Activity and Trafficking of Copper-Transporting ATPases in Tumor Development and Defense against Platinum-Based Drugs


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
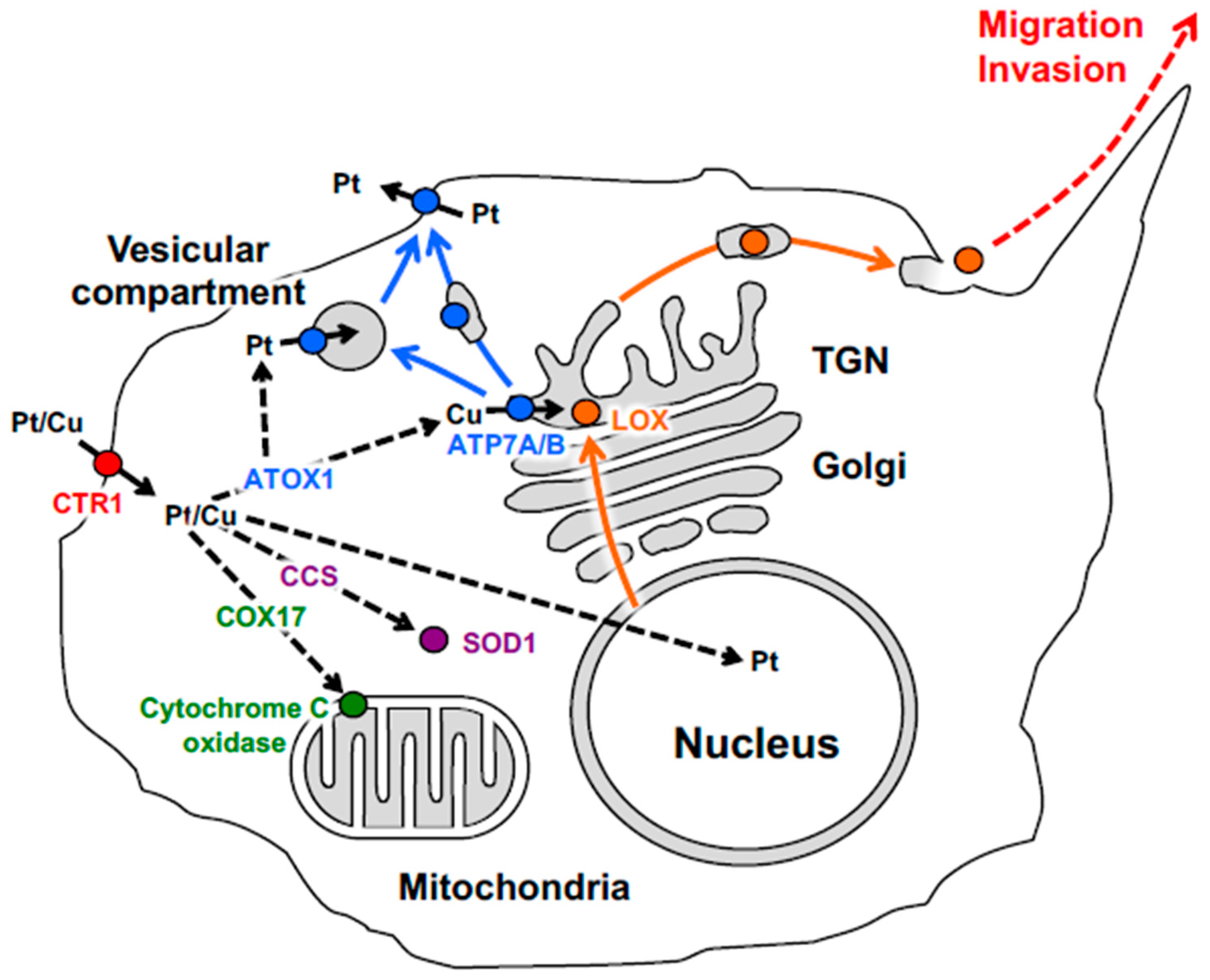
2. Cellular Pathways Operating in Copper and Platinum Distribution
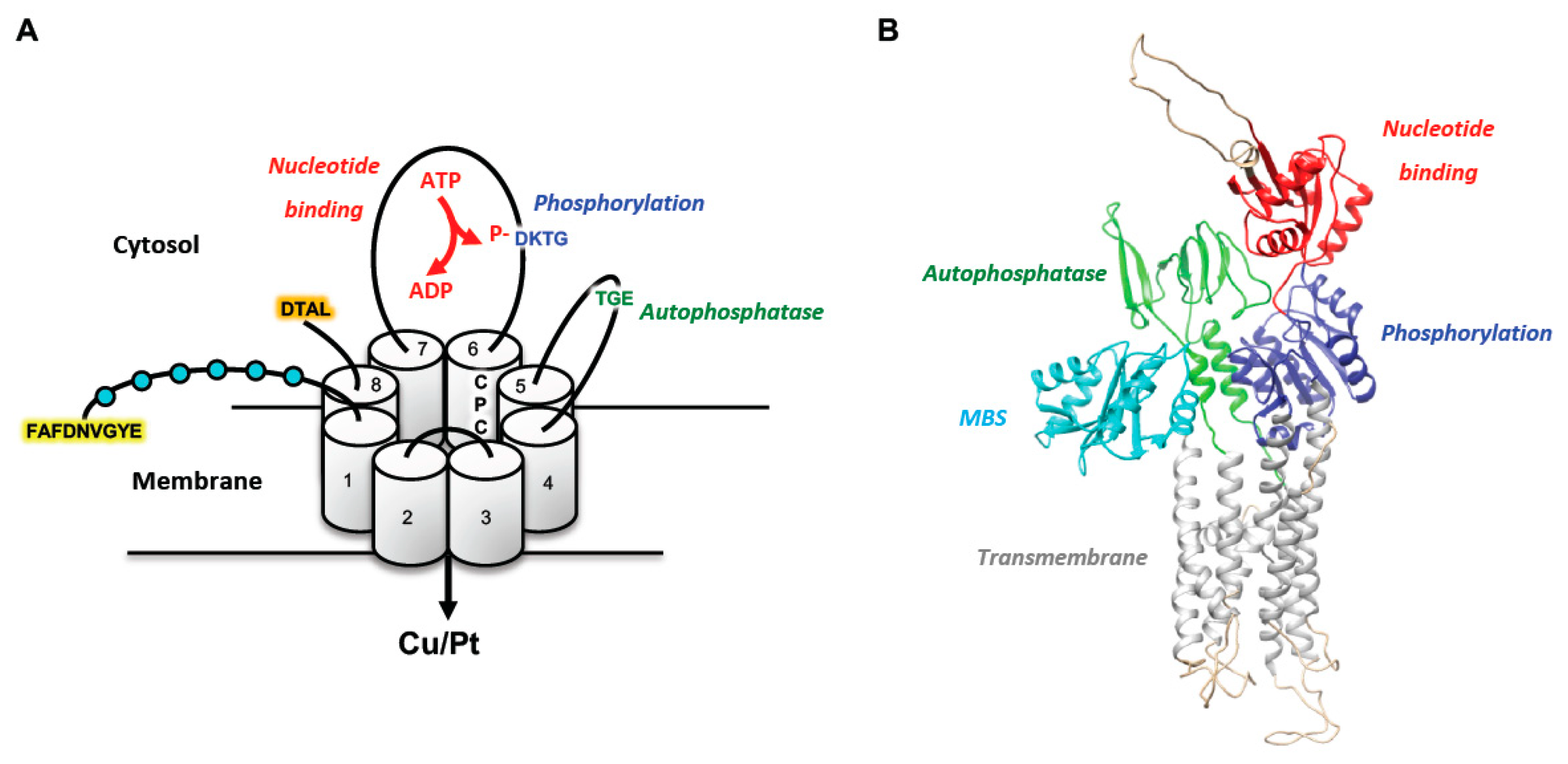
3. The Physiological Role and Trafficking of the Copper-Transporting ATPases
4. Copper-Binding Proteins and Multifactorial Mechanisms of Cisplatin Resistance
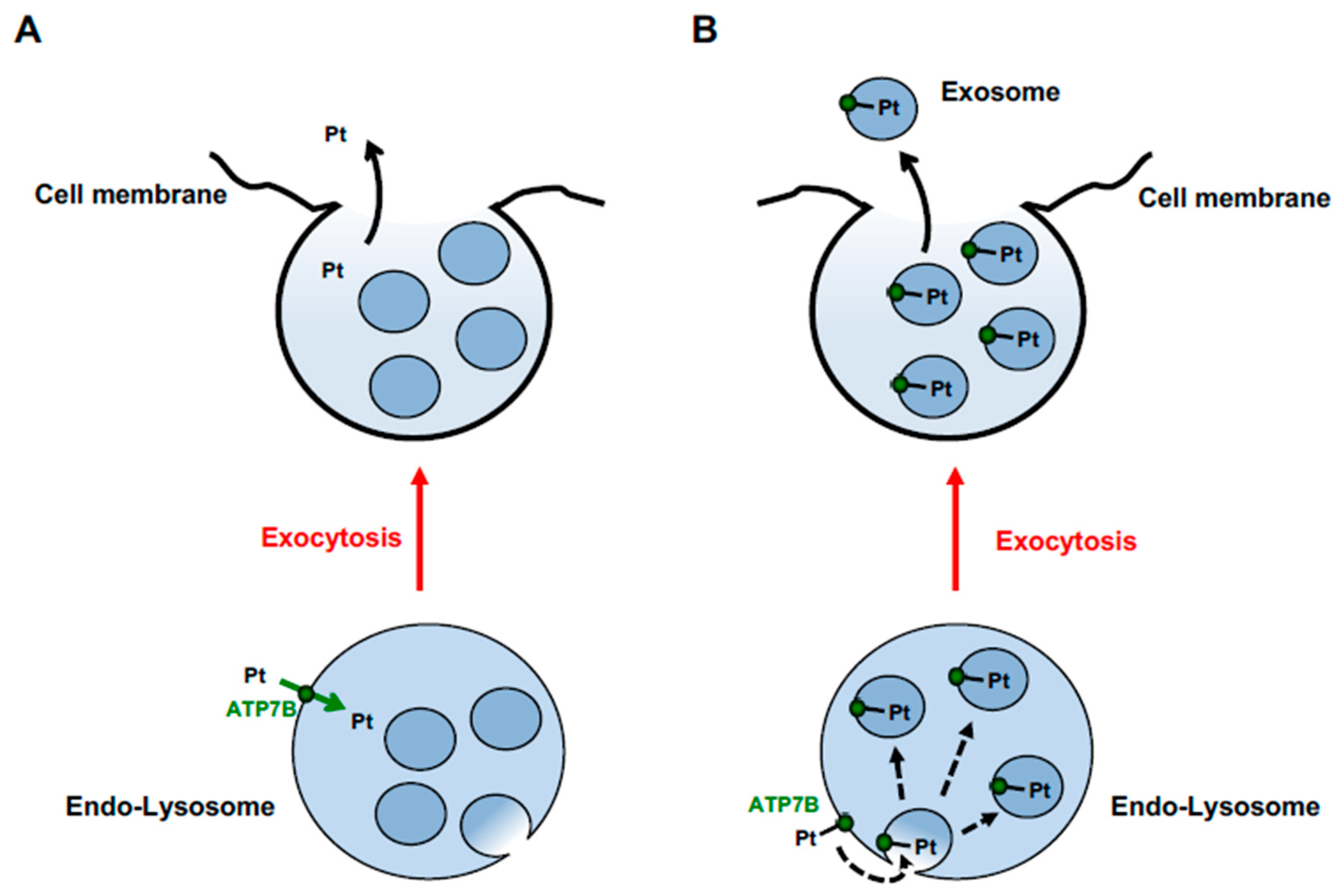
5. Trafficking and Activity of ATP7B and ATP7A in Cisplatin Resistance
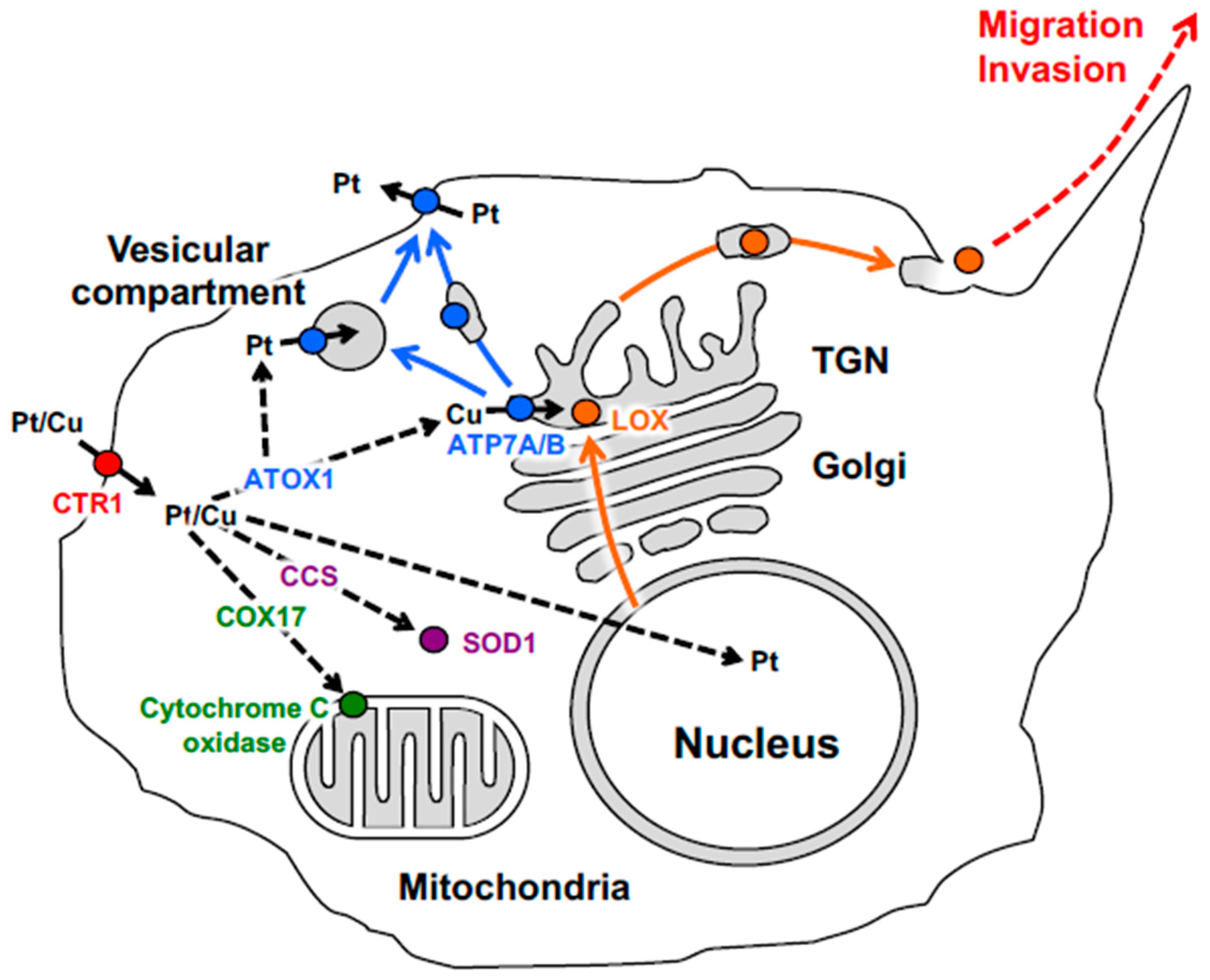
6. Role of ATP7A in Activation of LOX-Dependent Oncogenic Mechanisms
7. Prospective
Funding
Acknowledgments
Conflicts of Interest
References
- Zappa, F.; Failli, M.; De Matteis, M.A. The Golgi complex in disease and therapy. Curr. Opin. Cell Boil. 2018, 50, 102–116. [Google Scholar] [CrossRef] [PubMed]
- Baschieri, F.; Farhan, H. Endomembrane control of cell polarity: Relevance to cancer. Small GTPases 2015, 6, 104–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajiho, H.; Kajiho, Y.; Scita, G. Harnessing membrane trafficking to promote cancer spreading and invasion: The case of RAB2A. Small GTPases 2018, 9, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, M.; Baldassarre, M.; Beznoussenko, G.; Giacchetti, G.; Cao, J.; Zucker, S.; Luini, A.; Buccione, R. Intracellular processing and activation of membrane type 1 matrix metalloprotease depends on its partitioning into lipid domains. J. Cell Sci. 2004, 117, 6275–6287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etienne-Manneville, S. Polarity proteins in migration and invasion. Oncogene 2008, 27, 6970–6980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buccione, R.; Caldieri, G.; Ayala, I. Invadopodia: Specialized tumor cell structures for the focal degradation of the extracellular matrix. Cancer Metastasis Rev. 2009, 28, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Colanzi, A.; Corda, D. Mitosis controls the Golgi and the Golgi controls mitosis. Curr. Opin. Cell Boil. 2007, 19, 386–393. [Google Scholar] [CrossRef]
- Nevitt, T.; Ohrvik, H.; Thiele, D.J. Charting the travels of copper in eukaryotes from yeast to mammals. Biochim. Biophysica. Acta 2012, 1823, 1580–1593. [Google Scholar] [CrossRef] [Green Version]
- Brady, D.C.; Crowe, M.S.; Turski, M.L.; Hobbs, G.A.; Yao, X.; Chaikuad, A.; Knapp, S.; Xiao, K.; Campbell, S.L.; Thiele, D.J.; et al. Copper is required for oncogenic BRAF signalling and tumorigenesis. Nature 2014, 509, 492–496. [Google Scholar] [CrossRef] [Green Version]
- Turski, M.L.; Brady, D.C.; Kim, H.J.; Kim, B.E.; Nose, Y.; Counter, C.M.; Winge, D.R.; Thiele, D.J. A novel role for copper in Ras/mitogen-activated protein kinase signaling. Mol. Cell Biol. 2012, 32, 1284–1295. [Google Scholar] [CrossRef]
- Papa, L.; Manfredi, G.; Germain, D. SOD1, an unexpected novel target for cancer therapy. Genes Cancer 2014, 5, 15–21. [Google Scholar] [PubMed]
- Ishida, S.; Andreux, P.; Poitry-Yamate, C.; Auwerx, J.; Hanahan, D. Bioavailable copper modulates oxidative phosphorylation and growth of tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 19507–19512. [Google Scholar] [CrossRef] [Green Version]
- Barker, H.E.; Cox, T.R.; Erler, J.T. The rationale for targeting the LOX family in cancer. Nat. Rev. Cancer 2012, 12, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Díez, M.; Arroyo, M.; Cerdàn, F.; Muñoz, M.; Martin, M.; Balibrea, J. Serum and Tissue Trace Metal Levels in Lung Cancer. Oncology 1989, 46, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Zowczak, M.; Iskra, M.; Torlinski, L.; Cofta, S. Analysis of Serum Copper and Zinc Concentrations in Cancer Patients. Boil. Trace Element Res. 2001, 82, 1–8. [Google Scholar] [CrossRef]
- Tsai, C.-Y.; Finley, J.C.; Ali, S.S.; Patel, H.H.; Howell, S.B. Copper influx transporter 1 is required for FGF, PDGF and EGF-induced MAPK signaling. Biochem. Pharmacol. 2012, 84, 1007–1013. [Google Scholar] [CrossRef] [Green Version]
- Brady, D.C.; Crowe, M.S.; Greenberg, D.N.; Counter, C.M. Copper Chelation Inhibits BRAF(V600E)-Driven Melanomagenesis and Counters Resistance to BRAF(V600E) and MEK1/2 Inhibitors. Cancer Res. 2017, 77, 6240–6252. [Google Scholar] [CrossRef]
- Yoshii, J.; Yoshiji, H.; Kuriyama, S.; Ikenaka, Y.; Noguchi, R.; Okuda, H.; Tsujinoue, H.; Nakatani, T.; Kishida, H.; Nakae, D.; et al. The copper-chelating agent, trientine, suppresses tumor development and angiogenesis in the murine hepatocellular carcinoma cells. Int. J. Cancer 2001, 94, 768–773. [Google Scholar] [CrossRef]
- Chan, N.; Willis, A.; Kornhauser, N.; Ward, M.M.; Lee, S.B.; Nackos, E.; Seo, B.R.; Chuang, E.; Cigler, T.; Moore, A.; et al. Influencing the Tumor Microenvironment: A Phase II Study of Copper Depletion Using Tetrathiomolybdate in Patients with Breast Cancer at High Risk for Recurrence and in Preclinical Models of Lung Metastases. Clin. Cancer Res. 2017, 23, 666–676. [Google Scholar] [CrossRef]
- Komatsu, M.; Sumizawa, T.; Mutoh, M.; Chen, Z.S.; Terada, K.; Furukawa, T.; Yang, X.L.; Gao, H.; Miura, N.; Sugiyama, T.; et al. Copper-transporting P-type adenosine triphosphatase (ATP7B) is associated with cisplatin resistance. Cancer Res. 2000, 60, 1312–1316. [Google Scholar]
- Katano, K.; Kondo, A.; Safaei, R.; Holzer, A.; Samimi, G.; Mishima, M.; Kuo, Y.-M.; Rochdi, M.; Howell, S.B. Acquisition of resistance to cisplatin is accompanied by changes in the cellular pharmacology of copper. Cancer Res. 2002, 62, 6559–6565. [Google Scholar] [PubMed]
- Samimi, G.; Katano, K.; Holzer, A.K.; Safaei, R.; Howell, S.B. Modulation of the Cellular Pharmacology of Cisplatin and Its Analogs by the Copper Exporters ATP7A and ATP7B. Mol. Pharmacol. 2004, 66, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulikas, T.; Vougiouka, M. Cisplatin and platinum drugs at the molecular level. (Review). Oncol. Rep. 2003, 10, 1663–1682. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed]
- Safaei, R.; Howell, S.B. Copper transporters regulate the cellular pharmacology and sensitivity to Pt drugs. Crit. Rev. Oncol. 2005, 53, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.T.; Chen, H.H.; Song, I.S.; Savaraj, N.; Ishikawa, T. The roles of copper transporters in cisplatin resistance. Cancer Metastasis Rev. 2007, 26, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Howell, S.B.; Safaei, R.; Larson, C.A.; Sailor, M.J. Copper Transporters and the Cellular Pharmacology of the Platinum-Containing Cancer Drugs. Mol. Pharmacol. 2010, 77, 887–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samimi, G.; Varki, N.M.; Wilczynski, S.; Safaei, R.; Alberts, D.S.; Howell, S.B. Increase in expression of the copper transporter ATP7A during platinum drug-based treatment is associated with poor survival in ovarian cancer patients. Clin. Cancer Res. 2003, 9, 9. [Google Scholar]
- Nakayama, K.; Kanzaki, A.; Terada, K.; Mutoh, M.; Ogawa, K.; Sugiyama, T.; Takenoshita, S.; Itoh, K.; Yaegashi, N.; Miyazaki, K.; et al. Prognostic value of the Cu-transporting ATPase in ovarian carcinoma patients receiving cisplatin-based chemotherapy. Clin. Cancer Res. 2004, 10, 2804–2811. [Google Scholar] [CrossRef]
- Miyashita, H.; Nitta, Y.; Mori, S.; Kanzaki, A.; Nakayama, K.; Terada, K.; Sugiyama, T.; Kawamura, H.; Sato, A.; Morikawa, H.; et al. Expression of copper-transporting P-type adenosine triphosphatase (ATP7B) as a chemoresistance marker in human oral squamous cell carcinoma treated with cisplatin. Oral Oncol. 2003, 39, 157–162. [Google Scholar] [CrossRef]
- Higashimoto, M.; Kanzaki, A.; Shimakawa, T.; Konno, S.; Naritaka, Y.; Nitta, Y.; Mori, S.; Shirata, S.; Yoshida, A.; Terada, K.; et al. Expression of copper-transporting P-type adenosine triphosphatase in human esophageal carcinoma. Int. J. Mol. Med. 2003, 11, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Aida, T.; Takebayashi, Y.; Shimizu, T.; Okamura, C.; Higasimoto, M.; Kanzaki, A.; Nakayama, K.; Terada, K.; Sugiyama, T.; Miyazaki, K.; et al. Expression of copper-transporting P-type adenosine triphosphatase (ATP7B) as a prognostic factor in human endometrial carcinoma. Gynecol Oncol 2005, 97, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Inoue, Y.; Kodama, H.; Yamazaki, H.; Kawai, K.; Suemizu, H.; Masuda, R.; Iwazaki, M.; Yamada, S.; Ueyama, Y.; et al. Expression of copper-transporting P-type adenosine triphosphatase (ATP7B) correlates with cisplatin resistance in human non-small cell lung cancer xenografts. Oncol. Rep. 2008, 20, 265–270. [Google Scholar] [PubMed]
- Ohbu, M.; Ogawa, K.; Konno, S.; Kanzaki, A.; Terada, K.; Sugiyama, T.; Takebayashi, Y. Copper-transporting, P-type adenosine triphosphatase (ATP7B) is expressed in human gastric carcinoma. Cancer Lett. 2003, 189, 33–38. [Google Scholar] [CrossRef]
- Kanzaki, A.; Toi, M.; Neamati, N.; Miyashita, H.; Oubu, M.; Nakayama, K.; Bando, H.; Ogawa, K.; Mutoh, M.; Mori, S.; et al. Copper-transporting P-Type Adenosine Triphosphatase (ATP7B) Is Expressed in Human Breast Carcinoma. Jpn. J. Cancer Res. 2002, 93, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Chen, M.; Chen, T.; Thakur, A. Expression of the copper transporters hCtr1, ATP7A and ATP7B is associated with the response to chemotherapy and survival time in patients with resected non-small cell lung cancer. Oncol. Lett. 2015, 10, 2584–2590. [Google Scholar] [CrossRef] [Green Version]
- Lutsenko, S. Human copper homeostasis: A network of interconnected pathways. Curr. Opin. Chem. Boil. 2010, 14, 211–217. [Google Scholar] [CrossRef]
- Camakaris, J.; Voskoboinik, I.; Mercer, J. Molecular Mechanisms of Copper Homeostasis. Biochem. Biophys. Res. Commun. 1999, 261, 225–232. [Google Scholar] [CrossRef]
- Lee, J.; Prohaska, J.R.; Dagenais, S.L.; Glover, T.W.; Thiele, D.J. Isolation of a murine copper transporter gene, tissue specific expression and functional complementation of a yeast copper transport mutant. Gene 2000, 254, 87–96. [Google Scholar] [CrossRef]
- Öhrvik, H.; Nose, Y.; Wood, L.K.; Kim, B.E.; Gleber, S.C.; Ralle, M.; Thiele, D.J. Ctr2 regulates biogenesis of a cleaved form of mammalian Ctr1 metal transporter lacking the copper- and cisplatin-binding ecto-domain. Proc. Natl. Acad. Sci. USA 2013, 110, E4279–E4288. [Google Scholar] [CrossRef]
- van den Berghe, P.V.; Folmer, D.E.; Malingre, H.E.; van Beurden, E.; Klomp, A.E.; van de Sluis, B.; Merkx, M.; Berger, R.; Klomp, L.W. Human copper transporter 2 is localized in late endosomes and lysosomes and facilitates cellular copper uptake. Biochem. J. 2007, 407, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Culotta, V.C.; Klomp, L.W.J.; Strain, J.; Casareno, R.L.B.; Krems, B.; Gitlin, J.D. The Copper Chaperone for Superoxide Dismutase. J. Boil. Chem. 1997, 272, 23469–23472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, P.C.; Waggoner, D.; Subramaniam, J.R.; Tessarollo, L.; Bartnikas, T.B.; Culotta, V.C.; Price, D.L.; Rothstein, J.; Gitlin, J.D. Copper chaperone for superoxide dismutase is essential to activate mammalian Cu/Zn superoxide dismutase. Proc. Natl. Acad. Sci. USA 2000, 97, 2886–2891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glerum, D.M.; Shtanko, A.; Tzagoloff, A. Characterization of COX17, a yeast gene involved in copper metabolism and assembly of cytochrome oxidase. J. Biol. Chem. 1996, 271, 14504–14509. [Google Scholar] [CrossRef] [PubMed]
- Cobine, P.A.; Pierrel, F.; Winge, D.R. Copper trafficking to the mitochondrion and assembly of copper metalloenzymes. Biochim. Biophys. Acta. 2006, 1763, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Hamza, I.; Schaefer, M.; Klomp, L.W.J.; Gitlin, J.D. Interaction of the copper chaperone HAH1 with the Wilson disease protein is essential for copper homeostasis. Proc. Natl. Acad. Sci. USA 1999, 96, 13363–13368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamza, I.; Prohaska, J.; Gitlin, J.D. Essential role for Atox1 in the copper-mediated intracellular trafficking of the Menkes ATPase. Proc. Natl. Acad. Sci. USA 2003, 100, 1215–1220. [Google Scholar] [CrossRef] [Green Version]
- Ebadi, M.; Iversen, P. Metallothionein in carcinogenesis and cancer chemotherapy. Gen. Pharmacol. Vasc. Syst. 1994, 25, 1297–1310. [Google Scholar] [CrossRef]
- Bhattacharjee, A.; Chakraborty, K.; Shukla, A. Cellular copper homeostasis: Current concepts on its interplay with glutathione homeostasis and its implication in physiology and human diseases. Metallomics 2017, 9, 1376–1388. [Google Scholar] [CrossRef]
- Petris, M.J.; Mercer, J.F.; Culvenor, J.G.; Lockhart, P.; Gleeson, P.A.; Camakaris, J. Ligand-regulated transport of the Menkes copper P-type ATPase efflux pump from the Golgi apparatus to the plasma membrane: A novel mechanism of regulated trafficking. EMBO J. 1996, 15, 6084–6095. [Google Scholar] [CrossRef]
- La Fontaine, S.; Theophilos, M.B.; Firth, S.D.; Gould, R.; Parton, R.G.; Mercer, J.F. Effect of the toxic milk mutation (tx) on the function and intracellular localization of Wnd, the murine homologue of the Wilson copper ATPase. Hum. Mol. Genet. 2001, 10, 361–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, M.; Jones, C.; Solioz, M.; Dameron, C. Intracellular copper routing: The role of copper chaperones. Trends Biochem. Sci. 2000, 25, 29–32. [Google Scholar] [CrossRef]
- Huffman, D.L.; O’Halloran, T.V. Function, structure, and mechanism of intracellular copper trafficking proteins. Annu. Rev. Biochem. 2001, 70, 677–701. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Smith, K.; Petris, M.J. Cisplatin stabilizes a multimeric complex of the human Ctr1 copper transporter: requirement for the extracellular methionine-rich clusters. J. Boil. Chem. 2004, 279, 46393–46399. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Bertini, I.; Blaževitš, O.; Calderone, V.; Cantini, F.; Mao, J.; Trapananti, A.; Vieru, M.; Amori, I.; Cozzolino, M.; et al. Interaction of Cisplatin with Human Superoxide Dismutase. J. Am. Chem. Soc. 2012, 134, 7009–7014. [Google Scholar] [CrossRef] [PubMed]
- Palm, M.E.; Weise, C.F.; Lundin, C.; Wingsle, G.; Nygren, Y.; Björn, E.; Naredi, P.; Wolf-Watz, M.; Wittung-Stafshede, P. Cisplatin binds human copper chaperone Atox1 and promotes unfolding in vitro. Proc. Natl. Acad. Sci. USA 2011, 108, 6951–6956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palm-Espling, M.E.; Lundin, C.; Bjorn, E.; Naredi, P.; Wittung-Stafshede, P. Interaction between the anticancer drug Cisplatin and the copper chaperone Atox1 in human melanoma cells. Protein Pept. Lett. 2014, 21, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Cheng, Q.; Wang, Z.; Xi, Z.; Xu, D.; Liu, Y. Cisplatin binds to human copper chaperone Cox17: The mechanistic implication of drug delivery to mitochondria. Chem. Commun. 2014, 50, 2667–2669. [Google Scholar] [CrossRef]
- Dolgova, N.V.; Nokhrin, S.; Yu, C.H.; George, G.N.; Dmitriev, O.Y. Copper chaperone Atox1 interacts with the metal-binding domain of Wilson’s disease protein in cisplatin detoxification. Biochem. J. 2013, 454, 147–156. [Google Scholar] [CrossRef]
- Voskoboinik, I.; Strausak, D.; Greenough, M.; Brooks, H.; Petris, M.; Smith, S.; Mercer, J.F.; Camakaris, J. Functional analysis of the N-terminal CXXC metal-binding motifs in the human Menkes copper-transporting P-type ATPase expressed in cultured mammalian cells. J. Boil. Chem. 1999, 274, 22008–22012. [Google Scholar] [CrossRef]
- Voskoboinik, I.; Greenough, M.; La Fontaine, S.; Mercer, J.F.; Camakaris, J. Functional studies on the Wilson copper P-type ATPase and toxic milk mouse mutant. Biochem. Biophys. Res. Commun. 2001, 281, 966–970. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.F.B.; Livingston, J.; Hall, B.; Paynter, J.A.; Begy, C.; Chandrasekharappa, S.; Lockhart, P.; Grimes, A.; Bhave, M.; Siemieniak, D.; et al. Isolation of a partial candidate gene for Menkes disease by positional cloning. Nat. Genet. 1993, 3, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Chelly, J.; Tümer, Z.; Tønnesen, T.; Petterson, A.; Ishikawa-Brush, Y.; Tommerup, N.; Horn, N.; Monaco, A.P. Isolation of a candidate gene for Menkes disease that encodes a potential heavy metal binding protein. Nat. Genet. 1993, 3, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Bull, P.C.; Thomas, G.R.; Rommens, J.M.; Forbes, J.R.; Cox, D.W. The Wilson disease gene is a putative copper transporting P–type ATPase similar to the Menkes gene. Nat. Genet. 1993, 5, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Tanzi, R.; Petrukhin, K.; Chernov, I.; Pellequer, J.-L.; Wasco, W.; Ross, B.; Romano, D.; Parano, E.; Pavone, L.; Brzustowicz, L.; et al. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat. Genet. 1993, 5, 344–350. [Google Scholar] [CrossRef]
- Lutsenko, S.; Barnes, N.L.; Bartee, M.Y.; Dmitriev, O.Y. Function and Regulation of Human Copper-Transporting ATPases. Physiol. Rev. 2007, 87, 1011–1046. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hodgkinson, V.; Zhu, S.; Weisman, G.A.; Petris, M.J. Advances in the Understanding of Mammalian Copper Transporters12. Adv. Nutr. 2011, 2, 129–137. [Google Scholar] [CrossRef]
- Polishchuk, R.; Lutsenko, S. GOLGI IN COPPER HOMEOSTASIS: A VIEW FROM THE MEMBRANE TRAFFICKING FIELD. Histochem. Cell Boil. 2013, 140, 285–295. [Google Scholar] [CrossRef]
- Dmitriev, O.; Tsivkovskii, R.; Abildgaard, F.; Morgan, C.T.; Markley, J.L.; Lutsenko, S. Solution structure of the N-domain of Wilson disease protein: Distinct nucleotide-binding environment and effects of disease mutations. Proc. Natl. Acad. Sci. USA 2006, 103, 5302–5307. [Google Scholar] [CrossRef] [Green Version]
- Tsivkovskii, R.; Eisses, J.F.; Kaplan, J.H.; Lutsenko, S. Functional properties of the copper-transporting ATPase ATP7B (the Wilson’s disease protein) expressed in insect cells. J. Biol. Chem. 2002, 277, 976–983. [Google Scholar] [CrossRef]
- Gourdon, P.; Liu, X.; Skjorringe, T.; Morth, J.P.; Moller, L.B.; Pedersen, B.P.; Nissen, P. Crystal structure of a copper-transporting PIB-type ATPase. Nature 2011, 475, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Gourdon, P.; Sitsel, O.; Karlsen, J.L.; Møller, L.B.; Nissen, P. Structural models of the human copper P-type ATPases ATP7A and ATP7B. Boil. Chem. 2012, 393, 205–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattle, D.; Zhang, L.; Sitsel, O.; Pedersen, L.T.; Moncelli, M.R.; Tadini-Buoninsegni, F.; Gourdon, P.; Rees, D.C.; Nissen, P.; Meloni, G. A sulfur-based transport pathway in Cu+-ATPases. EMBO Rep. 2015, 16, 728–740. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.; Mattle, D.; Sitsel, O.; Klymchuk, T.; Nielsen, A.M.; Moller, L.B.; White, S.H.; Nissen, P.; Gourdon, P. Copper-transporting P-type ATPases use a unique ion-release pathway. Nat. Struct. Mol. Biol. 2014, 21, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Petris, M.J.; Voskoboinik, I.; Cater, M.; Smith, K.; Kim, B.E.; Llanos, R.M.; Strausak, D.; Camakaris, J.; Mercer, J.F. Copper-regulated Trafficking of the Menkes Disease Copper ATPase Is Associated with Formation of a Phosphorylated Catalytic Intermediate. J. Boil. Chem. 2002, 277, 46736–46742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banci, L.; Bertini, I.; Cantini, F.; Migliardi, M.; Natile, G.; Nushi, F.; Rosato, A. Solution Structures of the Actuator Domain of ATP7A and ATP7B, the Menkes and Wilson Disease Proteins. Biochemistry 2009, 48, 7849–7855. [Google Scholar] [CrossRef] [PubMed]
- Dagenais, S.L.; Adam, A.N.; Innis, J.W.; Glover, T.W. A novel frameshift mutation in exon 23 of ATP7A (MNK) results in occipital horn syndrome and not in Menkes disease. Am. J. Hum. Genet. 2001, 69, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Steveson, T.C.; Ciccotosto, G.D.; Ma, X.M.; Mueller, G.P.; Mains, R.E.; Eipper, B.A. Menkes protein contributes to the function of peptidylglycine alpha-amidating monooxygenase. Endocrinology 2003, 144, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Strausak, D.; Mercer, J.F.; Petris, M.J. The Menkes copper transporter is required for the activation of tyrosinase. Hum. Mol. Genet. 2000, 9, 2845–2851. [Google Scholar]
- Qin, Z.; Itoh, S.; Jeney, V.; Ushio-Fukai, M.; Fukai, T. Essential role for the Menkes ATPase in activation of extracellular superoxide dismutase: Implication for vascular oxidative stress. FASEB J. 2006, 20, 334–336. [Google Scholar] [CrossRef] [PubMed]
- Terada, K.; Nakako, T.; Yang, X.-L.; Iida, M.; Aiba, N.; Minamiya, Y.; Nakai, M.; Sakaki, T.; Miura, N.; Sugiyama, T. Restoration of Holoceruloplasmin Synthesis in LEC Rat after Infusion of Recombinant Adenovirus Bearing WND cDNA. J. Boil. Chem. 1998, 273, 1815–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roelofsen, H.; Wolters, H.; Van Luyn‡, M.J.; Miura§, N.; Kuipers, F.; Vonk, R.J. Copper-induced apical trafficking of ATP7B in polarized hepatoma cells provides a mechanism for biliary copper excretion. Gastroenterol. 2000, 119, 782–793. [Google Scholar] [CrossRef] [PubMed]
- Braiterman, L.; Nyasae, L.; Guo, Y.; Bustos, R.; Lutsenko, S.; Hubbard, A. Apical targeting and Golgi retention signals reside within a 9-amino acid sequence in the copper-ATPase, ATP7B. Am. J. Physiol. Liver Physiol. 2009, 296, G433–G444. [Google Scholar] [CrossRef] [PubMed]
- Polishchuk, E.V.; Concilli, M.; Iacobacci, S.; Chesi, G.; Pastore, N.; Piccolo, P.; Paladino, S.; Baldantoni, D.; Van Ijzendoorn, S.C.; Chan, J.; et al. Wilson Disease Protein ATP7B Utilizes Lysosomal Exocytosis to Maintain Copper Homeostasis. Dev. Cell 2014, 29, 686–700. [Google Scholar] [CrossRef] [Green Version]
- Gitlin, J.D. Wilson disease. Gastroenterology 2003, 125, 1868–1877. [Google Scholar] [CrossRef] [PubMed]
- Kaler, S.G. ATP7A-related copper transport diseases—emerging concepts and future trends. Nat. Rev. Neurol. 2011, 7, 15–29. [Google Scholar] [CrossRef]
- Monty, J.-F.; Llanos, R.M.; Mercer, J.F.B.; Kramer, D.R. Copper exposure induces trafficking of the menkes protein in intestinal epithelium of ATP7A transgenic mice. J. Nutr. 2005, 135, 2762–2766. [Google Scholar] [CrossRef]
- Greenough, M.; Pase, L.; Voskoboinik, I.; Petris, M.J.; O’Brien, A.W.; Camakaris, J. Signals regulating trafficking of Menkes (MNK; ATP7A) copper-translocating P-type ATPase in polarized MDCK cells. Am. J. Physiol. Physiol. 2004, 287, C1463–C1471. [Google Scholar] [CrossRef]
- Hellman, N.E.; Gitlin, J.D. Ceruloplasmin metabolism and function. Annu. Rev. Nutr 2002, 22, 439–458. [Google Scholar] [CrossRef]
- Polishchuk, E.V.; Polishchuk, R.S. The emerging role of lysosomes in copper homeostasis. Metallomics 2016, 8, 853–862. [Google Scholar] [CrossRef]
- Guo, Y.; Nyasae, L.; Braiterman, L.T.; Hubbard, A.L. NH2-terminal signals in ATP7B Cu-ATPase mediate its Cu-dependent anterograde traffic in polarized hepatic cells. Am. J. Physiol. Liver Physiol. 2005, 289, G904–G916. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Lutsenko, S. Human copper transporters: Mechanism, role in human diseases and therapeutic potential. Futur. Med. Chem. 2009, 1, 1125–1142. [Google Scholar] [CrossRef] [PubMed]
- Stornetta, A.; Zimmermann, M.; Cimino, G.D.; Henderson, P.T.; Sturla, S.J. DNA Adducts from Anticancer Drugs as Candidate Predictive Markers for Precision Medicine. Chem. Res. Toxicol. 2017, 30, 388–409. [Google Scholar] [CrossRef] [PubMed]
- Florea, A.M.; Busselberg, D. Cisplatin as an anti-tumor drug: Cellular mechanisms of activity, drug resistance and induced side effects. Cancers (Basel) 2011, 3, 1351–1371. [Google Scholar] [CrossRef] [PubMed]
- Amable, L. Cisplatin resistance and opportunities for precision medicine. Pharmacol Res. 2016, 106, 27–36. [Google Scholar] [CrossRef]
- Suzuki, T.; Nishio, K.; Tanabe, S. The MRP Family and Anticancer Drug Metabolism. Curr. Drug Metab. 2001, 2, 367–377. [Google Scholar] [CrossRef]
- Dolgova, N.V.; Yu, C.; Cvitkovic, J.P.; Hodak, M.; Nienaber, K.H.; Summers, K.L.; Cotelesage, J.J.H.; Bernholc, J.; Kaminski, G.A.; Pickering, I.J.; et al. Binding of Copper and Cisplatin to Atox1 Is Mediated by Glutathione through the Formation of Metal-Sulfur Clusters. Biochemistry 2017, 56, 3129–3141. [Google Scholar] [CrossRef]
- Ishida, S.; Lee, J.; Thiele, D.J.; Herskowitz, I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc. Natl. Acad. Sci. USA 2002, 99, 14298–14302. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Okuda, T.; Holzer, A.; Howell, S.B. The Copper Transporter CTR1 Regulates Cisplatin Uptake in Saccharomyces cerevisiae. Mol. Pharmacol. 2002, 62, 1154–1159. [Google Scholar] [CrossRef]
- Lee, Y.Y.; Choi, C.H.; Do, I.G.; Song, S.Y.; Lee, W.; Park, H.S.; Song, T.J.; Kim, M.K.; Kim, T.J.; Lee, J.W.; et al. Prognostic value of the copper transporters, CTR1 and CTR2, in patients with ovarian carcinoma receiving platinum-based chemotherapy. Gynecol. Oncol. 2011, 122, 361–365. [Google Scholar] [CrossRef]
- Ohrvik, H.; Logeman, B.; Turk, B.; Reinheckel, T.; Thiele, D.J. Cathepsin Protease Controls Copper and Cisplatin Accumulation via Cleavage of the Ctr1 Metal-binding Ectodomain. J. Boil. Chem. 2016, 291, 13905–13916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safaei, R.; Maktabi, M.H.; Blair, B.G.; Larson, C.A.; Howell, S.B. Effects of the loss of Atox1 on the cellular pharmacology of cisplatin. J. Inorg. Biochem. 2009, 103, 333–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samimi, G.; Katano, K.; Holzer, A.K.; Rochdi, M.; Tomioka, M.; Howell, S.B.; Safaei, R.; Goodman, M. Increased Expression of the Copper Efflux Transporter ATP7A Mediates Resistance to Cisplatin, Carboplatin, and Oxaliplatin in Ovarian Cancer Cells. Clin. Cancer Res. 2004, 10, 4661–4669. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Kanzaki, A.; Ogawa, K.; Miyazaki, K.; Neamati, N.; Takebayashi, Y. Copper-transporting P-type adenosine triphosphatase (ATP7B) as a cisplatin based chemoresistance marker in ovarian carcinoma: Comparative analysis with expression of MDR1, MRP1, MRP2, LRP and BCRP. Int. J. Cancer 2002, 101, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Mangala, L.S.; Zuzel, V.; Schmandt, R.; LeShane, E.S.; Halder, J.B.; Armaiz-Pena, G.N.; Spannuth, W.A.; Tanaka, T.; Shahzad, M.M.; Lin, Y.G.; et al. Therapeutic Targeting of ATP7B in Ovarian Carcinoma. Clin. Cancer Res. 2009, 15, 3770–3780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.H.; Qiu, M.Z.; Zeng, Z.L.; Luo, H.Y.; Wu, W.J.; Wang, F.; Wang, Z.Q.; Zhang, D.S.; Li, Y.H.; Xu, R.H. Copper-transporting P-type adenosine triphosphatase (ATP7A) is associated with platinum-resistance in non-small cell lung cancer (NSCLC). J. Transl. Med. 2012, 10, 21. [Google Scholar] [CrossRef]
- Zhu, S.; Shanbhag, V.; Wang, Y.; Lee, J.; Petris, M. A Role for The ATP7A Copper Transporter in Tumorigenesis and Cisplatin Resistance. J. Cancer 2017, 8, 1952–1958. [Google Scholar] [CrossRef] [Green Version]
- Safaei, R.; Otani, S.; Larson, B.J.; Rasmussen, M.L.; Howell, S.B. Transport of cisplatin by the copper efflux transporter ATP7B. Mol. Pharmacol. 2008, 73, 461–468. [Google Scholar] [CrossRef]
- Dmitriev, O.Y. Mechanism of tumor resistance to cisplatin mediated by the copper transporter ATP7B. Biochem. Cell Biol 2011, 89, 138–147. [Google Scholar] [CrossRef]
- Tadini-Buoninsegni, F.; Bartolommei, G.; Moncelli, M.R.; Inesi, G.; Galliani, A.; Sinisi, M.; Losacco, M.; Natile, G.; Arnesano, F. Translocation of platinum anticancer drugs by human copper ATPases ATP7A and ATP7B. Angew. Chem. Int. Ed. Engl. 2014, 53, 1297–1301. [Google Scholar] [CrossRef]
- Safaei, R.; Adams, P.L.; Maktabi, M.H.; Mathews, R.A.; Howell, S.B. The CXXC motifs in the metal binding domains are required for ATP7B to mediate resistance to cisplatin. J. Inorg. Biochem. 2012, 110, 8–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartwig, C.; Zlatic, S.A.; Wallin, M.; Vrailas-Mortimer, A.; Fahrni, C.J.; Faundez, V. Trafficking mechanisms of P-type ATPase copper transporters. Curr. Opin. Cell Boil. 2019, 59, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Katano, K.; Safaei, R.; Samimi, G.; Holzer, A.; Tomioka, M.; Goodman, M.; Howell, S.B. Confocal Microscopic Analysis of the Interaction between Cisplatin and the Copper Transporter ATP7B in Human Ovarian Carcinoma Cells. Clin. Cancer Res. 2004, 10, 4578–4588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalayda, G.V.; Zhang, G.; Abraham, T.; Tanke, H.J.; Reedijk, J. Application of Fluorescence Microscopy for Investigation of Cellular Distribution of Dinuclear Platinum Anticancer Drugs. J. Med. Chem. 2005, 48, 5191–5202. [Google Scholar] [CrossRef] [PubMed]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomes as mediators of drug resistance in cancer. Drug Resist. Updat. 2016, 24, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Safaei, R. Abnormal lysosomal trafficking and enhanced exosomal export of cisplatin in drug-resistant human ovarian carcinoma cells. Mol. Cancer Ther. 2005, 4, 1595–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Kagan, H.M.; Li, W. Lysyl oxidase: Properties, specificity, and biological roles inside and outside of the cell. J. Cell Biochem. 2003, 88, 660–672. [Google Scholar] [CrossRef]
- Lucero, H.A.; Kagan, H.M. Lysyl oxidase: An oxidative enzyme and effector of cell function. Cell. Mol. Life Sci. 2006, 63, 2304–2316. [Google Scholar] [CrossRef]
- Baker, A.M.; Bird, D.; Lang, G.; Cox, T.R.; Erler, J.T. Lysyl oxidase enzymatic function increases stiffness to drive colorectal cancer progression through FAK. Oncogene 2013, 32, 1863–1868. [Google Scholar] [CrossRef]
- Payne, S.L.; Fogelgren, B.; Hess, A.R.; Seftor, E.A.; Wiley, E.L.; Fong, S.F.; Csiszar, K.; Hendrix, M.J.; Kirschmann, D.A. Lysyl Oxidase Regulates Breast Cancer Cell Migration and Adhesion through a Hydrogen Peroxide-Mediated Mechanism. Cancer Res. 2005, 65, 11429–11436. [Google Scholar] [CrossRef] [PubMed]
- Salvador, F.; Martin, A.; López-Menéndez, C.; Moreno-Bueno, G.; Santos, V.; Vazquez-Naharro, A.; Santamaria, P.G.; Morales, S.; Dubus, P.R.; Muinelo-Romay, L.; et al. Lysyl oxidase-like protein LOXL2 promotes lung metastasis of breast cancer. Cancer Res. 2017, 77, 5846–5859. [Google Scholar] [CrossRef] [PubMed]
- Barker, H.E.; Chang, J.; Cox, T.R.; Lang, G.; Bird, D.; Nicolau, M.; Evans, H.R.; Gartland, A.; Erler, J.T. LOXL2-mediated matrix remodeling in metastasis and mammary gland involution. Cancer Res. 2011, 71, 1561–1572. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.G.; Dong, S.M.; Oshima, A.; Kim, W.H.; Lee, H.M.; Lee, S.A.; Kwon, S.H.; Lee, J.H.; Lee, J.M.; Jeong, J.; et al. LOXL2 expression is associated with invasiveness and negatively influences survival in breast cancer patients. Breast Cancer Res. Treat. 2013, 141, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Weise, J.B.; Rudolph, P.; Heiser, A.; Kruse, M.L.; Hedderich, J.; Cordes, C.; Hoffmann, M.; Brant, O.; Ambrosch, P.; Csiszar, K.; et al. LOXL4 is a selectively expressed candidate diagnostic antigen in head and neck cancer. Eur. J. Cancer 2008, 44, 1323–1331. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.T.; Kim, M.S.; Seo, J.Y.; Kim, H.C.; Kim, Y. Purification of enzymatically active human lysyl oxidase and lysyl oxidase-like protein from Escherichia coli inclusion bodies. Protein Expr. Purif. 2003, 31, 240–246. [Google Scholar] [CrossRef]
- Hajdu, I.; Kardos, J.; Major, B.; Fabo, G.; Lorincz, Z.; Cseh, S.; Dorman, G. Inhibition of the LOX enzyme family members with old and new ligands. Selectivity analysis revisited. Bioorg. Med. Chem. Lett. 2018, 28, 3113–3118. [Google Scholar] [CrossRef] [PubMed]
- Schilter, H.; Findlay, A.D.; Perryman, L.; Yow, T.T.; Moses, J.; Zahoor, A.; Turner, C.I.; Deodhar, M.; Foot, J.S.; Zhou, W.; et al. The lysyl oxidase like 2/3 enzymatic inhibitor, PXS-5153A, reduces crosslinks and ameliorates fibrosis. J. Cell Mol. Med. 2019, 23, 1759–1770. [Google Scholar] [PubMed]
- Amendola, P.G.; Reuten, R.; Erler, J.T. Interplay Between LOX Enzymes and Integrins in the Tumor Microenvironment. Cancers 2019, 11, 729. [Google Scholar] [CrossRef]
- Shanbhag, V.; Jasmer-McDonald, K.; Zhu, S.; Martin, A.L.; Gudekar, N.; Khan, A.; Ladomersky, E.; Singh, K.; Weisman, G.A.; Petris, M.J. ATP7A delivers copper to the lysyl oxidase family of enzymes and promotes tumorigenesis and metastasis. Proc. Natl. Acad. Sci. USA 2019, 116, 6836–6841. [Google Scholar] [CrossRef] [Green Version]
- Chesi, G.; Hegde, R.N.; Iacobacci, S.; Concilli, M.; Parashuraman, S.; Festa, B.P.; Polishchuk, E.V.; Di Tullio, G.; Carissimo, A.; Montefusco, S.; et al. Identification of p38 MAPK and JNK as new targets for correction of Wilson disease-causing ATP7B mutants. Hepatology 2016, 63, 1842–1859. [Google Scholar] [CrossRef] [PubMed]
- Comstra, H.S.; McArthy, J.; Rudin-Rush, S.; Hartwig, C.; Gokhale, A.; Zlatic, S.A.; Blackburn, J.B.; Werner, E.; Petris, M.; D’Souza, P.; et al. The interactome of the copper transporter ATP7A belongs to a network of neurodevelopmental and neurodegeneration factors. eLife 2017, 6, 1155. [Google Scholar] [CrossRef] [PubMed]
- Fang, T.; Chen, W.; Sheng, Y.; Yuan, S.; Tang, Q.; Li, G.; Huang, G.; Su, J.; Zhang, X.; Zang, J.; et al. Tetrathiomolybdate induces dimerization of the metal-binding domain of ATPase and inhibits platination of the protein. Nat. Commun. 2019, 10, 186. [Google Scholar] [CrossRef] [PubMed]
- Mariniello, M.; Petruzzelli, R.; Wanderlingh, L.G.; Montagna, R.L.; Carissimo, A.; Pane, F.; Amoresano, A.; Medina, D.; Polishchuk, R.S. Synthetic lethality screening reveals FDA-approved drugs that overcome ATP7B-mediated tolerance of tumor cells to cisplatin. BioRxiv 2019, 568535. [Google Scholar]
- Polishchuk, E.V.; Merolla, A.; Lichtmannegger, J.; Romano, A.; Indrieri, A.; Ilyechova, E.Y.; Concilli, M.; De Cegli, R.; Crispino, R.; Mariniello, M.; et al. Activation of Autophagy, Observed in Liver Tissues From Patients With Wilson Disease and From Atp7b-Deficient Animals, Protects Hepatocytes From Copper-Induced Apoptosis. Gastroenterology 2019, 156, 1173–1189. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Su, J.; Xu, Y.; Kang, J.; Li, H.; Zhang, L.; Yi, H.; Xiang, X.; Liu, F.; Sun, L. p62/SQSTM1 involved in cisplatin resistance in human ovarian cancer cells by clearing ubiquitinated proteins. Eur. J. Cancer 2011, 47, 1585–1594. [Google Scholar] [CrossRef]
- Wang, J.; Wu, G.S. Role of Autophagy in Cisplatin Resistance in Ovarian Cancer Cells. J. Boil. Chem. 2014, 289, 17163–17173. [Google Scholar] [CrossRef] [Green Version]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petruzzelli, R.; Polishchuk, R.S. Activity and Trafficking of Copper-Transporting ATPases in Tumor Development and Defense against Platinum-Based Drugs. Cells 2019, 8, 1080. https://doi.org/10.3390/cells8091080
Petruzzelli R, Polishchuk RS. Activity and Trafficking of Copper-Transporting ATPases in Tumor Development and Defense against Platinum-Based Drugs. Cells. 2019; 8(9):1080. https://doi.org/10.3390/cells8091080
Chicago/Turabian StylePetruzzelli, Raffaella, and Roman S. Polishchuk. 2019. "Activity and Trafficking of Copper-Transporting ATPases in Tumor Development and Defense against Platinum-Based Drugs" Cells 8, no. 9: 1080. https://doi.org/10.3390/cells8091080
APA StylePetruzzelli, R., & Polishchuk, R. S. (2019). Activity and Trafficking of Copper-Transporting ATPases in Tumor Development and Defense against Platinum-Based Drugs. Cells, 8(9), 1080. https://doi.org/10.3390/cells8091080