Transport of Ca2+ and Ca2+-Dependent Permeability Transition in Rat Liver Mitochondria under the Streptozotocin-Induced Type I Diabetes

 ,
, 
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Induction of Diabetes
2.3. Isolation of Rat Liver Mitochondria
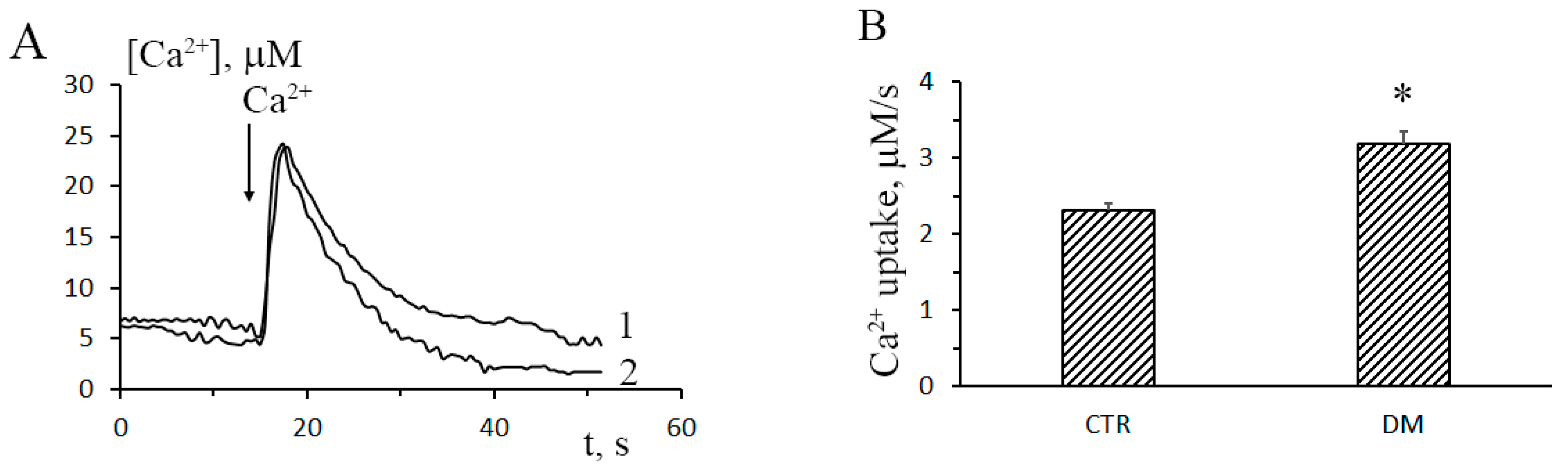
2.4. Ca2+ Uptake by Mitochondria
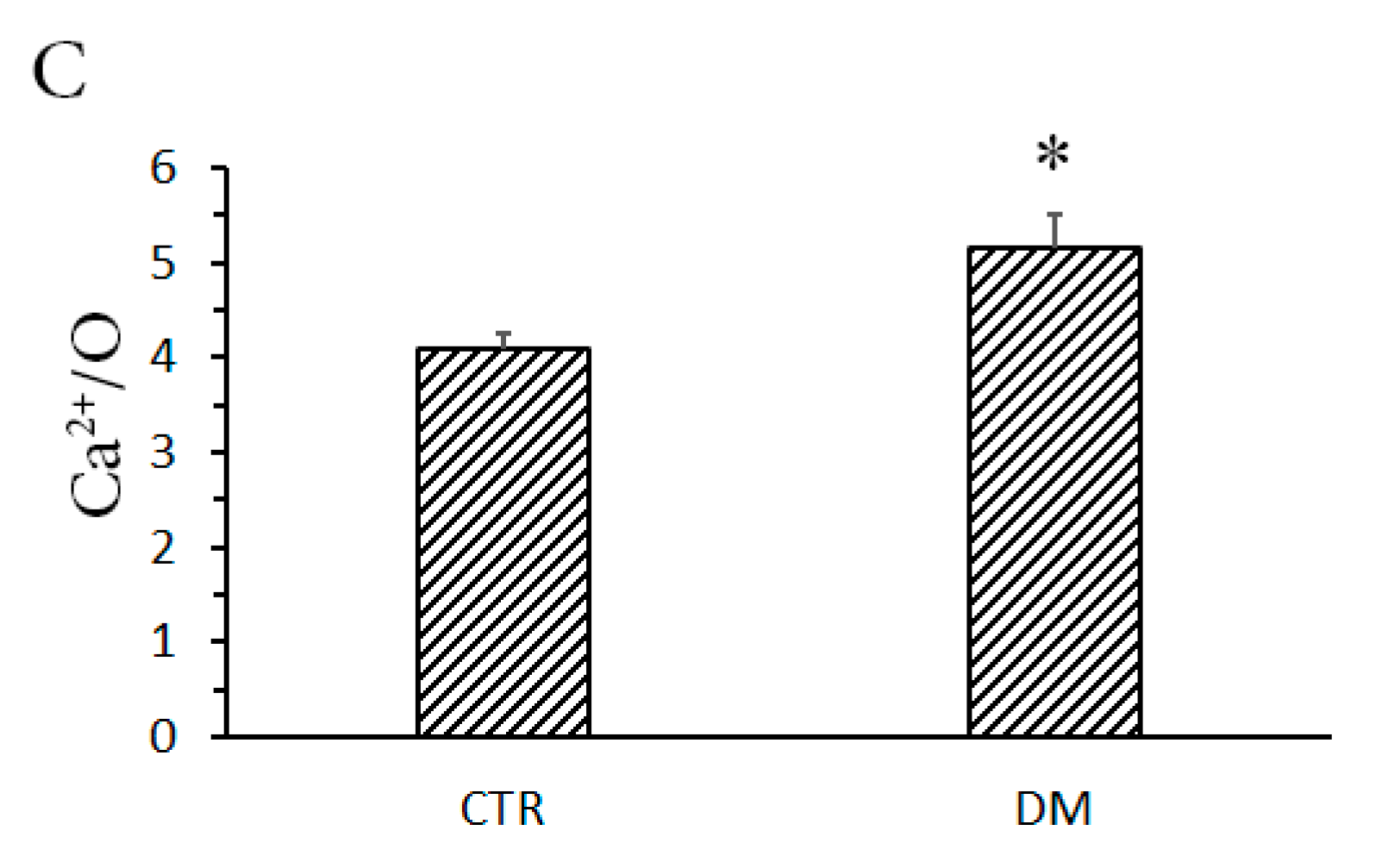
2.5. Mitochondrial Respiration and Ca2+/O
2.6. Mitochondrial Swelling
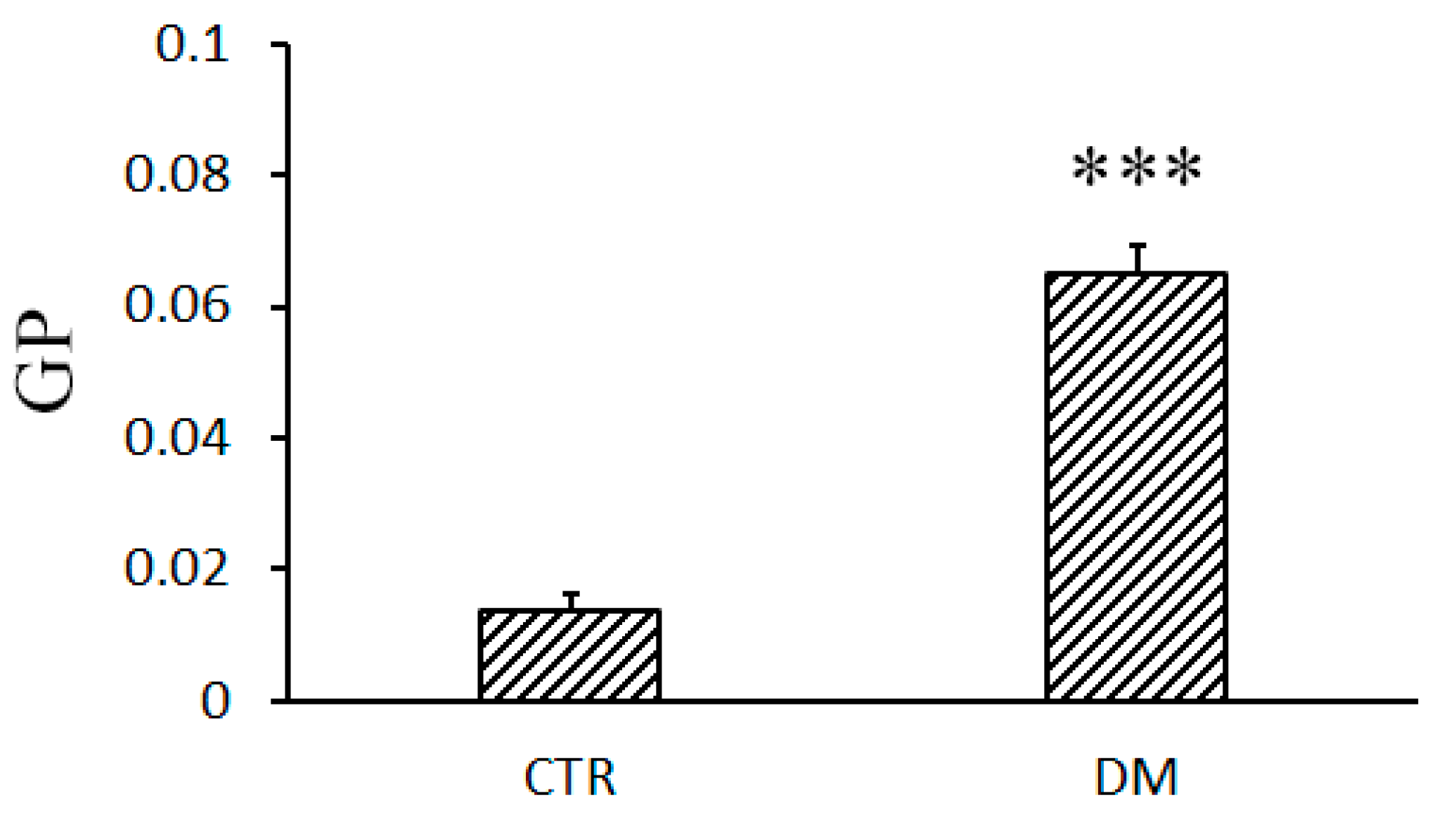
2.7. Fluidity of the Mitochondrial Membrane
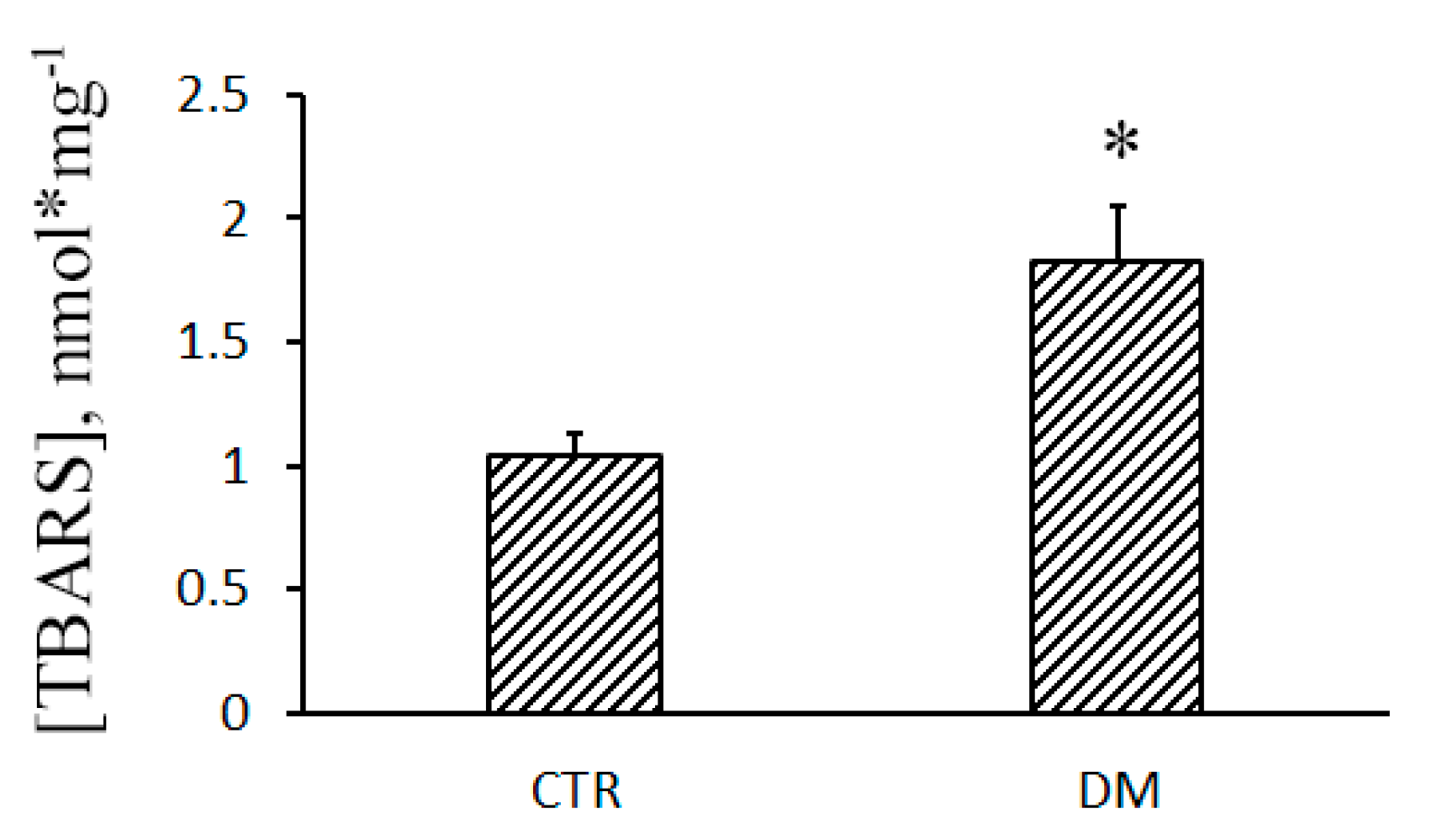
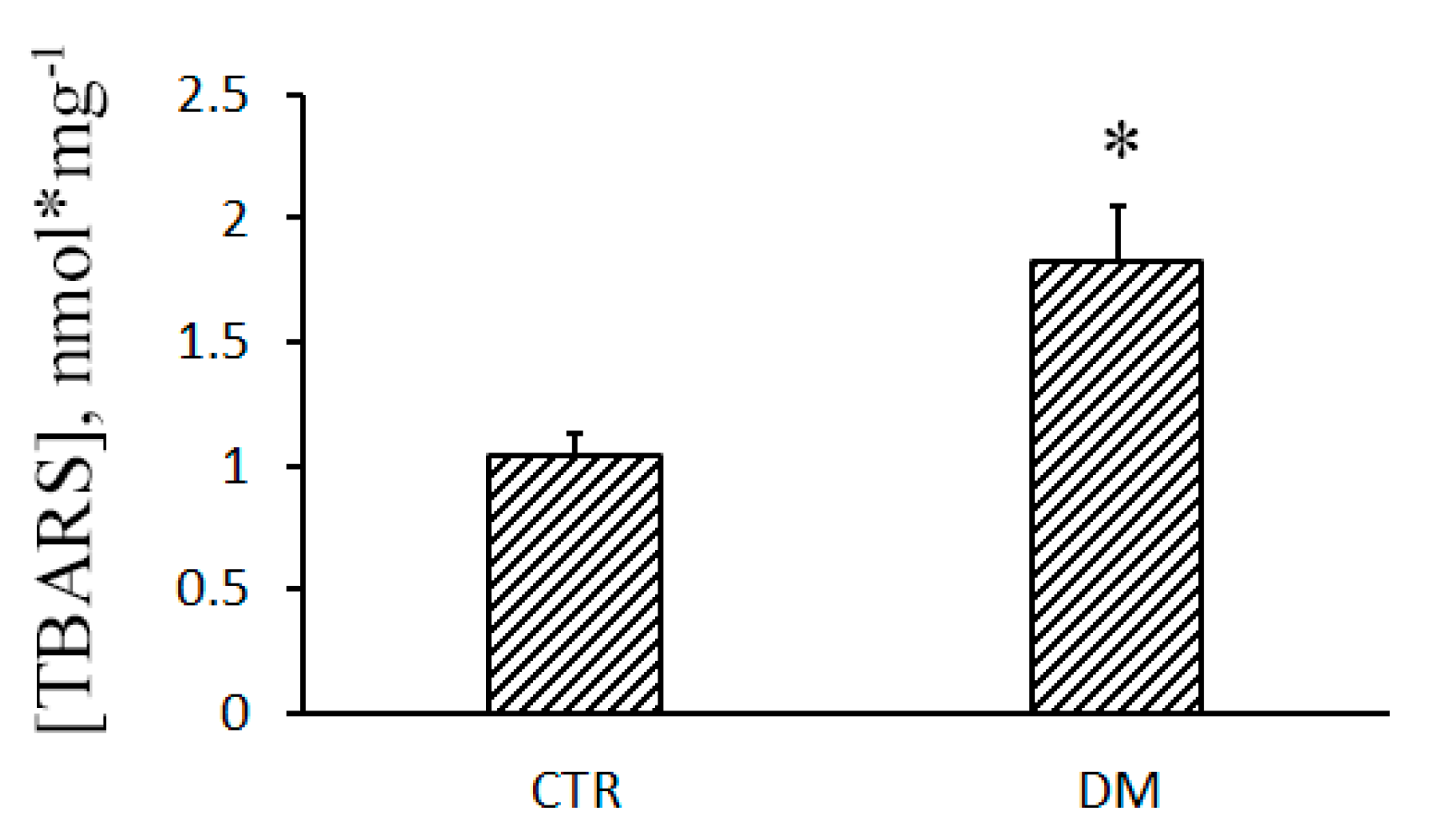
2.8. Lipid Peroxidation
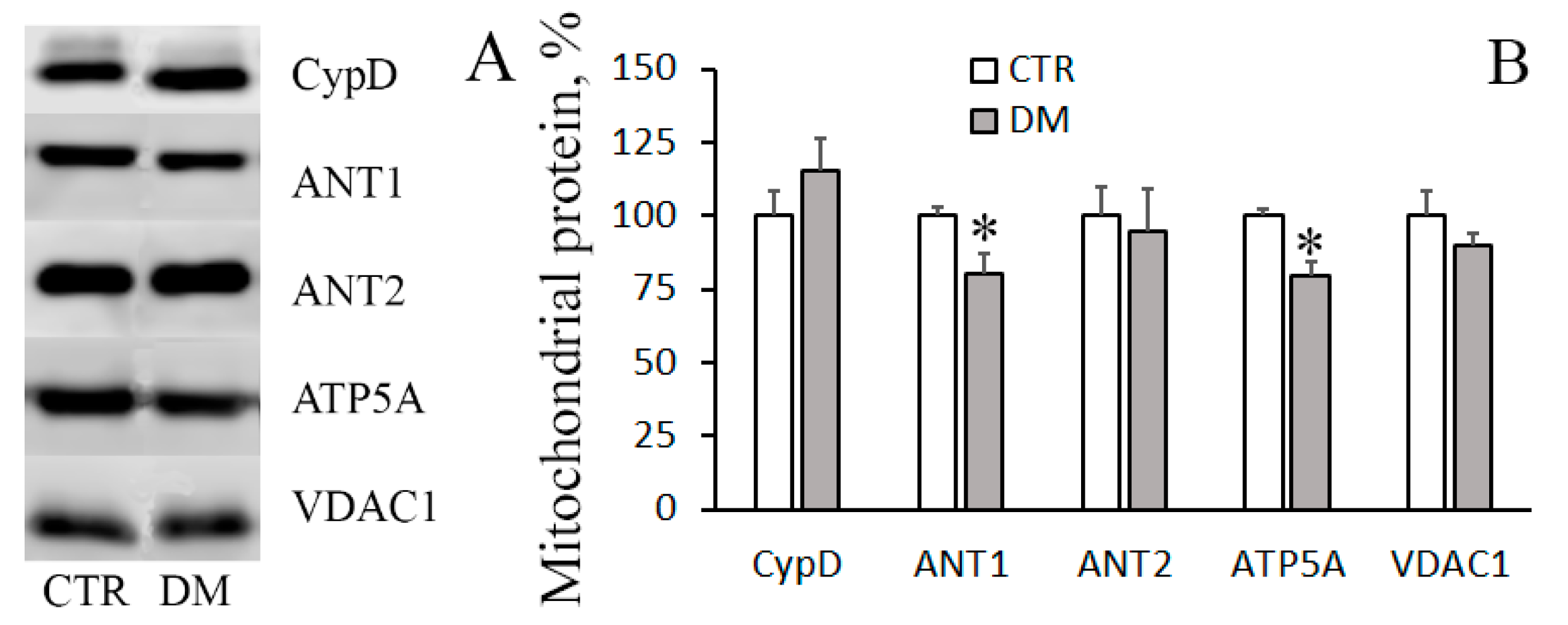
2.9. Electrophoresis and Immunoblotting of Mitochondrial Proteins
2.10. Fatty Acid Composition of Mitochondrial Phospholipids
2.11. Statistical Analysis
3. Results
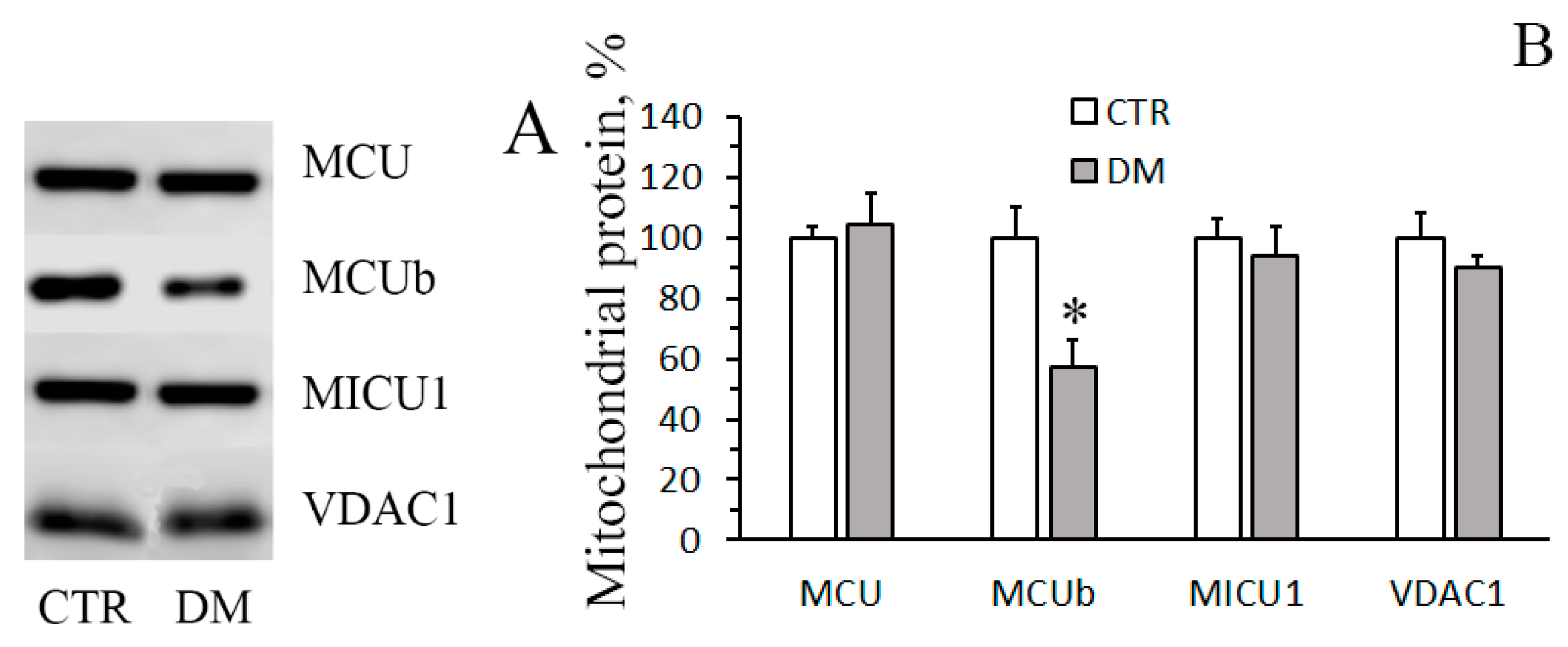
3.1. The Development of Diabetes Stimulates Ca2+ Uptake by Rat Liver Mitochondria
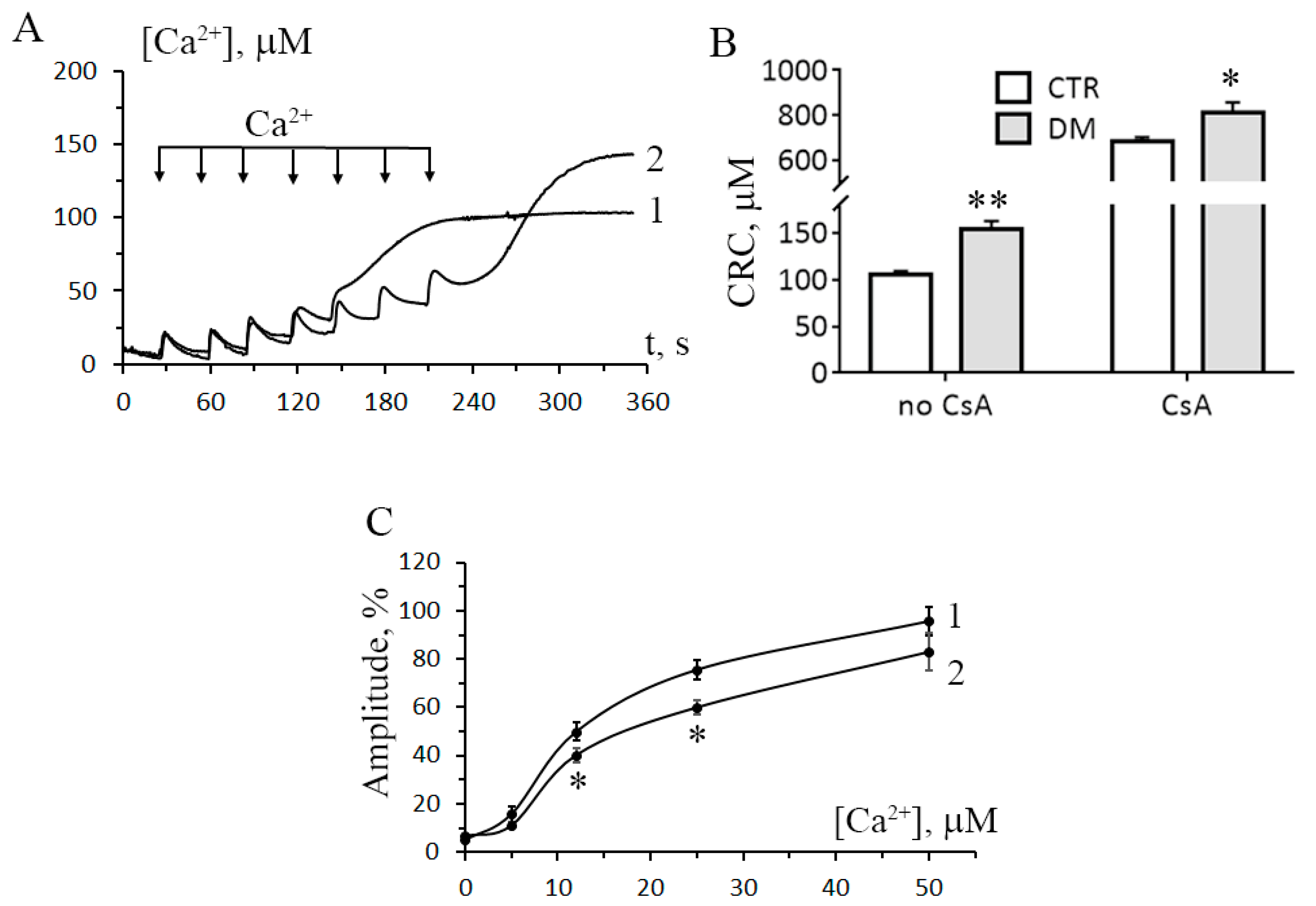
3.2. The Development of Diabetes Increases the Resistance of Liver Mitochondria to the Opening of MPT Pore
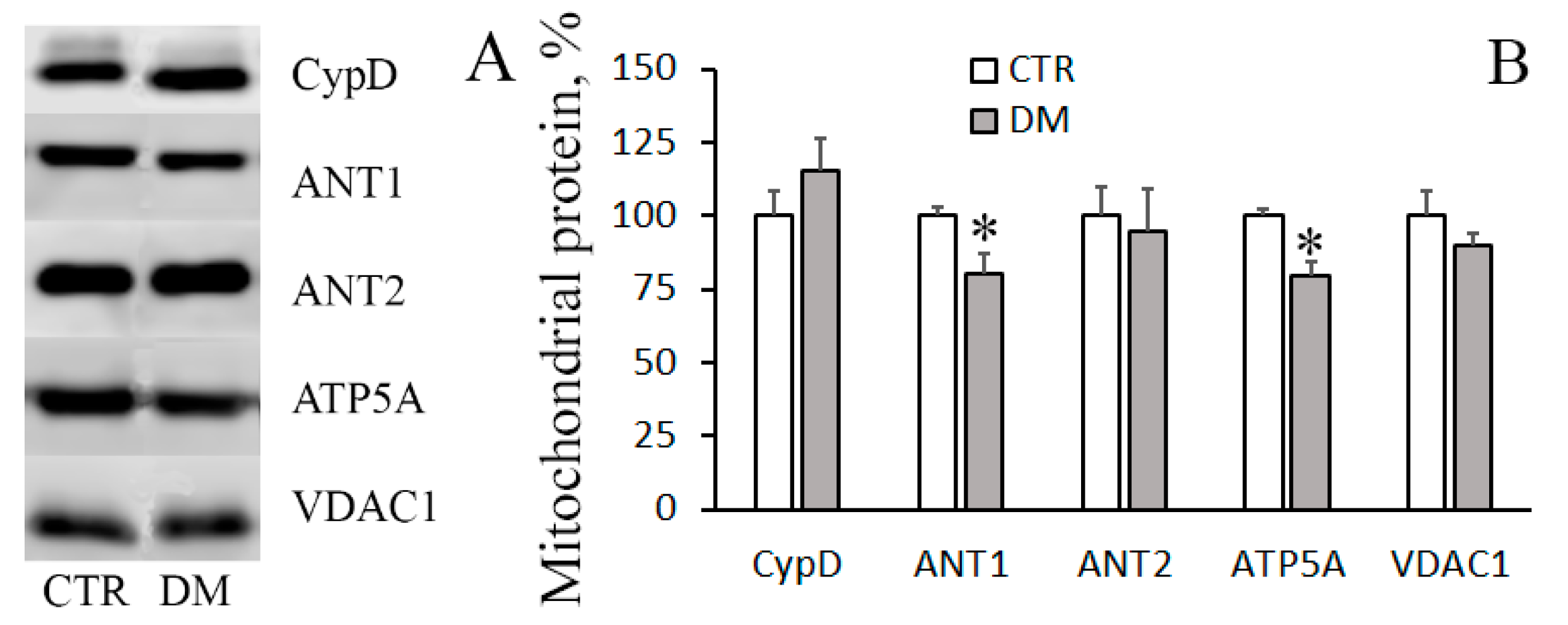
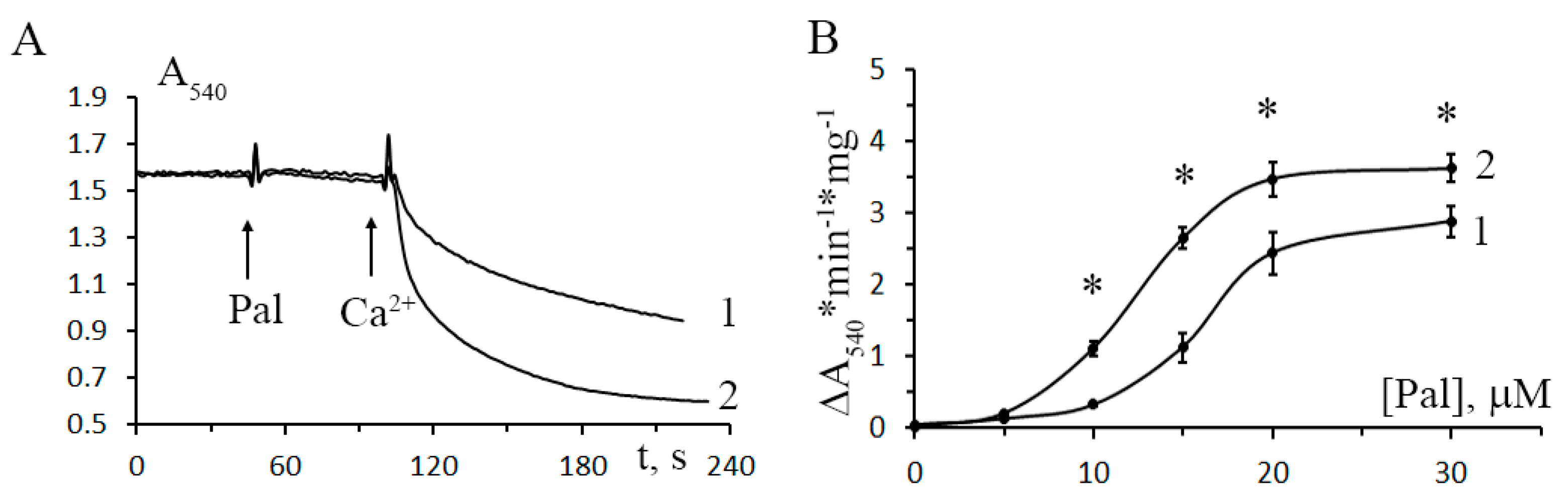
3.3. The Development of Diabetes Decreases the Resistance of Mitochondria to the Formation of CsA-Insensitive Palmitate/Ca2+-Dependent Pores
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fujimaki, S.; Kuwabara, T. Diabetes-induced dysfunction of mitochondria and stem cells in skeletal muscle and the nervous system. Int. J. Mol. Sci. 2017, 18, 2147. [Google Scholar] [CrossRef] [PubMed]
- Stump, C.S.; Short, K.R.; Bigelow, M.L.; Schimke, J.M.; Nair, K.S. Effect of insulin on human skeletal muscle mitochondrial ATP production, protein synthesis, and mRNA transcripts. Proc. Natl. Acad. Sci. USA 2003, 100, 7996–8001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, K.F.; Befroy, D.; Dufour, S.; Dziura, J.; Ariyan, C.; Rothman, D.L.; Di Pietro, L.; Cline, G.W.; Shulman, G.I. Mitochondrial dysfunction in the elderly: Possible role in insulin resistance. Science 2003, 300, 1140–1142. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef] [PubMed]
- Belosludtsev, K.N.; Dubinin, M.V.; Belosludtseva, N.V.; Mironova, G.D. Mitochondrial Ca2+ transport: Mechanisms, molecular structures, and role in cells. Biochemistry (Mosc) 2019, 84, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Raffaello, A.; De Stefani, D.; Sabbadin, D.; Teardo, E.; Merli, G.; Picard, A.; Checchetto, V.; Moro, S.; Szabo, I.; Rizzuto, R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013, 32, 2362–2376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paillard, M.; Csordas, G.; Szanda, G.; Golenar, T.; Debattisti, V.; Bartok, A.; Wang, N.; Moffat, C.; Seifert, E.L.; Spat, A.; et al. Tissue-specific mitochondrial decoding of cytoplasmic Ca2+ signals is controlled by the stoichiometry of MICU1/2 and MCU. Cell Rep. 2017, 18, 2291–2300. [Google Scholar] [CrossRef]
- Briston, T.; Selwood, D.L.; Szabadkai, G.; Duchen, M.R. Mitochondrial permeability transition: A molecular lesion with multiple drug targets. Trends Pharmacol. Sci. 2019, 40, 50–70. [Google Scholar] [CrossRef]
- Agafonov, A.; Gritsenko, E.; Belosludtsev, K.; Kovalev, A.; Gateau-Roesch, O.; Saris, N.-E.L.; Mironova, G.D. A permeability transition in liposomes induced by the formation of Ca2+/palmitic acid complexes. Biochim. Biophys. Acta. 2003, 1609, 153–160. [Google Scholar] [CrossRef]
- Chan, K.M.; Junger, K.D. The effect of streptozocin-induced diabetes on the plasma membrane calcium uptake activity of rat liver. Diabetes 1984, 33, 1072–1077. [Google Scholar] [CrossRef]
- Boquist, L.; Boquist, S.; Alehagen, U. Mitochondrial changes and associated alterations induced in mice by streptozotocin administered in vivo and in vitro. Diabetes Res. Clin. Pract. 1987, 3, 179–190. [Google Scholar] [CrossRef]
- Garrick, R.A.; Hall, J.C. Adenosine diphosphate and calcium stimulation of respiration in mitochondria from alloxan diabetic rats. J. Cell Physiol. 1974, 84, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Juarez, J.; Suarez, J.; Cividini, F.; Scott, B.T.; Diemer, T.; Dai, A.; Dillmann, W.H. Expression of the mitochondrial calcium uniporter in cardiac myocytes improves impaired mitochondrial calcium handling and metabolism in simulated hyperglycemia. Am. J. Physiol. Cell Physiol. 2016, 311, C1005–C1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, M.F.; Natali, A.J.; Da Silva, E.; Gomes, G.J.; Teodoro, B.G.; Cunha, D.N.; Drummond, L.R.; Drummond, F.R.; Moura, A.G.; Belfort, F.G.; et al. Attenuation of Ca2+ homeostasis, oxidative stress, and mitochondrial dysfunctions in diabetic rat heart: Insulin therapy or aerobic exercise? J. Appl. Physiol. 2015, 119, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Kristal, B.S.; Matsuda, M.; Yu, B.P. Abnormalities in the mitochondrial permeability transition in diabetic rats. Biochem. Biophys. Res. Commun. 1996, 222, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Gvazava, I.G.; Rogovaya, O.S.; Borisov, M.A.; Vorotelyak, E.A.; Vasiliev, A.V. Pathogenesis of type 1 diabetes mellitus and rodent experimental models. Acta. Naturae 2018, 10, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Kume, E.; Fujimura, H.; Matsuki, N.; Ito, M.; Aruga, C.; Toriumi, W.; Kitamura, K.; Doi, K. Hepatic changes in the acute phase of streptozotocin (SZ)-induced diabetes in mice. Exp. Toxicol. Pathol. 2004, 55, 467–480. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Agafonov, A.V.; Astashev, M.E.; Kazakov, A.S.; Saris, N.-E.L.; Mironova, G.D. Ca2+-dependent permeabilization of mitochondria and liposomes by palmitic and oleic acids: A comparative study. Biochim. Biophys. Acta. 2014, 1838, 2600–2606. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Agafonov, A.V.; Pavlik, L.L.; Dubinin, M.V. Effect of bedaquiline on the functions of rat liver mitochondria. Biochim. Biophys. Acta. Biomembr. 2019, 1861, 288–297. [Google Scholar] [CrossRef]
- Katyare, S.S.; Challberg, M.D.; Howland, J.L. Energy coupling in liver mitochondria from dystrophic mice: Differential sensitivity of oxidative phosphorylation and Ca2+ uptake to K+. Metabolism 1978, 27, 761–769. [Google Scholar] [CrossRef]
- Amaral, A.U.; Cecatto, C.; da Silva, J.C.; Wajner, A.; Godoy, K.D.S.; Ribeiro, R.T.; Wajner, M. cis-4-Decenoic and decanoic acids impair mitochondrial energy, redox and Ca2+ homeostasis and induce mitochondrial permeability transition pore opening in rat brain and liver: Possible implications for the pathogenesis of MCAD deficiency. Biochim. Biophys. Acta. 2016, 1857, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Ohkawa, H.; Ohishi, N.; Yagi, K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 1979, 95, 351–358. [Google Scholar] [CrossRef]
- Bligh, E.; Dyer, W. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Pinton, P. A New Current for the Mitochondrial Permeability Transition. Trends Biochem. Sci. 2019, 44, 559–561. [Google Scholar] [CrossRef] [PubMed]
- Ly, L.D.; Xu, S.; Choi, S.K.; Ha, C.M.; Thoudam, T.; Cha, S.K.; Wiederkehr, A.; Wollheim, C.B.; Lee, I.K.; Park, K.S. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp. Mol. Med. 2017, 49, e291. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, R.H.; Zerenturk, E.J.; Prakoso, D.; Calkin, A.C. Lipid metabolism and its implications for type 1 diabetes-associated cardiomyopathy. J. Mol. Endocrinol. 2017, 58, R225–R240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suarez, J.; Cividini, F.; Scott, B.T.; Lehmann, K.; Diaz-Juarez, J.; Diemer, T.; Dai, A.; Suarez, J.A.; Jain, M.; Dillmann, W.H. Restoring mitochondrial calcium uniporter expression in diabetic mouse heart improves mitochondrial calcium handling and cardiac function. J. Biol. Chem. 2018, 293, 8182–8195. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, F.M.; Seiça, R.; Oliveira, P.J.; Coxito, P.M.; Moreno, A.J.; Palmeira, C.M.; Santos, M.S. Diabetes induces metabolic adaptations in rat liver mitochondria: Role of coenzyme Q and cardiolipin contents. Biochim. Biophys. Acta. 2003, 1639, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Belosludtseva, N.V.; Belosludtsev, K.N.; Agafonov, A.V.; Mironova, G.D. Effect of cholesterol on the formation of palmitate/Ca2+-activated pore in mitochondria and liposomes. Biofizika 2009, 54, 464–470. [Google Scholar]
- Belosludtsev, K.N.; Saris, N.E.; Belosludtseva, N.V.; Trudovishnikov, A.S.; Lukyanova, L.D.; Mironova, G.D. Physiological aspects of the mitochondrial cyclosporin A-insensitive palmitate/Ca2+-induced pore: Tissue specificity, age profile and dependence on the animal’s adaptation to hypoxia. J. Bioenerg. Biomembr. 2009, 41, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Dobretsov, G.E.; Borschevskaya, T.A.; Petrov, V.A.; Vladimirov, Y.U. The increase of phospholipid bilayer rigidity after lipid peroxidation. FEBS Lett. 1977, 84, 125–128. [Google Scholar] [CrossRef] [Green Version]
- Mironova, G.D.; Saris, N.E.; Belosludtseva, N.V.; Agafonov, A.V.; Elantsev, A.B.; Belosludtsev, K.N. Involvement of palmitate/Ca2+(Sr2+)-induced pore in the cycling of ions across the mitochondrial membrane. Biochim. Biophys. Acta. 2015, 1848, 488–495. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | DM | |
|---|---|---|
| Initial BW, g | 90.2 ± 6.6 | 86.5 ± 5.5 |
| Final BW, g (after two weeks) | 197.7 ± 3.6 | 135.1 ± 6.8 **** |
| BG (mg/dL) | 125.9 ± 8.9 | 489.1 ± 16.5 **** |
| Fatty Acids | Control | DM1 |
|---|---|---|
| C16:0 | 19.98 ± 1.05 | 25.98 ± 2.32 * |
| C18:0 | 15.07 ± 0.865 | 27.71 ± 2.72 ** |
| C18:1 | 6.68 ± 0.24 | 10.49 ± 0.63 ** |
| C18:2 | 13.99 ± 1.04 | 27.60 ± 3.59 * |
| C20:4 | 18.35 ± 1.35 | 26.71 ± 1.24 ** |
| C24:1 | 0.71 ± 0.08 | 0.61 ± 0.05 |
| C22:6 | 8.32 ± 0.84 | 12.18 ± 1.34 |
| C24:0 | 0.53 ± 0.16 | 0.41 ± 0.12 |
| TFA | 83.01 ± 4.45 | 132.2 ± 14.77 * |
| SFA | 35.31 ± 1.81 | 53.31 ± 5.67 * |
| PUFA | 40.66 ± 2.76 | 69.44 ± 7.87 * |
| UI | 158.3 ± 11.07 | 258.6 ± 28.92 * |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belosludtsev, K.N.; Talanov, E.Y.; Starinets, V.S.; Agafonov, A.V.; Dubinin, M.V.; Belosludtseva, N.V. Transport of Ca2+ and Ca2+-Dependent Permeability Transition in Rat Liver Mitochondria under the Streptozotocin-Induced Type I Diabetes. Cells 2019, 8, 1014. https://doi.org/10.3390/cells8091014
Belosludtsev KN, Talanov EY, Starinets VS, Agafonov AV, Dubinin MV, Belosludtseva NV. Transport of Ca2+ and Ca2+-Dependent Permeability Transition in Rat Liver Mitochondria under the Streptozotocin-Induced Type I Diabetes. Cells. 2019; 8(9):1014. https://doi.org/10.3390/cells8091014
Chicago/Turabian StyleBelosludtsev, Konstantin N., Eugeny Yu. Talanov, Vlada S. Starinets, Alexey V. Agafonov, Mikhail V. Dubinin, and Natalia V. Belosludtseva. 2019. "Transport of Ca2+ and Ca2+-Dependent Permeability Transition in Rat Liver Mitochondria under the Streptozotocin-Induced Type I Diabetes" Cells 8, no. 9: 1014. https://doi.org/10.3390/cells8091014
APA StyleBelosludtsev, K. N., Talanov, E. Y., Starinets, V. S., Agafonov, A. V., Dubinin, M. V., & Belosludtseva, N. V. (2019). Transport of Ca2+ and Ca2+-Dependent Permeability Transition in Rat Liver Mitochondria under the Streptozotocin-Induced Type I Diabetes. Cells, 8(9), 1014. https://doi.org/10.3390/cells8091014