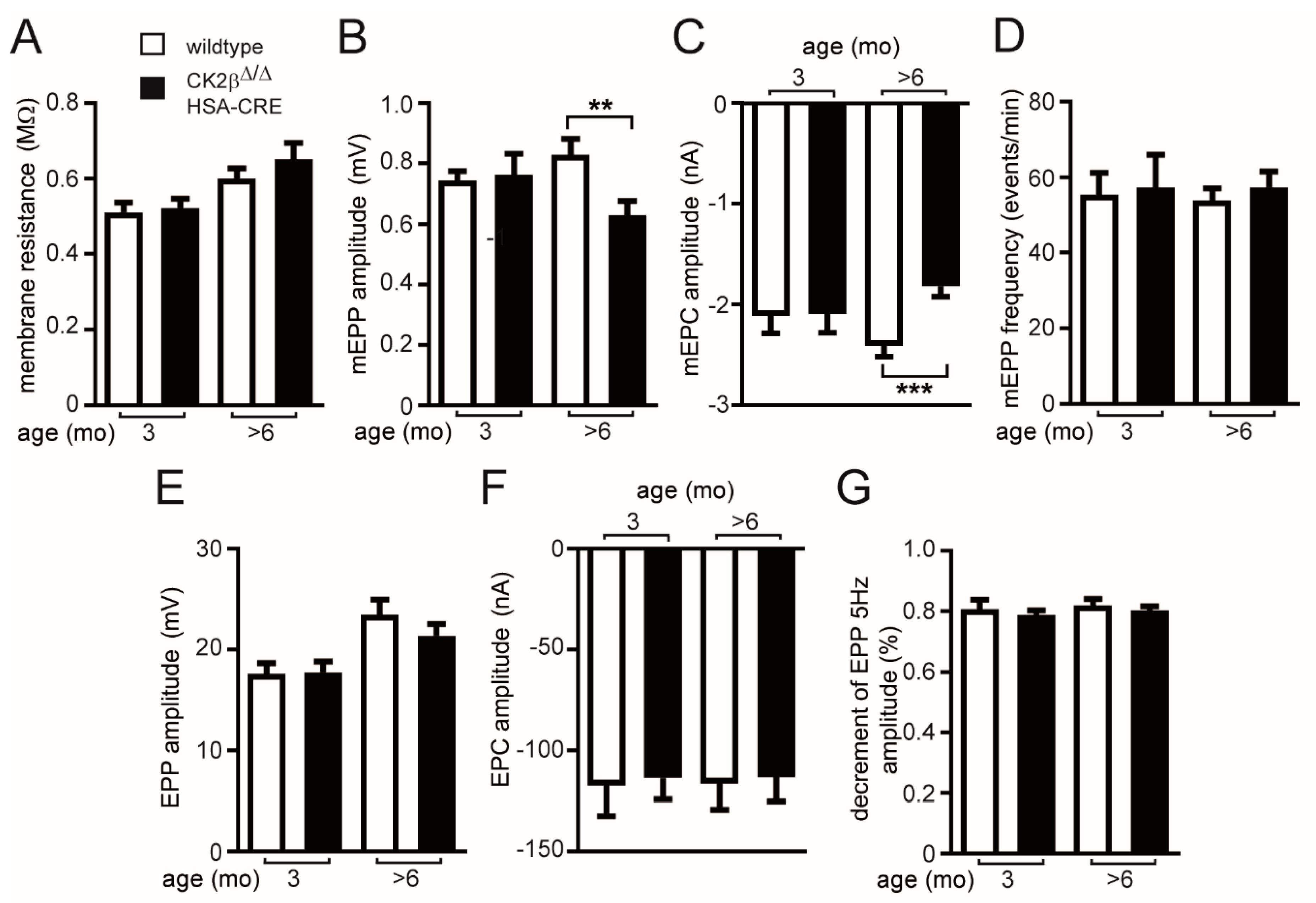
3.2. In the Absence of the CK2β Subunit the Function of the NMJ Is Impaired
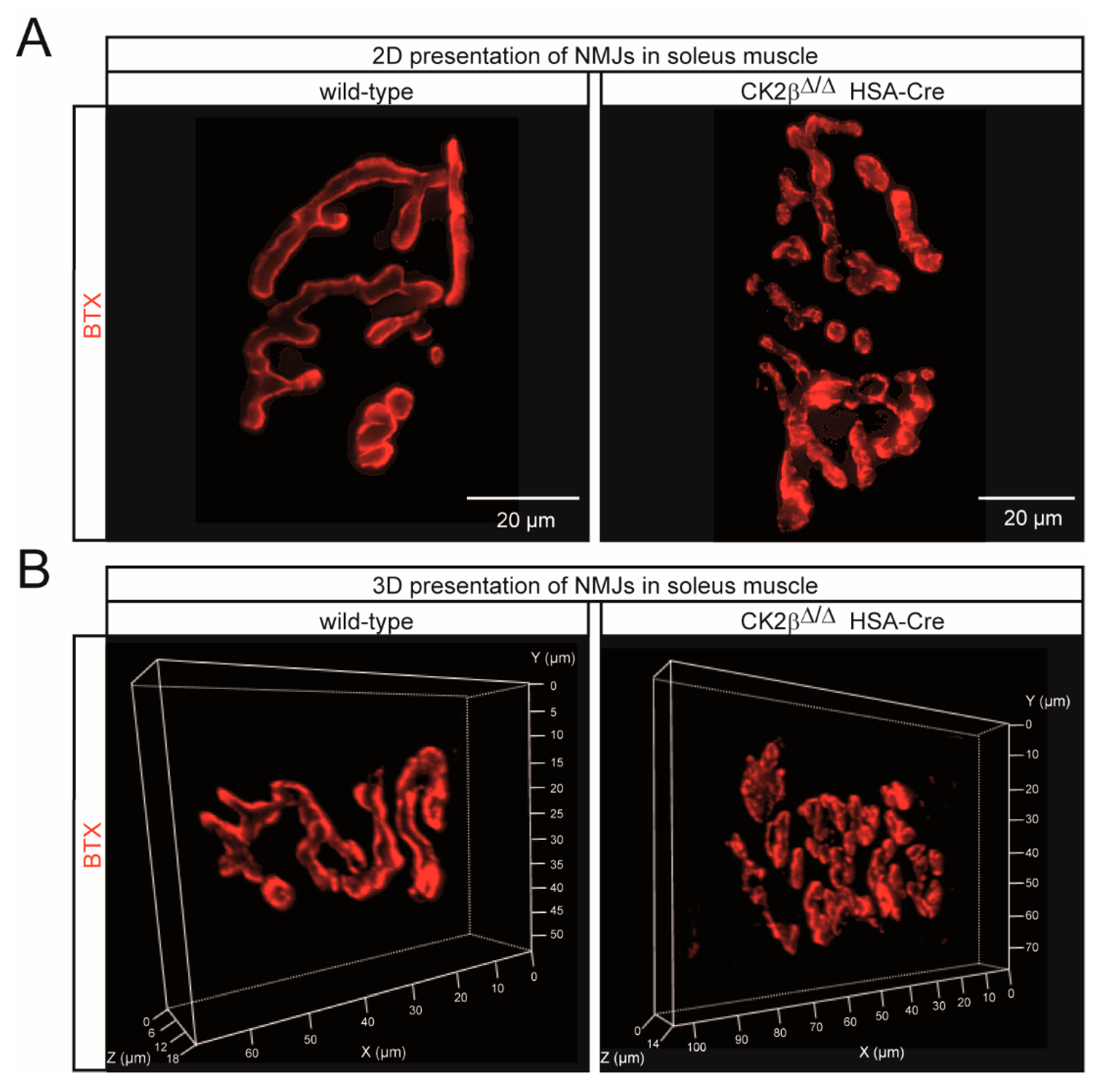
3.3. Structural Changes of Clustered AChRs in CK2β-Deficient Myofibers and Cultured Myotubes
3.4. Changes of Postsynaptic Gene Expression Unraveled in CK2β-Deficient Muscle Fibers
3.5. The Turnover of Neurotransmitter Receptor Clusters Is Impressively Increased in CK2β-Deficient Muscles
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Li, L.; Xiong, W.C.; Mei, L. Neuromuscular Junction Formation, Aging, and Disorders. Annu. Rev. Physiol 2018, 80, 159–188. [Google Scholar] [CrossRef] [PubMed]
- Weatherbee, S.D.; Anderson, K.V.; Niswander, L.A. LDL-receptor-related protein 4 is crucial for formation of the neuromuscular junction. Development 2006, 133, 4993–5000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Luo, S.; Wang, Q.; Suzuki, T.; Xiong, W.C.; Mei, L. LRP4 serves as a coreceptor of agrin. Neuron 2008, 60, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Stiegler, A.L.; Cameron, T.O.; Hallock, P.T.; Gomez, A.M.; Huang, J.H.; Hubbard, S.R.; Dustin, M.L.; Burden, S.J. Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell 2008, 135, 334–342. [Google Scholar] [CrossRef] [PubMed]
- DeChiara, T.M.; Bowen, D.C.; Valenzuela, D.M.; Simmons, M.V.; Poueymirou, W.T.; Thomas, S.; Kinetz, E.; Compton, D.L.; Rojas, E.; Park, J.S.; et al. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell 1996, 85, 501–512. [Google Scholar] [CrossRef]
- Glass, D.J.; DeChiara, T.M.; Stitt, T.N.; DiStefano, P.S.; Valenzuela, D.M.; Yancopoulos, G.D. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation and is a functional receptor for agrin. Cold Spring Harb. Symp. Quant. Biol. 1996, 61, 435–444. [Google Scholar] [PubMed]
- Simeone, L.; Straubinger, M.; Khan, M.A.; Nalleweg, N.; Cheusova, T.; Hashemolhosseini, S. Identification of Erbin interlinking MuSK and ErbB2 and its impact on acetylcholine receptor aggregation at the neuromuscular junction. J. Neurosci. 2010, 30, 6620–6634. [Google Scholar] [CrossRef] [PubMed]
- Cheusova, T.; Khan, M.A.; Schubert, S.W.; Gavin, A.C.; Buchou, T.; Jacob, G.; Sticht, H.; Allende, J.; Boldyreff, B.; Brenner, H.R.; et al. Casein kinase 2-dependent serine phosphorylation of MuSK regulates acetylcholine receptor aggregation at the neuromuscular junction. Genes Dev. 2006, 20, 1800–1816. [Google Scholar] [CrossRef] [Green Version]
- Okada, K.; Inoue, A.; Okada, M.; Murata, Y.; Kakuta, S.; Jigami, T.; Kubo, S.; Shiraishi, H.; Eguchi, K.; Motomura, M.; et al. The muscle protein Dok-7 is essential for neuromuscular synaptogenesis. Science 2006, 312, 1802–1805. [Google Scholar] [CrossRef]
- Luo, Z.G.; Wang, Q.; Zhou, J.Z.; Wang, J.; Luo, Z.; Liu, M.; He, X.; Wynshaw-Boris, A.; Xiong, W.C.; Lu, B.; et al. Regulation of AChR clustering by Dishevelled interacting with MuSK and PAK1. Neuron 2002, 35, 489–505. [Google Scholar] [CrossRef]
- Luo, Z.G.; Je, H.S.; Wang, Q.; Yang, F.; Dobbins, G.C.; Yang, Z.H.; Xiong, W.C.; Lu, B.; Mei, L. Implication of geranylgeranyltransferase I in synapse formation. Neuron 2003, 40, 703–717. [Google Scholar] [CrossRef]
- Herrmann, D.; Straubinger, M.; Hashemolhosseini, S. Protein kinase CK2 interacts at the neuromuscular synapse with Rapsyn, Rac1, 14-3-3gamma, and Dok-7 proteins and phosphorylates the latter two. J. Biol. Chem. 2015, 290, 22370–22384. [Google Scholar] [CrossRef]
- Litchfield, D.W. Protein kinase CK2: Structure, regulation and role in cellular decisions of life and death. Biochem. J. 2003, 369, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Buchou, T.; Vernet, M.; Blond, O.; Jensen, H.H.; Pointu, H.; Olsen, B.B.; Cochet, C.; Issinger, O.G.; Boldyreff, B. Disruption of the regulatory beta subunit of protein kinase CK2 in mice leads to a cell-autonomous defect and early embryonic lethality. Mol. Cell. Biol. 2003, 23, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Seldin, D.C.; Lou, D.Y.; Toselli, P.; Landesman-Bollag, E.; Dominguez, I. Gene targeting of CK2 catalytic subunits. Mol. Cell. Biochem. 2008, 316, 141–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kravic, B.; Harbauer, A.B.; Romanello, V.; Simeone, L.; Vogtle, F.N.; Kaiser, T.; Straubinger, M.; Huraskin, D.; Bottcher, M.; Cerqua, C.; et al. In mammalian skeletal muscle, phosphorylation of TOMM22 by protein kinase CSNK2/CK2 controls mitophagy. Autophagy 2018, 14, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Eiber, N.; Simeone, L.; Hashemolhosseini, S. Ablation of Protein Kinase CK2beta in Skeletal Muscle Fibers Interferes with Their Oxidative Capacity. Pharmaceuticals 2017, 10, 13. [Google Scholar] [CrossRef] [PubMed]
- Heuss, D.; Klascinski, J.; Schubert, S.W.; Moriabadi, T.; Lochmuller, H.; Hashemolhosseini, S. Examination of transcript amounts and activity of protein kinase CK2 in muscle lysates of different types of human muscle pathologies. Mol. Cell. Biochem. 2008, 316, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Toyka, K.V.; Brachman, D.B.; Pestronk, A.; Kao, I. Myasthenia gravis: Passive transfer from man to mouse. Science 1975, 190, 397–399. [Google Scholar] [CrossRef]
- Ruvinsky, I.; Katz, M.; Dreazen, A.; Gielchinsky, Y.; Saada, A.; Freedman, N.; Mishani, E.; Zimmerman, G.; Kasir, J.; Meyuhas, O. Mice deficient in ribosomal protein S6 phosphorylation suffer from muscle weakness that reflects a growth defect and energy deficit. PLoS ONE 2009, 4, e5618. [Google Scholar] [CrossRef]
- Liley, A.W. An investigation of spontaneous activity at the neuromuscular junction of the rat. J. Physiol. 1956, 132, 650–666. [Google Scholar] [CrossRef] [Green Version]
- Sandrock, A.W., Jr.; Dryer, S.E.; Rosen, K.M.; Gozani, S.N.; Kramer, R.; Theill, L.E.; Fischbach, G.D. Maintenance of acetylcholine receptor number by neuregulins at the neuromuscular junction in vivo. Science 1997, 276, 599–603. [Google Scholar] [CrossRef]
- Plomp, J.J.; van Kempen, G.T.; Molenaar, P.C. Adaptation of quantal content to decreased postsynaptic sensitivity at single endplates in alpha-bungarotoxin-treated rats. J. Physiol. 1992, 458, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Rogozhin, A.A.; Pang, K.K.; Bukharaeva, E.; Young, C.; Slater, C.R. Recovery of mouse neuromuscular junctions from single and repeated injections of botulinum neurotoxin A. J. Physiol. 2008, 586, 3163–3182. [Google Scholar] [CrossRef] [PubMed]
- Kroger, S. Differential distribution of agrin isoforms in the developing and adult avian retina. Mol. Cell. Neurosci. 1997, 10, 149–161. [Google Scholar] [CrossRef]
- Gesemann, M.; Denzer, A.J.; Ruegg, M.A. Acetylcholine receptor-aggregating activity of agrin isoforms and mapping of the active site. J. Cell. Biol. 1995, 128, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Roder, I.V.; Petersen, Y.; Choi, K.R.; Witzemann, V.; Hammer, J.A.; Rudolf, R. Role of Myosin Va in the plasticity of the vertebrate neuromuscular junction in vivo, 3rd ed. PLoS ONE 2008, 3, e3871. [Google Scholar] [CrossRef] [PubMed]
- Yampolsky, P.; Pacifici, P.G.; Lomb, L.; Giese, G.; Rudolf, R.; Roder, I.V.; Witzemann, V. Time lapse in vivo visualization of developmental stabilization of synaptic receptors at neuromuscular junctions. J. Biol. Chem. 2010, 285, 34589–34596. [Google Scholar] [CrossRef]
- Kravic, B.; Huraskin, D.; Frick, A.D.; Jung, J.; Redai, V.; Palmisano, R.; Marchetto, S.; Borg, J.P.; Mei, L.; Hashemolhosseini, S. LAP proteins are localized at the post-synaptic membrane of neuromuscular junctions and appear to modulate synaptic morphology and transmission. J. Neurochem. 2016, 139, 381–395. [Google Scholar] [CrossRef] [Green Version]
- Banker, B.Q.; Kelly, S.S.; Robbins, N. Neuromuscular transmission and correlative morphology in young and old mice. J. Physiol. 1983, 339, 355–377. [Google Scholar] [CrossRef]
- Wood, S.J.; Slater, C.R. The contribution of postsynaptic folds to the safety factor for neuromuscular transmission in rat fast- and slow-twitch muscles. J. Physiol. 1997, 500, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Andonian, M.H.; Fahim, M.A. Effects of endurance exercise on the morphology of mouse neuromuscular junctions during ageing. J. Neurocytol. 1987, 16, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Rudolf, R.; Khan, M.M.; Labeit, S.; Deschenes, M.R. Degeneration of Neuromuscular Junction in Age and Dystrophy. Front. Aging Neurosci. 2014, 6, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, M.; Witkin, K.L.; Cohen-Fix, O. Sizing up the nucleus: Nuclear shape, size and nuclear-envelope assembly. J. Cell. Sci. 2009, 122, 1477–1486. [Google Scholar] [CrossRef] [PubMed]
- Witzemann, V.; Barg, B.; Criado, M.; Stein, E.; Sakmann, B. Developmental regulation of five subunit specific mRNAs encoding acetylcholine receptor subtypes in rat muscle. FEBS Lett. 1989, 242, 419–424. [Google Scholar] [CrossRef] [Green Version]
- Cescon, M.; Gregorio, I.; Eiber, N.; Borgia, D.; Fusto, A.; Sabatelli, P.; Scorzeto, M.; Megighian, A.; Pegoraro, E.; Hashemolhosseini, S.; et al. Collagen VI is required for the structural and functional integrity of the neuromuscular junction. Acta Neuropathol. 2018, 136, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Yampolsky, P.; Gensler, S.; McArdle, J.; Witzemann, V. AChR channel conversion and AChR-adjusted neuronal survival during embryonic development. Mol. Cell. Neurosci. 2008, 37, 634–645. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence |
|---|---|
| AChRα | 5′-CTACCTGCCCACAGACTCAG-3′ 5′-TTGGACTCCTGGTCTGACTT-3′ |
| AChRγ | 5′-GGTCAATGTCAGCCTGAAGC-3′ 5′-GCACATGCATCCGTAACAGC-3′ |
| AChRβ | 5′-TCTCCAACTATGATAGCTCGGT-3′ 5′-CATTGATGTCCAGGGCAACGTC-3′ |
| AChRε | 5′-TGTATGGCTGCCAGAGATTG-3′ 5′-GCGGATGATGAGCGTATAGA-3′ |
| AChRδ | 5′-ATGAGGAACAAAGGCTGATCCA-3′ 5′-ACAGTGATGTTCCCGAAGTCGT-3′ |
| Rapsyn | 5′-CCGCTACAGGCACTCTGTCT-3′ 5′-TCAGTCTCCTCCACGCACTC-3′ |
| MuSK | 5′-GCCTTGGTTGAAGAAGTAGC -3′ 5′-CTTGATCCAGGACACAGATG -3′ |
| RPL8 | 5′-GTTCGTGTACTGCGGCAAGA-3′ 5′-ACAGGATTCATGGCCACACC-3′ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eiber, N.; Rehman, M.; Kravic, B.; Rudolf, R.; Sandri, M.; Hashemolhosseini, S. Loss of Protein Kinase Csnk2b/CK2β at Neuromuscular Junctions Affects Morphology and Dynamics of Aggregated Nicotinic Acetylcholine Receptors, Neuromuscular Transmission, and Synaptic Gene Expression. Cells 2019, 8, 940. https://doi.org/10.3390/cells8080940
Eiber N, Rehman M, Kravic B, Rudolf R, Sandri M, Hashemolhosseini S. Loss of Protein Kinase Csnk2b/CK2β at Neuromuscular Junctions Affects Morphology and Dynamics of Aggregated Nicotinic Acetylcholine Receptors, Neuromuscular Transmission, and Synaptic Gene Expression. Cells. 2019; 8(8):940. https://doi.org/10.3390/cells8080940
Chicago/Turabian StyleEiber, Nane, Michael Rehman, Bojana Kravic, Rüdiger Rudolf, Marco Sandri, and Said Hashemolhosseini. 2019. "Loss of Protein Kinase Csnk2b/CK2β at Neuromuscular Junctions Affects Morphology and Dynamics of Aggregated Nicotinic Acetylcholine Receptors, Neuromuscular Transmission, and Synaptic Gene Expression" Cells 8, no. 8: 940. https://doi.org/10.3390/cells8080940
APA StyleEiber, N., Rehman, M., Kravic, B., Rudolf, R., Sandri, M., & Hashemolhosseini, S. (2019). Loss of Protein Kinase Csnk2b/CK2β at Neuromuscular Junctions Affects Morphology and Dynamics of Aggregated Nicotinic Acetylcholine Receptors, Neuromuscular Transmission, and Synaptic Gene Expression. Cells, 8(8), 940. https://doi.org/10.3390/cells8080940