Trends and Challenges in Tumor Anti-Angiogenic Therapies


Abstract
1. Introduction
2. Mechanisms of Angiogenesis
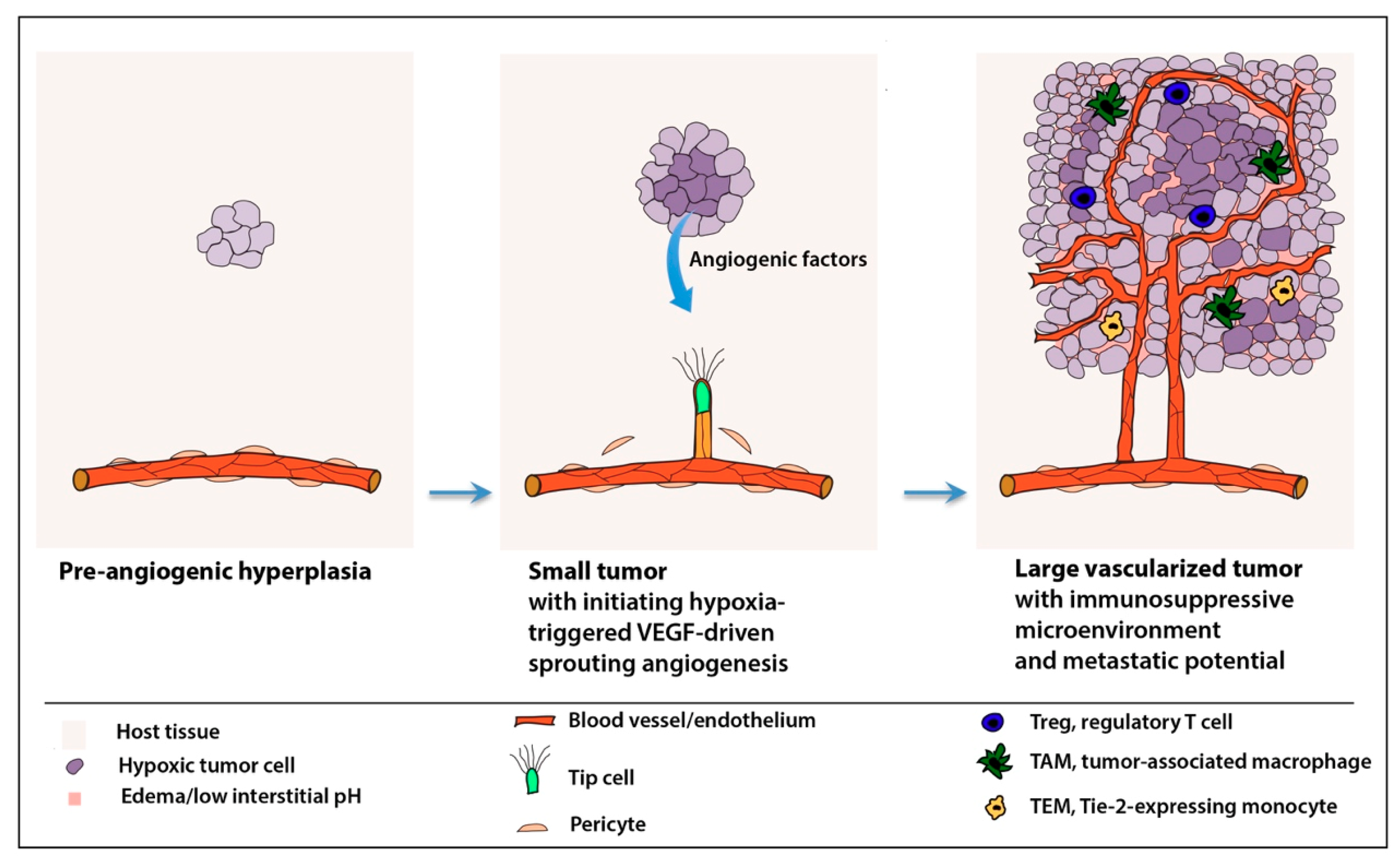
2.1. “Angiogenic Switch”
2.2. Normal vs. Pathological Angiogenesis—Similar, yet Distinct
3. Angiogenic Factors and Signaling Pathways of Tumor Angiogenesis
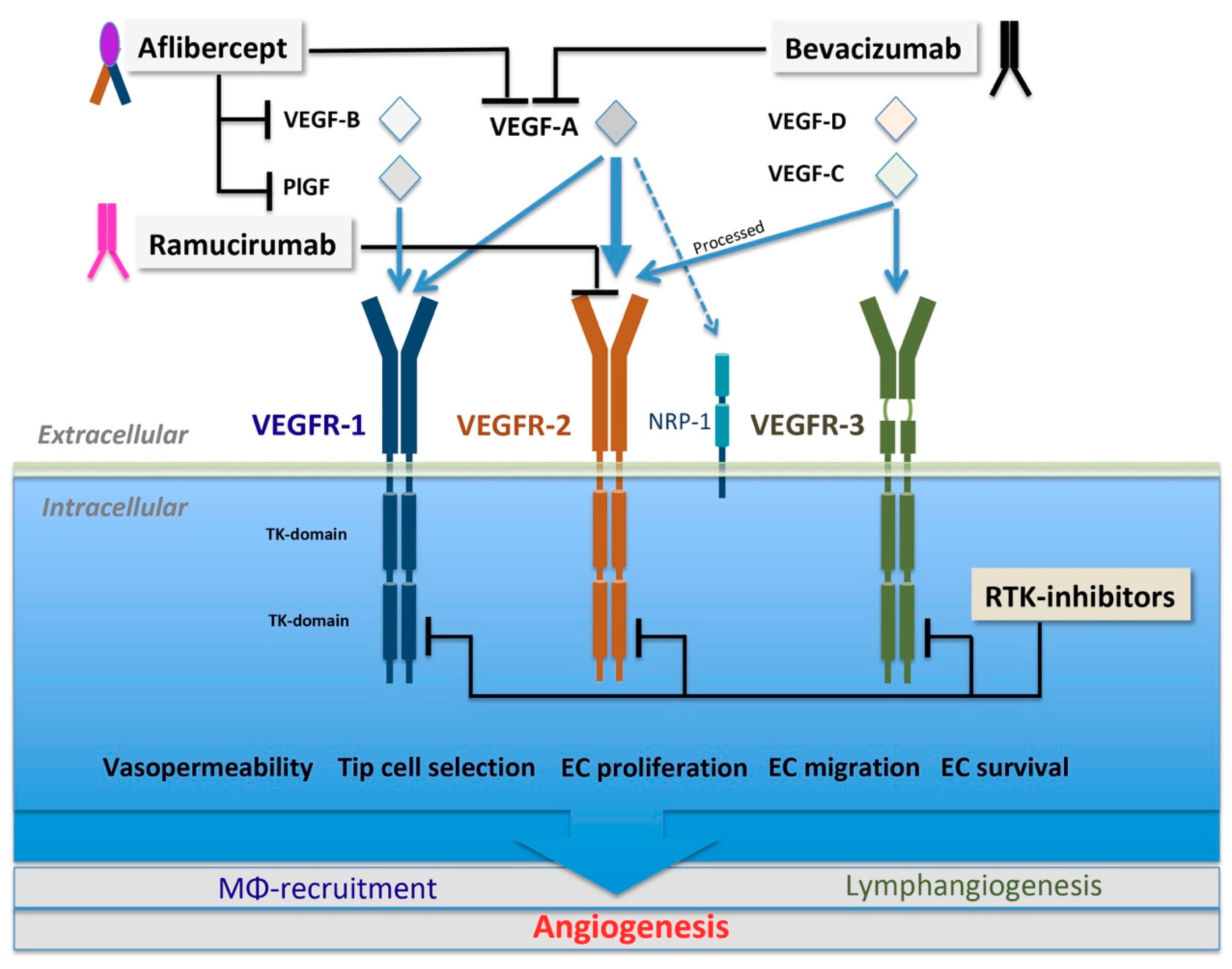
3.1. VEGF Family and VEGFR Signaling
3.2. Alternative Angiogenic Factors
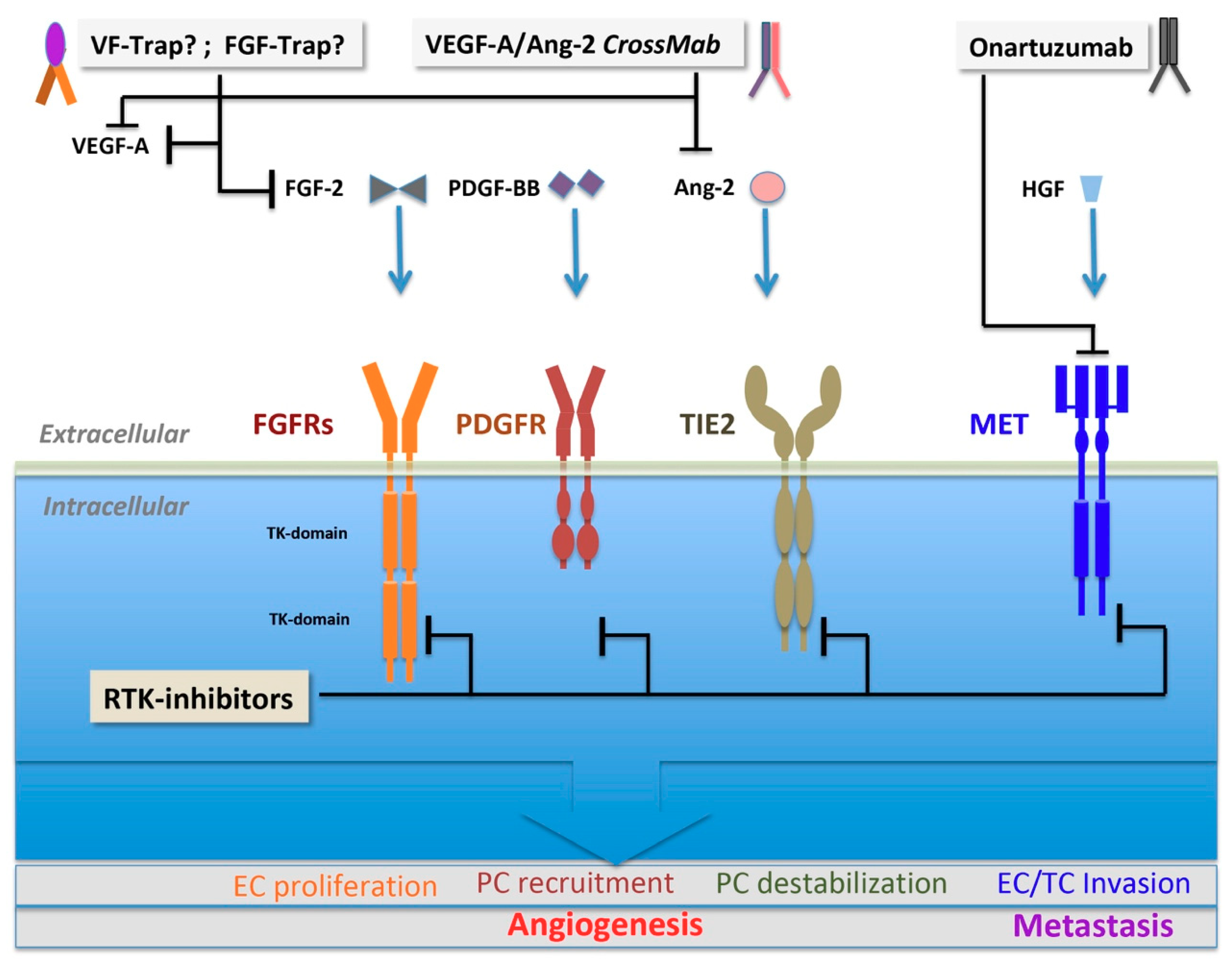
3.2.1. FGF Family and FGF Receptors
3.2.2. PDGF Family and PDGF Receptors
3.2.3. Angiopoietin and TIE2
3.2.4. HGF and c-MET
4. Microenvironmental Confounder Factors in Anti-Angiogenesis
4.1. Abnormal Vessel Structure and Differential Sensitivity
4.2. Metabolic Switch and Extracellular Acidosis
4.2.1. Metabolic Reprogramming
Tumor Cells
Cancer-Associated Fibroblasts
ECs
4.2.2. Extracellular Acidosis
4.3. Tumor Microenvironment and Deregulated Inflammatory Responses
5. Therapeutic Modalities
5.1. Large Molecules
5.1.1. Biologics Targeting VEGF Ligands
Bevacizumab
VEGF-Trap
5.1.2. Biologics Targeting VEGFR2
Ramucirumab
5.2. Small-Molecule Multikinase Inhibitors
6. Future Perspectives—Possible Answers to Therapy Resistance
6.1. Metronomic Therapy Regimes
6.2. Drug Repurposing
6.3. Reprogramming the Immunosuppressive Microenvironment
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yamamoto, T. Oncogenes and tumor suppressor genes. In Molecular Biology in Cancer Medicine; Kurzrock, R., Talpaz, M., Eds.; Oxford University Press: Oxford, UK, 1995; pp. 98–112. [Google Scholar]
- Sherwood, L.M.; Parris, E.E.; Folkman, J. Tumor Angiogenesis: Therapeutic Implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Successful Treatment of an Angiogenic Disease. N. Engl. J. Med. 1989, 320, 1211–1212. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Javaherian, K.; Lo, K.M.; Folkman, J.; Hanahan, D. Effects of Angiogenesis Inhibitors on Multistage Carcinogenesis in Mice. Science 1999, 284, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.S.; Lee, J.; Ferrara, N. Targeting the tumour vasculature: Insights from physiological angiogenesis. Nat. Rev. Cancer 2010, 10, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Boehm-Viswanathan, T. Is angiogenesis inhibition the Holy Grail of cancer therapy? Curr. Opin. Oncol. 2000, 12, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Baluk, P.; Hashizume, H.; McDonald, D.M. Cellular abnormalities of blood vessels as targets in cancer. Curr. Opin. Genet. Dev. 2005, 15, 102–111. [Google Scholar] [CrossRef]
- Ferrara, N.; Kerbel, R.S. Angiogenesis as a therapeutic target. Nature 2005, 438, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Grothey, A.; Galanis, E. Targeting angiogenesis: Progress with anti-VEGF treatment with large molecules. Nat. Rev. Clin. Oncol. 2009, 6, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Angiogenesis: An organizing principle for drug discovery? Nat. Rev. Drug Discov. 2007, 6, 273–286. [Google Scholar] [CrossRef]
- Carmeliet, P. VEGF as a Key Mediator of Angiogenesis in Cancer. Oncology 2005, 69, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Yancopoulos, G.D.; Davis, S.; Gale, N.W.; Rudge, J.S.; Wiegand, S.J.; Holash, J.; Yancopoulos, G.D.; Davis, S.; Gale, N.W.; Rudge, J.S.; et al. Vascular-specific growth factors and blood vessel formation. Nature 2000, 407, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Olsson, A.-K.; Dimberg, A.; Kreuger, J.; Claesson-Welsh, L. VEGF receptor signalling? in control of vascular function. Nat. Rev. Mol. Cell Biol. 2006, 7, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Adamis, A.P. Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Discov. 2016, 15, 385–403. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. VEGF and the quest for tumour angiogenesis factors. Nat. Rev. Cancer 2002, 2, 795–803. [Google Scholar] [CrossRef]
- Dancey, J.; Sausville, E.A. Issues and progress with protein kinase inhibitors for cancer treatment. Nat. Rev. Drug Discov. 2003, 2, 296–313. [Google Scholar] [CrossRef] [PubMed]
- Yancopoulos, G.D. Clinical Application of Therapies Targeting VEGF. Cell 2010, 143, 13–16. [Google Scholar] [CrossRef]
- Miller, J.W. The Harvard angiogenesis story. Surv. Ophthalmol. 2014, 59, 361–364. [Google Scholar] [CrossRef]
- De Bock, K.; Cauwenberghs, S.; Carmeliet, P. Vessel abnormalization: Another hallmark of cancer? Molecular mechanisms and therapeutic implications. Curr. Opin. Genet. Dev. 2011, 21, 73–79. [Google Scholar] [CrossRef]
- Gacche, R.N. Compensatory angiogenesis and tumor refractoriness. Oncogenesis 2015, 4, e153. [Google Scholar] [CrossRef]
- Stapor, P.; Wang, X.; Goveia, J.; Moens, S.; Carmeliet, P. Angiogenesis revisited—Role and therapeutic potential of targeting endothelial metabolism. J. Cell Sci. 2014, 127, 4331–4341. [Google Scholar] [CrossRef] [PubMed]
- Casanovas, O.; Hicklin, D.J.; Bergers, G.; Hanahan, D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005, 8, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Crawford, Y.; Kasman, I.; Yu, L.; Zhong, C.; Wu, X.; Modrusan, Z.; Kaminker, J.; Ferrara, N. PDGF-C Mediates the Angiogenic and Tumorigenic Properties of Fibroblasts Associated with Tumors Refractory to Anti-VEGF Treatment. Cancer Cell 2009, 15, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Xie, K.; Ding, G.; Li, J.; Chen, K.; Li, H.; Qian, J.; Jiang, C.; Fang, J. Tumor resistance to anti-VEGF therapy through up-regulation of VEGF-C expression. Cancer Lett. 2014, 346, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, F.; Wu, X.; Malik, A.K.; Zhong, C.; Baldwin, M.E.; Schanz, S.; Fuh, G.; Gerber, H.P.; Ferrara, N. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat. Biotechnol. 2007, 25, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, F.; Wu, X.; Zhong, C.; Yu, L.; Liang, X.-H.; Yao, J.; Blanchard, D.; Bais, C.; Peale, F.V.; Van Bruggen, N.; et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature 2007, 450, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.P.; Rodríguez, M.; Adams, K.; Goldenberg, D.M.; Blumenthal, R.D. Altered tumor vessel maturation and proliferation in placenta growth factor-producing tumors: Potential relationship to post-therapy tumor angiogenesis and recurrence. Int. J. Cancer 2003, 105, 158–164. [Google Scholar] [CrossRef]
- Michaelsen, S.R.; Staberg, M.; Pedersen, H.; Jensen, K.E.; Majewski, W.; Broholm, H.; Nedergaard, M.K.; Meulengracht, C.; Urup, T.; Villingshøj, M.; et al. VEGF-C sustains VEGFR2 activation under bevacizumab therapy and promotes glioblastoma maintenance. Neuro-Oncology 2018, 20, 1462–1474. [Google Scholar] [CrossRef]
- Pahler, J.C.; Tazzyman, S.; Erez, N.; Chen, Y.-Y.; Murdoch, C.; Nozawa, H.; Lewis, C.E.; Hanahan, D. Plasticity in Tumor-Promoting Inflammation: Impairment of Macrophage Recruitment Evokes a Compensatory Neutrophil Response. Neoplasia 2008, 10, 329–340. [Google Scholar] [CrossRef]
- Carmeliet, P. Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 2000, 6, 389–395. [Google Scholar] [CrossRef]
- Ribatti, D.; Nico, B.; Crivellato, E.; Roccaro, A.M.; Vacca, A. The history of the angiogenic switch concept. Leukemia 2007, 21, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, F.; Ferrara, N. Role of the microenvironment in tumor growth and in refractoriness/resistance to anti-angiogenic therapies. Drug Resist. Updat. 2008, 11, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Folkman, J. Patterns and Emerging Mechanisms of the Angiogenic Switch during Tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef]
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer 2003, 3, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Baeriswyl, V.; Christofori, G. The angiogenic switch in carcinogenesis. Semin. Cancer Biol. 2009, 19, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995, 1, 27–30. [Google Scholar] [CrossRef]
- Folkman, J.; Klagsbrun, M. Angiogenic factors. Science 1987, 235, 442–447. [Google Scholar] [CrossRef]
- Rust, R.; Gantner, C.; Schwab, M.E. Pro- and antiangiogenic therapies: Current status and clinical implications. FASEB J. 2019, 33, 34–48. [Google Scholar] [CrossRef]
- Shweiki, D.; Itin, A.; Soffer, D.; Keshet, E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 1992, 359, 843–845. [Google Scholar] [CrossRef] [PubMed]
- Chan-Ling, T.; Gock, B.; Stone, J. The effect of oxygen on vasoformative cell division. Evidence that ’physiological hypoxia’ is the stimulus for normal retinal vasculogenesis. Investig. Ophthalmol. Vis. Sci. 1995, 36, 1201–1214. [Google Scholar]
- Goel, S.; Duda, D.G.; Xu, L.; Munn, L.L.; Boucher, Y.; Fukumura, D.; Jain, R.K. Normalization of the Vasculature for Treatment of Cancer and Other Diseases. Physiol. Rev. 2011, 91, 1071–1121. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.A.; Chang, S.-H.; Dvorak, A.M.; Dvorak, H.F. Why are tumour blood vessels abnormal and why is it important to know? Br. J. Cancer 2009, 100, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [PubMed]
- Alfarouk, K.O.; Verduzco, D.; Rauch, C.; Muddathir, A.K.; Bashir, A.H.H.; Elhassan, G.O.; Ibrahim, M.E.; Orozco, J.D.P.; Cardone, R.A.; Reshkin, S.J.; et al. Glycolysis, tumor metabolism, cancer growth and dissemination. A new pH-based etiopathogenic perspective and therapeutic approach to an old cancer question. Oncoscience 2014, 1, 777–802. [Google Scholar] [CrossRef] [PubMed]
- Payne, S.J.; Jones, L. Influence of the tumor microenvironment on angiogenesis. Future Oncol. 2011, 7, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Nico, B.; Crivellato, E.; Vacca, A. Macrophages and tumor angiogenesis. Leukemia 2007, 21, 2085–2089. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, F.; Zhong, C.; Wu, X.; Yu, L.; Ferrara, N. Role of myeloid cells in tumor angiogenesis and growth. Trends Cell Biol. 2008, 18, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.-M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; Von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau Ubiquitylation Complex by O2-Regulated Prolyl Hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Pugh, C.W.; Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: Role of the HIF system. Nat. Med. 2003, 9, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, N. The Ubiquitin-Proteasome System Meets Angiogenesis. Mol. Cancer Ther. 2012, 11, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Eelen, G.; De Zeeuw, P.; Simons, M.; Carmeliet, P. Endothelial Cell Metabolism in Normal and Diseased Vasculature. Circ. Res. 2015, 116, 1231–1244. [Google Scholar] [CrossRef] [PubMed]
- Yana, I.; Sagara, H.; Takaki, S.; Takatsu, K.; Nakamura, K.; Nakao, K.; Katsuki, M.; Taniguchi, S.-I.; Aoki, T.; Sato, H.; et al. Crosstalk between neovessels and mural cells directs the site-specific expression of MT1-MMP to endothelial tip cells. J. Cell Sci. 2007, 120, 1607–1614. [Google Scholar] [CrossRef] [PubMed]
- Gaengel, K.; Armulik, A.; Betsholtz, C.; Genové, G.; Keller, A. Endothelial-Mural Cell Signaling in Vascular Development and Angiogenesis. Arter. Thromb. Vasc. Biol. 2009, 29, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, L.; Franco, C.A.; Bentley, K.; Collins, R.T.; Ponsioen, B.; Aspalter, I.M.; Rosewell, I.; Busse, M.; Thurston, G.; Medvinsky, A.; et al. Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nature 2010, 12, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, H.; Golding, M.; Fruttiger, M.; Ruhrberg, C.; Lundkvist, A.; Abramsson, A.; Jeltsch, M.; Mitchell, C.; Alitalo, K.; Shima, D.; et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 2003, 161, 1163–1177. [Google Scholar] [CrossRef]
- Siemerink, M.J.; Klaassen, I.; Van Noorden, C.J.; Schlingemann, R.O. Endothelial tip cells in ocular angiogenesis: Potential target for anti-angiogenesis therapy. J. Histochem. Cytochem. 2013, 61, 101–115. [Google Scholar] [CrossRef]
- De Smet, F.; Segura, I.; De Bock, K.; Hohensinner, P.J.; Carmeliet, P. Mechanisms of vessel branching: Filopodia on endothelial tip cells lead the way. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Hellstrom, M.; Phng, L.K.; Gerhardt, H. VEGF and Notch signaling: The yin and yang of angiogenic sprouting. Cell. Adh. Migr. 2007, 1, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Hellström, M.; Phng, L.-K.; Hofmann, J.J.; Wallgard, E.; Coultas, L.; Lindblom, P.; Alva, J.; Nilsson, A.-K.; Karlsson, L.; Gaiano, N.; et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 2007, 445, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, L.; Bentley, K.; Gerhardt, H. VEGFRs and Notch: A dynamic collaboration in vascular patterning. Biochem. Soc. Trans. 2009, 37 Pt 6, 1233–1236. [Google Scholar] [CrossRef]
- Tung, J.J.; Tattersall, I.W.; Kitajewski, J. Tips, stalks, tubes: Notch-mediated cell fate determination and mechanisms of tubulogenesis during angiogenesis. Cold Spring Harb. Perspect. Med. 2012, 2, a006601. [Google Scholar] [CrossRef] [PubMed]
- Moya, I.M.; Umans, L.; Maas, E.; Pereira, P.N.G.; Beets, K.; Francis, A.; Sents, W.; Robertson, E.J.; Mummery, C.L.; Huylebroeck, D.; et al. Stalk cell phenotype depends on integration of Notch and Smad1/5 signaling cascades. Dev. Cell 2012, 22, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and Therapeutic Aspects of Angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Ruan, Q.; Chen, Z.J.; Han, S. Connection of pericyte–angiopoietin-Tie-2 system in diabetic retinopathy: Friend or foe? Future Med. Chem. 2012, 4, 2163–2176. [Google Scholar] [CrossRef]
- Fantin, A.; Vieira, J.M.; Gestri, G.; Denti, L.; Schwarz, Q.; Prykhozhij, S.; Peri, F.; Wilson, S.W.; Ruhrberg, C. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood 2010, 116, 829–840. [Google Scholar] [CrossRef]
- Adams, R.H.; Eichmann, A. Axon Guidance Molecules in Vascular Patterning. Cold Spring Harb. Perspect. Biol. 2010, 2, a001875. [Google Scholar] [CrossRef]
- Öhlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Moons, L.; Luttun, A.; Vincenti, V.; Compernolle, V.; De Mol, M.; Wu, Y.; Bono, F.; Devy, L.; Beck, H.; et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat. Med. 2001, 7, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Dudvarski Stankovic, N.; Bicker, F.; Keller, S.; Jones, D.T.; Harter, P.N.; Kienzle, A.; Gillmann, C.; Arnold, P.; Golebiewska, A.; Keunen, O.; et al. EGFL7 enhances surface expression of integrin alpha5beta1 to promote angiogenesis in malignant brain tumors. EMBO Mol. Med. 2018, 10, e8420. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, I.; Stankovic, N.D.; Bicker, F.; Meister, J.; Braun, H.; Awwad, K.; Baumgart, J.; Simon, K.; Thal, S.C.; Patra, C.; et al. EGFL7 ligates alphavbeta3 integrin to enhance vessel formation. Blood 2013, 121, 3041–3050. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Pettersson, A.; Nagy, J.A.; Brown, L.F.; Sundberg, C.; Morgan, E.; Jungles, S.; Carter, R.; Krieger, J.E.; Manseau, E.J.; Harvey, V.S.; et al. Heterogeneity of the Angiogenic Response Induced in Different Normal Adult Tissues by Vascular Permeability Factor/Vascular Endothelial Growth Factor. Lab. Investig. 2000, 80, 99–115. [Google Scholar] [CrossRef]
- Rocha, S.F.; Schiller, M.; Jing, D.; Li, H.; Butz, S.; Vestweber, D.; Biljes, D.; Drexler, H.C.; Nieminen-Kelhä, M.; Vajkoczy, P.; et al. Esm1 Modulates Endothelial Tip Cell Behavior and Vascular Permeability by Enhancing VEGF Bioavailability. Circ. Res. 2014, 115, 581–590. [Google Scholar] [CrossRef]
- Koch, S.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb. Perspect. Med. 2012, 2, a006502. [Google Scholar] [CrossRef]
- Pellet-Many, C.; Frankel, P.; Jia, H.; Zachary, I. Neuropilins: Structure, function and role in disease. Biochem. J. 2008, 411, 211–226. [Google Scholar] [CrossRef]
- Senger, D.; Galli, S.; Dvorak, A.; Perruzzi, C.; Harvey, V.; Dvorak, H. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 1983, 219, 983–985. [Google Scholar] [CrossRef]
- Ferrara, N.; Henzel, W.J. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem. Biophys. Res. Commun. 1989, 161, 851–858. [Google Scholar] [CrossRef]
- Keck, P.; Hauser, S.; Krivi, G.; Sanzo, K.; Warren, T.; Feder, J.; Connolly, D. Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science 1989, 246, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.; Cachianes, G.; Kuang, W.; Goeddel, D.; Ferrara, N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989, 246, 1306–1309. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.W. VEGF: From Discovery to Therapy: The Champalimaud Award Lecture. Transl. Vis. Sci. Technol. 2016, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Alon, T.; Hemo, I.; Itin, A.; Pe’Er, J.; Stone, J.; Keshet, E. Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat. Med. 1995, 1, 1024–1028. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. Role of vascular endothelial growth factor in regulation of physiological angiogenesis. Am. J. Physiol. Physiol. 2001, 280, C1358–C1366. [Google Scholar] [CrossRef]
- Zubilewicz, A.; Hecquet, C.; Jeanny, J.-C.; Soubrane, G.; Courtois, Y.; Mascarelli, F. Two distinct signalling pathways are involved in FGF2-stimulated proliferation of choriocapillary endothelial cells: A comparative study with VEGF. Oncogene 2001, 20, 1403–1413. [Google Scholar] [CrossRef]
- Dvorak, H.F.; Brown, L.F.; Detmar, M.; Dvorak, A.M. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am. J. Pathol. 1995, 146, 1029–1039. [Google Scholar]
- Ferrara, N.; Gerber, H.-P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Rennel, E.S.; Harper, S.J.; Bates, D.O. Therapeutic potential of manipulating VEGF splice isoforms in oncology. Future Oncol. 2009, 5, 703–712. [Google Scholar] [CrossRef]
- Houck, K.A.; Ferrara, N.; Winer, J.; Cachianes, G.; Li, B.; Leung, D.W. The Vascular Endothelial Growth Factor Family: Identification of a Fourth Molecular Species and Characterization of Alternative Splicing of RNA. Mol. Endocrinol. 1991, 5, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Keller, G.A.; Ferrara, N. The vascular endothelial growth factor (VEGF) isoforms: Differential deposition into the subepithelial extracellular matrix and bioactivity of extracellular matrix-bound VEGF. Mol. Biol. Cell 1993, 4, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Peach, C.J.; Mignone, V.W.; Arruda, M.A.; Alcobia, D.C.; Hill, S.J.; Kilpatrick, L.E.; Woolard, J. Molecular Pharmacology of VEGF-A Isoforms: Binding and Signalling at VEGFR2. Int. J. Mol. Sci. 2018, 19, 1264. [Google Scholar] [CrossRef] [PubMed]
- Bry, M.; Kivelä, R.; Leppänen, V.-M.; Alitalo, K. Vascular Endothelial Growth Factor-B in Physiology and Disease. Physiol. Rev. 2014, 94, 779–794. [Google Scholar] [CrossRef] [PubMed]
- Joukov, V.; Pajusola, K.; Kaipainen, A.; Chilov, D.; Lahtinen, I.; Kukk, E.; Saksela, O.; Kalkkinen, N.; Alitalo, K. A novel vascular endothelial growth factor, VEGF-C, is a ligand for the Flt4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. EMBO J. 1996, 15, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M. Vascular endothelial growth factor receptor-1 (VEGFR-1/Flt-1): A dual regulator for angiogenesis. Angiogenesis 2006, 9, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Van de Veire, S.; Stalmans, I.; Heindryckx, F.; Oura, H.; Tijeras-Raballand, A.; Schmidt, T.; Loges, S.; Albrecht, I.; Jonckx, B.; Vinckier, S.; et al. Further pharmacological and genetic evidence for the efficacy of PlGF inhibition in cancer and eye disease. Cell 2010, 141, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Kowalczuk, L.; Touchard, E.; Omri, S.; Jonet, L.; Klein, C.; Valamanes, F.; Berdugo, M.; Bigey, P.; Massin, P.; Jeanny, J.-C.; et al. Placental Growth Factor Contributes to Micro-Vascular Abnormalization and Blood-Retinal Barrier Breakdown in Diabetic Retinopathy. PLoS ONE 2011, 6, e17462. [Google Scholar] [CrossRef]
- Maglione, D.; Guerriero, V.; Viglietto, G.; Delli-Bovi, P.; Persico, M.G. Isolation of a human placenta cDNA coding for a protein related to the vascular permeability factor. Proc. Natl. Acad. Sci. USA 1991, 88, 9267–9271. [Google Scholar] [CrossRef]
- Fischer, C.; Mazzone, M.; Jonckx, B.; Carmeliet, P. FLT1 and its ligands VEGFB and PlGF: Drug targets for anti-angiogenic therapy? Nat. Rev. Cancer 2008, 8, 942–956. [Google Scholar] [CrossRef]
- Marrony, S.; Bassilana, F.; Seuwen, K.; Keller, H. Bone morphogenetic protein 2 induces placental growth factor in mesenchymal stem cells. Bone 2003, 33, 426–433. [Google Scholar] [CrossRef]
- Dewerchin, M.; Carmeliet, P. PlGF: A Multitasking Cytokine with Disease-Restricted Activity. Cold Spring Harb. Perspect. Med. 2012, 2, a011056. [Google Scholar] [CrossRef] [PubMed]
- Autiero, M.; Waltenberger, J.; Communi, D.; Kranz, A.; Moons, L.; Lambrechts, D.; Kroll, J.; Plaisance, S.; De Mol, M.; Bono, F.; et al. Role of PlGF in the intra- and intermolecular cross talk between the VEGF receptors Flt1 and Flk1. Nat. Med. 2003, 9, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Chen, H.H.; Winer, J.; Houck, K.A.; Ferrara, N. Placenta growth factor. Potentiation of vascular endothelial growth factor bioactivity, in vitro and in vivo, and high affinity binding to Flt-1 but not to Flk-1/KDR. J. Biol. Chem. 1994, 269, 25646–25654. [Google Scholar] [PubMed]
- Roy, H.; Bhardwaj, S.; Babu, M.; Jauhiainen, S.; Herzig, K.-H.; Bellu, A.R.; Haisma, H.J.; Carmeliet, P.; Alitalo, K.; Ylä-Herttuala, S.; et al. Adenovirus-Mediated Gene Transfer of Placental Growth Factor to Perivascular Tissue Induces Angiogenesis via Upregulation of the Expression of Endogenous Vascular Endothelial Growth Factor-A. Hum. Gene Ther. 2005, 16, 1422–1428. [Google Scholar] [CrossRef] [PubMed]
- Landgren, E.; Schiller, P.; Cao, Y.; Claesson-Welsh, L. Placenta growth factor stimulates MAP kinase and mitogenicity but not phospholipase C-gamma and migration of endothelial cells expressing Flt 1. Oncogene 1998, 16, 359–367. [Google Scholar] [CrossRef]
- Sawano, A.; Takahashi, T.; Yamaguchi, S.; Aonuma, M.; Shibuya, M. Flt-1 but not KDR/Flk-1 tyrosine kinase is a receptor for placenta growth factor, which is related to vascular endothelial growth factor. Cell Growth Differ. 1996, 7, 213–221. [Google Scholar]
- Terman, B.I.; Khandke, L.; Dougher-Vermazan, M.; Maglione, D.; Lassam, N.J.; Gospodarowicz, D.; Persico, M.G.; Böhlen, P.; Eisinger, M. VEGF Receptor Subtypes KDR and FLT1 Show Different Sensitivities to Heparin and Placenta Growth Factor. Growth Factors 1994, 11, 187–195. [Google Scholar] [CrossRef]
- Murakami, M.; Zheng, Y.; Hirashima, M.; Suda, T.; Morita, Y.; Ooehara, J.; Ema, H.; Fong, G.-H.; Shibuya, M. VEGFR1 Tyrosine Kinase Signaling Promotes Lymphangiogenesis as Well as Angiogenesis Indirectly via Macrophage Recruitment. Arter. Thromb. Vasc. Biol. 2008, 28, 658–664. [Google Scholar] [CrossRef]
- Pipp, F.; Heil, M.; Issbrücker, K.; Ziegelhoeffer, T.; Martin, S.; van den Heuvel, J.; Weich, H.; Fernandez, B.; Golomb, G.; Carmeliet, P.; et al. VEGFR-1-selective VEGF homologue PlGF is arteriogenic: Evidence for a monocyte-mediated mechanism. Circ. Res. 2003, 92, 378–385. [Google Scholar] [CrossRef]
- Selvaraj, S.K.; Hiramatsu, H.; Nishikomori, R.; Heike, T.; Ito, M.; Kobayashi, K.; Katamura, K.; Nakahata, T. Mechanism of monocyte activation and expression of proinflammatory cytochemokines by placenta growth factor. Blood 2003, 102, 1515–1524. [Google Scholar] [CrossRef] [PubMed]
- Alitalo, K.; Tammela, T.; Petrova, T.V. Lymphangiogenesis in development and human disease. Nature 2005, 438, 946–953. [Google Scholar] [CrossRef] [PubMed]
- Tammela, T.; Zarkada, G.; Wallgard, E.; Murtomäki, A.; Suchting, S.; Wirzenius, M.; Waltari, M.; Hellström, M.; Schomber, T.; Peltonen, R.; et al. Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature 2008, 454, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Valtola, R.; Salven, P.; Heikkilä, P.; Taipale, J.; Joensuu, H.; Rehn, M.; Pihlajaniemi, T.; Weich, H.; DeWaal, R.; Alitalo, K. VEGFR-3 and Its Ligand VEGF-C Are Associated with Angiogenesis in Breast Cancer. Am. J. Pathol. 1999, 154, 1381–1390. [Google Scholar] [CrossRef]
- Regenfuss, B.; Cursiefen, C. Concept of Angiogenic Privilege. In Encyclopedia of the Eye; Dartt, D.A., Besharse, J., Dana, R., Eds.; Academic Press: San Diego, CA, USA, 2010; pp. 334–338. [Google Scholar]
- Puddu, A.; Sanguineti, R.; Durante, A.; Nicolò, M.; Viviani, G.L. Vascular endothelial growth factor-C secretion is increased by advanced glycation end-products: Possible implication in ocular neovascularization. Mol. Vis. 2012, 18, 2509–2517. [Google Scholar] [PubMed]
- Regenfuss, B.; Cursiefen, C. Angiogenesis in the Eye. In Encyclopedia of the Eye; Dartt, D.A., Besharse, J., Dana, R., Eds.; Academic Press: San Diego, CA, USA, 2010; pp. 93–98. [Google Scholar]
- Shibuya, M.; Claesson-Welsh, L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp. Cell Res. 2006, 312, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Drinkwater, C.; Gotley, D.; Ward, L.; Hayward, N.; Grimmond, S.; Lagercrantz, J.; Silins, G.; Townson, S.; Pollock, P.; Carson, E.; et al. Cloning and characterization of a novel human gene related to vascular endothelial growth factor. Genome Res. 1996, 6, 124–131. [Google Scholar]
- Zhang, F.; Tang, Z.; Hou, X.; Lennartsson, J.; Li, Y.; Koch, A.W.; Scotney, P.; Lee, C.; Arjunan, P.; Dong, L.; et al. VEGF-B is dispensable for blood vessel growth but critical for their survival, and VEGF-B targeting inhibits pathological angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 6152–6157. [Google Scholar] [CrossRef] [PubMed]
- Guttmann-Raviv, N.; Kessler, O.; Shraga-Heled, N.; Lange, T.; Herzog, Y.; Neufeld, G. The neuropilins and their role in tumorigenesis and tumor progression. Cancer Lett. 2006, 231, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Ronca, R.; Giacomini, A.; Rusnati, M.; Presta, M. The potential of fibroblast growth factor/fibroblast growth factor receptor signaling as a therapeutic target in tumor angiogenesis. Expert Opin. Ther. Targets 2015, 19, 1361–1377. [Google Scholar] [CrossRef] [PubMed]
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Vacca, A.; Rusnati, M.; Presta, M. The discovery of basic fibroblast growth factor/fibroblast growth factor-2 and its role in haematological malignancies. Cytokine Growth Factor Rev. 2007, 18, 327–334. [Google Scholar] [CrossRef]
- Jászai, J.; Reifers, F.; Picker, A.; Langenberg, T.; Brand, M. Isthmus-to-midbrain transformation in the absence of midbrain-hindbrain organizer activity. Development 2003, 130, 6611–6623. [Google Scholar] [CrossRef][Green Version]
- Gospodarowicz, D.; Schweigerer, L.; Neufeld, G.; Ferrara, N. Structural Characterization and Biological Functions of Fibroblast Growth Factor. Endocr. Rev. 1987, 8, 95–114. [Google Scholar] [CrossRef]
- Presta, M.; Dell’Era, P.; Mitola, S.M.F.; Moroni, E.; Ronca, R.; Rusnati, M.; Rusnati, M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005, 16, 159–178. [Google Scholar] [CrossRef]
- Rusnati, M.; Presta, M. Fibroblast growth factors/fibroblast growth factor receptors as targets for the development of anti-angiogenesis strategies. Curr. Pharm. Des. 2007, 13, 2025–2044. [Google Scholar] [CrossRef]
- Allen, E.; Walters, I.B.; Hanahan, D. Brivanib, a dual FGF/VEGF inhibitor, is active both first and second line against mouse pancreatic neuroendocrine tumors developing adaptive/evasive resistance to VEGF inhibition. Clin. Cancer Res. 2011, 17, 5299–5310. [Google Scholar] [CrossRef]
- Chan, J.K.; Deng, W.; Higgins, R.V.; Tewari, K.S.; Bonebrake, A.J.; Hicks, M.; Gaillard, S.; Ramirez, P.T.; Chafe, W.; Monk, B.J.; et al. A phase II evaluation of brivanib in the treatment of persistent or recurrent carcinoma of the cervix: An NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol. 2017, 146, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Bolos, D.; Finn, R.S. Systemic therapy in HCC: Lessons from brivanib. J. Hepatol. 2014, 61, 947–950. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, D.; Xie, K.; Zhang, L.; Yao, X.; Li, H.; Xu, Q.; Wang, X.; Jiang, J.; Fang, J. Dual blockade of vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (FGF-2) exhibits potent anti-angiogenic effects. Cancer Lett. 2016, 377, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.-H.; Lennartsson, J. Structural and Functional Properties of Platelet-Derived Growth Factor and Stem Cell Factor Receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a009100. [Google Scholar] [CrossRef] [PubMed]
- Abramsson, A.; Lindblom, P.; Betsholtz, C. Endothelial and nonendothelial sources of PDGF-B regulate pericyte recruitment and influence vascular pattern formation in tumors. J. Clin. Investig. 2003, 112, 1142–1151. [Google Scholar] [CrossRef] [PubMed]
- Lindblom, P.; Gerhardt, H.; Liebner, S.; Abramsson, A.; Enge, M.; Hellstrom, M.; Bäckström, G.; Fredriksson, S.; Landegren, U.; Nyström, H.C.; et al. Endothelial PDGF-B retention is required for proper investment of pericytes in the microvessel wall. Genome Res. 2003, 17, 1835–1840. [Google Scholar] [CrossRef]
- Song, S.; Ewald, A.J.; Stallcup, W.; Werb, Z.; Bergers, G. PDGFRbeta+ perivascular progenitor cells in tumours regulate pericyte differentiation and vascular survival. Nat. Cell Biol. 2005, 7, 870–879. [Google Scholar] [CrossRef]
- Heldin, C.-H.; Rubin, K.; Pietras, K.; Östman, A. High interstitial fluid pressure—An obstacle in cancer therapy. Nat. Rev. Cancer 2004, 4, 806–813. [Google Scholar] [CrossRef]
- Song, N.; Huang, Y.; Shi, H.; Yuan, S.; Ding, Y.; Song, X.; Fu, Y.; Luo, Y. Overexpression of platelet-derived growth factor-BB increases tumor pericyte content via stromal-derived factor-1alpha/CXCR4 axis. Cancer Res. 2009, 69, 6057–6064. [Google Scholar] [CrossRef]
- Benjamin, L.E.; Golijanin, D.; Itin, A.; Pode, D.; Keshet, E. Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J. Clin. Investig. 1999, 103, 159–165. [Google Scholar] [CrossRef]
- Benjamin, L.E.; Hemo, I.; Keshet, E. A plasticity window for blood vessel remodelling is defined by pericyte coverage of the preformed endothelial network and is regulated by PDGF-B and VEGF. Development 1998, 125, 1591–1598. [Google Scholar] [PubMed]
- Erber, R.; Thurnher, A.; Katsen, A.D.; Groth, G.; Kerger, H.; Hammes, H.-P.; Menger, M.D.; Ullrich, A.; Vajkoczy, P. Combined inhibition of VEGF and PDGF signaling enforces tumor vessel regression by interfering with pericyte-mediated endothelial cell survival mechanisms. FASEB J. 2004, 18, 338–340. [Google Scholar] [CrossRef] [PubMed]
- Xian, X.; Hakansson, J.; Ståhlberg, A.; Lindblom, P.; Betsholtz, C.; Gerhardt, H.; Semb, H. Pericytes limit tumor cell metastasis. J. Clin. Investig. 2006, 116, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Ma, W.; Xu, H.; Huang, M.; Zhang, D.; He, Z.; Zhang, L.; Brem, S.; O’Rourke, D.M.; Gong, Y.; et al. PDGF-mediated mesenchymal transformation renders endothelial resistance to anti-VEGF treatment in glioblastoma. Nat. Commun. 2018, 9, 3439. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Bhat, A.; Woodnutt, G.; Lappe, R. Targeting the ANGPT–TIE2 pathway in malignancy. Nat. Rev. Cancer 2010, 10, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, D.M.; Griffiths, J.A.; Rojas, J.; Aldrich, T.H.; Jones, P.F.; Zhou, H.; McClain, J.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; et al. Angiopoietins 3 and 4: Diverging gene counterparts in mice and humans. Proc. Natl. Acad. Sci. USA 1999, 96, 1904–1909. [Google Scholar] [CrossRef]
- Gamble, J.R.; Drew, J.; Trezise, L.; Underwood, A.; Parsons, M.; Kasminkas, L.; Rudge, J.; Yancopoulos, G.; Vadas, M.A. Angiopoietin-1 Is an Antipermeability and Anti-Inflammatory Agent In Vitro and Targets Cell Junctions. Circ. Res. 2000, 87, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, S.; Sako, K.; Noda, K.; Zhang, J.; Minami, M.; Mochizuki, N. Angiopoietin-1/Tie2 receptor signaling in vascular quiescence and angiogenesis. Histol. Histopathol. 2010, 25, 387–396. [Google Scholar] [PubMed]
- Felcht, M.; Luck, R.; Schering, A.; Seidel, P.; Srivastava, K.; Hu, J.; Bartol, A.; Kienast, Y.; Vettel, C.; Loos, E.K.; et al. Angiopoietin-2 differentially regulates angiogenesis through TIE2 and integrin signaling. J. Clin. Investig. 2012, 122, 1991–2005. [Google Scholar] [CrossRef]
- Murdoch, C.; Tazzyman, S.; Webster, S.; Lewis, C.E. Expression of Tie-2 by human monocytes and their responses to angiopoietin-2. J. Immunol. 2007, 178, 7405–7411. [Google Scholar] [CrossRef]
- Scharpfenecker, M.; Fiedler, U.; Reiss, Y.; Augustin, H.G. The Tie-2 ligand Angiopoietin-2 destabilizes quiescent endothelium through an internal autocrine loop mechanism. J. Cell Sci. 2005, 118 Pt 4, 771–780. [Google Scholar] [CrossRef]
- Shen, J.; Frye, M.; Lee, B.L.; Reinardy, J.L.; McClung, J.M.; Ding, K.; Kojima, M.; Xia, H.; Seidel, C.; Silva, R.L.E.; et al. Targeting VE-PTP activates TIE2 and stabilizes the ocular vasculature. J. Clin. Investig. 2014, 124, 4564–4576. [Google Scholar] [CrossRef] [PubMed]
- Duh, E.J.; Sun, J.K.; Stitt, A.W. Diabetic retinopathy: Current understanding, mechanisms, and treatment strategies. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Molecular regulation of vessel maturation. Nat. Med. 2003, 9, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Antiangiogenesis Strategies Revisited: From Starving Tumors to Alleviating Hypoxia. Cancer Cell 2014, 26, 605–622. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Nico, B.; Crivellato, E. The role of pericytes in angiogenesis. Int. J. Dev. Biol. 2011, 55, 261–268. [Google Scholar] [CrossRef] [PubMed]
- De Palma, M.; Venneri, M.A.; Galli, R.; Sergi, L.S.; Politi, L.S.; Sampaolesi, M.; Naldini, L. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell 2005, 8, 211–226. [Google Scholar] [CrossRef]
- Gerald, D.; Chintharlapalli, S.; Augustin, H.; Benjamin, L.E. Angiopoietin-2: An Attractive Target for Improved Antiangiogenic Tumor Therapy. Cancer Res. 2013, 73, 1649–1657. [Google Scholar] [CrossRef]
- Park, J.-S.; Kim, I.-K.; Han, S.; Park, I.; Kim, C.; Bae, J.; Oh, S.J.; Lee, S.; Kim, J.H.; Woo, D.-C.; et al. Normalization of Tumor Vessels by Tie2 Activation and Ang2 Inhibition Enhances Drug Delivery and Produces a Favorable Tumor Microenvironment. Cancer Cell 2016, 30, 953–967. [Google Scholar] [CrossRef]
- Peters, S.; Cree, I.A.; Alexander, R.; Turowski, P.; Ockrim, Z.; Patel, J.; Boyd, S.R.; Joussen, A.M.; Ziemssen, F.; Hykin, P.G.; et al. Angiopoietin modulation of vascular endothelial growth factor: Effects on retinal endothelial cell permeability. Cytokine 2007, 40, 144–150. [Google Scholar] [CrossRef]
- Rigamonti, N.; Kadioglu, E.; Keklikoglou, I.; Rmili, C.W.; Leow, C.C.; De Palma, M. Role of Angiopoietin-2 in Adaptive Tumor Resistance to VEGF Signaling Blockade. Cell Rep. 2014, 8, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, H.; Falcón, B.L.; Kuroda, T.; Baluk, P.; Coxon, A.; Yu, D.; Bready, J.V.; Oliner, J.D.; McDonald, D.M. Complementary Actions of Inhibitors of Angiopoietin-2 and VEGF on Tumor Angiogenesis and Growth. Cancer Res. 2010, 70, 2213–2223. [Google Scholar] [CrossRef] [PubMed]
- Coutelle, O.; Schiffmann, L.M.; Liwschitz, M.; Brunold, M.; Goede, V.; Hallek, M.; Kashkar, H.; Hacker, U.T. Dual targeting of Angiopoetin-2 and VEGF potentiates effective vascular normalisation without inducing empty basement membrane sleeves in xenograft tumours. Br. J. Cancer 2015, 112, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Regula, J.T.; Lundh von Leithner, P.; Foxton, R.; Barathi, V.A.; Cheung, C.M.; Bo Tun, S.B.; Wey, Y.S.; Iwata, D.; Dostalek, M.; Moelleken, J.; et al. Targeting key angiogenic pathways with a bispecific CrossMAb optimized for neovascular eye diseases. EMBO Mol. Med. 2016, 8, 1265–1288. [Google Scholar] [CrossRef] [PubMed]
- Kloepper, J.; Riedemann, L.; Amoozgar, Z.; Seano, G.; Susek, K.; Yu, V.; Dalvie, N.; Amelung, R.L.; Datta, M.; Song, J.W.; et al. Ang-2/VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc. Natl. Acad. Sci. USA 2016, 113, 4476–4481. [Google Scholar] [CrossRef] [PubMed]
- Peterson, T.E.; Kirkpatrick, N.D.; Huang, Y.; Farrar, C.T.; Marijt, K.A.; Kloepper, J.; Datta, M.; Amoozgar, Z.; Seano, G.; Jung, K.; et al. Dual inhibition of Ang-2 and VEGF receptors normalizes tumor vasculature and prolongs survival in glioblastoma by altering macrophages. Proc. Natl. Acad. Sci. USA 2016, 113, 4470–4475. [Google Scholar] [CrossRef] [PubMed]
- Organ, S.L.; Tsao, M.-S. An overview of the c-MET signaling pathway. Ther. Adv. Med Oncol. 2011, 3 (Suppl. 1), S7–S19. [Google Scholar] [CrossRef]
- Matsumoto, K.; Nakamura, T. Hepatocyte growth factor and the Met system as a mediator of tumor–stromal interactions. Int. J. Cancer 2006, 119, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Bussolino, F. Hepatocyte growth factor is a potent angiogenic factor which stimulates endothelial cell motility and growth. J. Cell Biol. 1992, 119, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Sulpice, E.; Ding, S.; Bergé, M.; Tobelem, G.; Muscatelli-Groux, B.; Merkulova-Rainon, T.; Han, Z.C.; Plouet, J. Cross-talk between the VEGF-A and HGF signalling pathways in endothelial cells. Biol. Cell 2009, 101, 525–539. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-W.; Su, Y.; Volpert, O.V.; Woude, G.F.V. Hepatocyte growth factor/scatter factor mediates angiogenesis through positive VEGF and negative thrombospondin 1 regulation. Proc. Natl. Acad. Sci. USA 2003, 100, 12718–12723. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.V.; Chang, J.P.; Parachoniak, C.A.; Pandika, M.M.; Aghi, M.K.; Meyronet, D.; Isachenko, N.; Fouse, S.D.; Phillips, J.J.; Cheresh, D.A.; et al. VEGF Inhibits Tumor Cell Invasion and Mesenchymal Transition Through a MET/VEGFR2 Complex. Cancer Cell 2012, 22, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Adjei, A.A. MET: A promising anticancer therapeutic target. Nat. Rev. Clin. Oncol. 2012, 9, 314–326. [Google Scholar] [CrossRef] [PubMed]
- Sennino, B.; Ishiguro-Oonuma, T.; Wei, Y.; Naylor, R.M.; Williamson, C.W.; Bhagwandin, V.; Tabruyn, S.P.; You, W.-K.; Chapman, H.A.; Christensen, J.G.; et al. Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discov. 2012, 2, 270–287. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.; Finocchiaro, G.; Belda-Iniesta, C.; Recht, L.; Brandes, A.A.; Pineda, E.; Mikkelsen, T.; Chinot, O.L.; Balana, C.; Macdonald, D.R.; et al. Randomized, Double-Blind, Placebo-Controlled, Multicenter Phase II Study of Onartuzumab Plus Bevacizumab Versus Placebo Plus Bevacizumab in Patients With Recurrent Glioblastoma: Efficacy, Safety, and Hepatocyte Growth Factor and O6-Methylguanine–DNA Methyltransferase Biomarker Analyses. J. Clin. Oncol. 2017, 35, 343–351. [Google Scholar] [PubMed]
- Nagy, J.A.; Chang, S.-H.; Shih, S.-C.; Dvorak, A.M.; Dvorak, H.F. Heterogeneity of the Tumor Vasculature. Semin. Thromb. Hemost. 2010, 36, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Sitohy, B.; Nagy, J.A.; Shih, S.-C.; Dvorak, H.F. Tumor surrogate blood vessel subtypes exhibit differential susceptibility to anti-VEGF therapy. Cancer Res. 2011, 71, 7021–7028. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Waxman, D.J. Combination of Anti-angiogenesis with Chemotherapy for More Effective Cancer Treatment*. Mol. Cancer Ther. 2008, 7, 3670–3684. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yuan, J.; Righi, E.; Kamoun, W.S.; Ancukiewicz, M.; Nezivar, J.; Santosuosso, M.; Martin, J.D.; Martin, M.R.; Vianello, F.; et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 17561–17566. [Google Scholar] [CrossRef]
- Kim, I.-K.; Kim, K.; Lee, E.; Oh, D.S.; Park, C.S.; Park, S.; Yang, J.M.; Kim, J.-H.; Kim, H.-S.; Shima, D.T.; et al. Sox7 promotes high-grade glioma by increasing VEGFR2-mediated vascular abnormality. J. Exp. Med. 2018, 215, 963–983. [Google Scholar] [CrossRef]
- Li, W.; Quan, Y.-Y.; Li, Y.; Lu, L.; Cui, M. Monitoring of tumor vascular normalization: The key points from basic research to clinical application. Cancer Manag. Res. 2018, 10, 4163–4172. [Google Scholar] [CrossRef] [PubMed]
- North, S.; Moenner, M.; Bikfalvi, A. Recent developments in the regulation of the angiogenic switch by cellular stress factors in tumors. Cancer Lett. 2005, 218, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sormendi, S.; Wielockx, B. Hypoxia Pathway Proteins As Central Mediators of Metabolism in the Tumor Cells and Their Microenvironment. Front. Immunol. 2018, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Pelicano, H.; Martin, D.S.; Xu, R.-H.; Huang, P. Glycolysis inhibition for anticancer treatment. Oncogene 2006, 25, 4633–4646. [Google Scholar] [CrossRef] [PubMed]
- Al Tameemi, W.; Dale, T.P.; Al-Jumaily, R.M.K.; Forsyth, N.R. Hypoxia-Modified Cancer Cell Metabolism. Front. Cell Dev. Biol. 2019, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- López-Lázaro, M. The warburg effect: Why and how do cancer cells activate glycolysis in the presence of oxygen? Anti-Cancer Agents Med. Chem. 2008, 8, 305–312. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1 and tumor progression: Pathophysiology and therapeutics. Trends Mol. Med. 2002, 8 (Suppl. 4), S62–S67. [Google Scholar] [CrossRef]
- Heiden, M.G.V. Exploiting tumor metabolism: Challenges for clinical translation. J. Clin. Investig. 2013, 123, 3648–3651. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Robey, I.F.; Lien, A.D.; Welsh, S.J.; Baggett, B.K.; Gillies, R.J. Hypoxia-inducible factor-1alpha and the glycolytic phenotype in tumors. Neoplasia 2005, 7, 324–330. [Google Scholar] [CrossRef]
- Alfarouk, K.O. Tumor metabolism, cancer cell transporters, and microenvironmental resistance. J. Enzym. Inhib. Med. Chem. 2016, 31, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Lu, M.; Jung, K.H.; Park, J.H.; Yu, L.; Onuchic, J.N.; Kaipparettu, B.A.; Levine, H. Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc. Natl. Acad. Sci. USA 2019, 116, 3909–3918. [Google Scholar] [CrossRef] [PubMed]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy-Kanniappan, S.; Geschwind, J.-F.H. Tumor glycolysis as a target for cancer therapy: Progress and prospects. Mol. Cancer 2013, 12, 152. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.; Cairns, R.A.; Chaudary, N.; Vellanki, R.N.; Kalliomaki, T.; Moriyama, E.H.; Mujcic, H.; Wilson, B.C.; Wouters, B.G.; Hill, R.; et al. Metabolic targeting of HIF-dependent glycolysis reduces lactate, increases oxygen consumption and enhances response to high-dose single-fraction radiotherapy in hypoxic solid tumors. BMC Cancer 2017, 17, 418. [Google Scholar] [CrossRef] [PubMed]
- Augsten, M. Cancer-Associated Fibroblasts as Another Polarized Cell Type of the Tumor Microenvironment. Front. Oncol. 2014, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Tuveson, D.A. Deciphering cancer fibroblasts. J. Exp. Med. 2018, 215, 2967–2968. [Google Scholar] [CrossRef]
- Erez, N.; Glanz, S.; Raz, Y.; Avivi, C.; Barshack, I. Cancer Associated Fibroblasts express pro-inflammatory factors in human breast and ovarian tumors. Biochem. Biophys. Res. Commun. 2013, 437, 397–402. [Google Scholar] [CrossRef]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef]
- Schoors, S.; Cantelmo, A.R.; Georgiadou, M.; Stapor, P.; Wang, X.; Quaegebeur, A.; Cauwenberghs, S.; Wong, B.W.; Bifari, F.; Decimo, I.; et al. Incomplete and transitory decrease of glycolysis: A new paradigm for anti-angiogenic therapy? Cell Cycle 2014, 13, 16–22. [Google Scholar] [CrossRef]
- Raghunand, N.; Gillies, R.J. pH and drug resistance in tumors. Drug Resist. Updat. 2000, 3, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Wojtkowiak, J.W.; Verduzco, D.; Schramm, K.J.; Gillies, R.J. Drug resistance and cellular adaptation to tumor acidic pH microenvironment. Mol. Pharm. 2011, 8, 2032–2038. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Le, X.; Wang, B.; Abbruzzese, J.L.; Xiong, Q.; He, Y.; Xie, K. Regulation of vascular endothelial growth factor expression by acidosis in human cancer cells. Oncogene 2001, 20, 3751–3756. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Fukumura, D.; Jain, R.K. Acidic extracellular pH induces vascular endothelial growth factor (VEGF) in human glioblastoma cells via ERK1/2 MAPK signaling pathway: Mechanism of low pH-induced VEGF. J. Biol. Chem. 2002, 277, 11368–11374. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Xu, L.; Chen, Y.; Gohongi, T.; Seed, B.; Jain, R.K. Hypoxia and acidosis independently up-regulate vascular endothelial growth factor transcription in brain tumors in vivo. Cancer Res. 2001, 61, 6020–6024. [Google Scholar] [PubMed]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Gocheva, V.; Zeng, W.; Ke, D.; Klimstra, D.; Reinheckel, T.; Peters, C.; Hanahan, D.; Joyce, J.A. Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genome Res. 2006, 20, 543–556. [Google Scholar] [CrossRef]
- Robey, I.F.; Baggett, B.K.; Kirkpatrick, N.D.; Roe, D.J.; Dosescu, J.; Sloane, B.F.; Hashim, A.I.; Morse, D.L.; Raghunand, N.; Gatenby, R.A.; et al. Bicarbonate Increases Tumor pH and Inhibits Spontaneous Metastases. Cancer Res. 2009, 69, 2260–2268. [Google Scholar] [CrossRef]
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity 2014, 41, 866. [Google Scholar] [CrossRef]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- De Palma, M.; Murdoch, C.; Venneri, M.A.; Naldini, L.; Lewis, C.E. Tie2-expressing monocytes: Regulation of tumor angiogenesis and therapeutic implications. Trends Immunol. 2007, 28, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Sharabi, A.; Tsokos, M.G.; Ding, Y.; Malek, T.R.; Klatzmann, D.; Tsokos, G.C. Regulatory T cells in the treatment of disease. Nat. Rev. Drug Discov. 2018, 17, 823–844. [Google Scholar] [CrossRef]
- Dewhirst, M.W.; Mowery, Y.M.; Mitchell, J.B.; Cherukuri, M.K.; Secomb, T.W. Rationale for hypoxia assessment and amelioration for precision therapy and immunotherapy studies. J. Clin. Investig. 2019, 129, 489–491. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Westendorf, A.M.; Skibbe, K.; Adamczyk, A.; Buer, J.; Geffers, R.; Hansen, W.; Pastille, E.; Jendrossek, V. Hypoxia Enhances Immunosuppression by Inhibiting CD4+ Effector T Cell Function and Promoting Treg Activity. Cell. Physiol. Biochem. 2017, 41, 1271–1284. [Google Scholar] [CrossRef]
- Ben-Shoshan, J.; Maysel-Auslender, S.; Mor, A.; Keren, G.; George, J. Hypoxia controls CD4+CD25+ regulatory T-cell homeostasis via hypoxia-inducible factor-1alpha. Eur. J. Immunol. 2008, 38, 2412–2418. [Google Scholar] [CrossRef]
- Comito, G.; Giannoni, E.; Segura, C.P.; Barcellos-de-Souza, P.; Raspollini, M.R.; Baroni, G.; Lanciotti, M.; Serni, S.; Chiarugi, P. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene 2014, 33, 2423–2431. [Google Scholar] [CrossRef]
- De Palma, M.; Lewis, C.E. Macrophage Regulation of Tumor Responses to Anticancer Therapies. Cancer Cell 2013, 23, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Dewhirst, M.W.; Secomb, T.W. Transport of drugs from blood vessels to tumour tissue. Nat. Rev. Cancer 2017, 17, 738–750. [Google Scholar]
- Bonavita, E.; Pelly, V.S.; Zelenay, S. Resolving the dark side of therapy-driven cancer cell death. J. Exp. Med. 2018, 215, 9–11. [Google Scholar]
- Bhargava, P.; Robinson, M.O. Development of Second-Generation VEGFR Tyrosine Kinase Inhibitors: Current Status. Curr. Oncol. Rep. 2011, 13, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Gschwind, A.; Fischer, O.M.; Ullrich, A. The discovery of receptor tyrosine kinases: Targets for cancer therapy. Nat. Rev. Cancer 2004, 4, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Easwaran, H.; Tsai, H.-C.; Baylin, S.B. Cancer Epigenetics: Tumor Heterogeneity, Plasticity of Stem-like States, and Drug Resistance. Mol. Cell 2014, 54, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhu, M.-S.; Wu, W.-R.; Shi, X.-D.; Xu, L.-B. Role of anti-angiogenesis therapy in the management of hepatocellular carcinoma: The jury is still out. World J. Hepatol. 2014, 6, 830–835. [Google Scholar] [CrossRef]
- Holash, J.; Davis, S.; Papadopoulos, N.; Croll, S.D.; Ho, L.; Russell, M.; Boland, P.; Leidich, R.; Hylton, D.; Burova, E.; et al. VEGF-Trap: A VEGF blocker with potent antitumor effects. Proc. Natl. Acad. Sci. USA 2002, 99, 11393–11398. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, F.; Ferrara, N. Refractoriness to Antivascular Endothelial Growth Factor Treatment: Role of Myeloid Cells. Cancer Res. 2008, 68, 5501–5504. [Google Scholar] [CrossRef]
- Angiogenesis Inhibitors. Available online: www.cancer.gov/about-cancer/treatment/types/immunotherapy/angiogenesis-inhibitors-fact-sheet (accessed on 7 April 2018).
- Ferrara, N.; Hillan, K.J.; Novotny, W. Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem. Biophys. Res. Commun. 2005, 333, 328–335. [Google Scholar] [CrossRef]
- Kim, K.J.; Li, B.; Winer, J.; Armanini, M.; Gillett, N.; Phillips, H.S.; Ferrara, N. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 1993, 362, 841–844. [Google Scholar] [CrossRef]
- Sennino, B.; McDonald, D.M. Controlling escape from angiogenesis inhibitors. Nat. Rev. Cancer 2012, 12, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Taugourdeau-Raymond, S.; Centers, T.F.N.O.T.P.; Rouby, F.; Default, A.; Jean-Pastor, M.-J. Bevacizumab-induced serious side-effects: A review of the French pharmacovigilance database. Eur. J. Clin. Pharmacol. 2012, 68, 1103–1107. [Google Scholar] [CrossRef] [PubMed]
- Ciombor, K.K.; Berlin, J. Aflibercept—A decoy VEGF receptor. Curr. Oncol. Rep. 2014, 16, 368. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.M.; Hurwitz, H.I. Ziv-aflibercept: Binding to more than VEGF-A—Does more matter? Nat. Rev. Clin. Oncol. 2013, 10, 10–11. [Google Scholar] [CrossRef] [PubMed]
- Gaya, A.; Tse, V. A preclinical and clinical review of aflibercept for the management of cancer. Cancer Treat. Rev. 2012, 38, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Economides, A.N.; Carpenter, L.R.; Rudge, J.S.; Wong, V.; Koehler-Stec, E.M.; Hartnett, C.; Pyles, E.A.; Xu, X.; Daly, T.J.; Young, M.R.; et al. Cytokine traps: Multi-component, high-affinity blockers of cytokine action. Nat. Med. 2003, 9, 47–52. [Google Scholar] [CrossRef]
- Papadopoulos, N.; Martin, J.; Ruan, Q.; Rafique, A.; Rosconi, M.P.; Shi, E.; Pyles, E.A.; Yancopoulos, G.D.; Stahl, N.; Wiegand, S.J. Binding and neutralization of vascular endothelial growth factor (VEGF) and related ligands by VEGF Trap, ranibizumab and bevacizumab. Angiogenesis 2012, 15, 171–185. [Google Scholar] [CrossRef]
- Rudge, J.S.; Holash, J.; Hylton, D.; Russell, M.; Jiang, S.; Leidich, R.; Papadopoulos, N.; Pyles, E.A.; Torri, A.; Wiegand, S.J.; et al. VEGF Trap complex formation measures production rates of VEGF, providing a biomarker for predicting efficacious angiogenic blockade. Proc. Natl. Acad. Sci. USA 2007, 104, 18363–18370. [Google Scholar] [CrossRef]
- Macdonald, D.A.; Martin, J.; Muthusamy, K.K.; Luo, J.-K.; Pyles, E.; Rafique, A.; Huang, T.; Potocky, T.; Liu, Y.; Cao, J.; et al. Aflibercept exhibits VEGF binding stoichiometry distinct from bevacizumab and does not support formation of immune-like complexes. Angiogenesis 2016, 19, 389–406. [Google Scholar] [CrossRef]
- Meyer, T.; Robson, T.; Langer, F.; Desai, H.; Davila, M.; Amaya, M.; Francis, J.L.; Amirkhosravi, A.; Robles-Carrillo, L.; Robles-Carrillo, L. Bevacizumab immune complexes activate platelets and induce thrombosis in FCGR2A transgenic mice. J. Thromb. Haemost. 2009, 7, 171–181. [Google Scholar] [CrossRef]
- Hollanders, K.; Van Bergen, T.; Van de Velde, S.; Sijnave, D.; Vandewalle, E.; Moons, L.; Stalmans, I. Bevacizumab revisited: Its use in different mouse models of ocular pathologies. Curr. Eye Res. 2015, 40, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Kadenhe-Chiweshe, A.; Papa, J.; McCrudden, K.W.; Frischer, J.; Bae, J.-O.; Huang, J.; Fisher, J.; Lefkowitch, J.H.; Feirt, N.; Rudge, J.; et al. Sustained VEGF Blockade Results in Microenvironmental Sequestration of VEGF by Tumors and Persistent VEGF Receptor-2 Activation. Mol. Cancer Res. 2008, 6, 1–9. [Google Scholar] [CrossRef]
- Rolny, C.; Mazzone, M.; Tugues, S.; Laoui, D.; Johansson, I.; Coulon, C.; Squadrito, M.L.; Segura, I.; Li, X.; Knevels, E.; et al. HRG Inhibits Tumor Growth and Metastasis by Inducing Macrophage Polarization and Vessel Normalization through Downregulation of PlGF. Cancer Cell 2011, 19, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Incio, J.; Tam, J.; Rahbari, N.N.; Suboj, P.; McManus, D.T.; Chin, S.M.; Vardam, T.D.; Batista, A.; Babykutty, S.; Jung, K.; et al. PlGF/VEGFR-1 signaling promotes macrophage polarization and accelerated tumor progression in obesity. Clin. Cancer Res. 2016, 22, 2993–3004. [Google Scholar] [CrossRef] [PubMed]
- Kanda, A.; Noda, K.; Saito, W.; Ishida, S. Aflibercept Traps Galectin-1, an Angiogenic Factor Associated with Diabetic Retinopathy. Sci. Rep. 2015, 5, 17946. [Google Scholar] [CrossRef] [PubMed]
- Croci, D.O.; Cerliani, J.P.; Dalotto-Moreno, T.; Méndez-Huergo, S.P.; Mascanfroni, I.D.; Dergan-Dylon, S.; Toscano, M.A.; Caramelo, J.J.; García-Vallejo, J.J.; Ouyang, J.; et al. Glycosylation-Dependent Lectin-Receptor Interactions Preserve Angiogenesis in Anti-VEGF Refractory Tumors. Cell 2014, 156, 744–758. [Google Scholar] [CrossRef] [PubMed]
- Croci, D.O.; Rabinovich, G.A. Linking tumor hypoxia with VEGFR2 signaling and compensatory angiogenesis: Glycans make the difference. Oncoimmunology 2014, 3, e29380. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Tabernero, J.; Lakomy, R.; Prenen, H.; Prausová, J.; Macarulla, T.; Ruff, P.; Van Hazel, G.A.; Moiseyenko, V.; Ferry, D.; et al. Addition of Aflibercept to Fluorouracil, Leucovorin, and Irinotecan Improves Survival in a Phase III Randomized Trial in Patients With Metastatic Colorectal Cancer Previously Treated With an Oxaliplatin-Based Regimen. J. Clin. Oncol. 2012, 30, 3499–3506. [Google Scholar] [CrossRef] [PubMed]
- Spratlin, J. Ramucirumab (IMC-1121B): Monoclonal antibody inhibition of vascular endothelial growth factor receptor-2. Curr. Oncol. Rep. 2011, 13, 97–102. [Google Scholar] [CrossRef]
- Calvetti, L.; Pilotto, S.; Carbognin, L.; Ferrara, R.; Caccese, M.; Tortora, G.; Bria, E. The coming of ramucirumab in the landscape of anti-angiogenic drugs: Potential clinical and translational perspectives. Expert Opin. Biol. Ther. 2015, 15, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Falcon, B.L.; Chintharlapalli, S.; Uhlik, M.T.; Pytowski, B. Antagonist antibodies to vascular endothelial growth factor receptor 2 (VEGFR-2) as anti-angiogenic agents. Pharmacol. Ther. 2016, 164, 204–225. [Google Scholar] [CrossRef] [PubMed]
- Lohela, M.; Bry, M.; Tammela, T.; Alitalo, K. VEGFs and receptors involved in angiogenesis versus lymphangiogenesis. Curr. Opin. Cell Biol. 2009, 21, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Vennepureddy, A.; Singh, P.; Rastogi, R.; Atallah, J.P.; Terjanian, T. Evolution of ramucirumab in the treatment of cancer—A review of literature. J. Oncol. Pharm. Pract. 2017, 23, 525–539. [Google Scholar] [CrossRef] [PubMed]
- Bocci, G. Increased Plasma Vascular Endothelial Growth Factor (VEGF) as a Surrogate Marker for Optimal Therapeutic Dosing of VEGF Receptor-2 Monoclonal Antibodies. Cancer Res. 2004, 64, 6616–6625. [Google Scholar] [CrossRef]
- Gotink, K.J.; Verheul, H.M. Anti-angiogenic tyrosine kinase inhibitors: What is their mechanism of action? Angiogenesis 2010, 13, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Levitzki, A. Tyrosine Kinase Inhibitors: Views of Selectivity, Sensitivity, and Clinical Performance. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 161–185. [Google Scholar] [CrossRef] [PubMed]
- Raymond, E.; Dahan, L.; Raoul, J.-L.; Bang, Y.-J.; Borbath, I.; Lombard-Bohas, C.; Valle, J.; Metrakos, P.; Smith, D.; Vinik, A.; et al. Sunitinib Malate for the Treatment of Pancreatic Neuroendocrine Tumors. N. Engl. J. Med. 2011, 364, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Schmieder, R.; Hoffmann, J.; Becker, M.; Bhargava, A.; Müller, T.; Kahmann, N.; Ellinghaus, P.; Adams, R.; Rosenthal, A.; Thierauch, K.H.; et al. Regorafenib (BAY 73-4506): Antitumor and antimetastatic activities in preclinical models of colorectal cancer. Int. J. Cancer 2014, 135, 1487–1496. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Dumas, J.; Adnane, L.; Lynch, M.; Carter, C.A.; Schütz, G.; Thierauch, K.H.; Zopf, D. Regorafenib (BAY 73-4506): A new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int. J. Cancer 2011, 129, 245–255. [Google Scholar] [CrossRef]
- Meille, C.; Serdjebi, C.; Giacometti, S.; Padovani, L.; Ciccolini, J.; Barbolosi, D.; Lombard, A.; Pasquier, E.; André, N. Pharmacokinetics and Pharmacodynamics-Based Mathematical Modeling Identifies an Optimal Protocol for Metronomic Chemotherapy. Cancer Res. 2017, 77, 4723–4733. [Google Scholar]
- Kim, J.Y.; Kim, Y.-M. Tumor endothelial cells as a potential target of metronomic chemotherapy. Arch. Pharm. Res. 2019, 42, 1–13. [Google Scholar] [CrossRef]
- Romiti, A.; Cox, M.C.; Sarcina, I.; Di Rocco, R.; D’Antonio, C.; Barucca, V.; Marchetti, P. Metronomic chemotherapy for cancer treatment: A decade of clinical studies. Cancer Chemother. Pharmacol. 2013, 72, 13–33. [Google Scholar] [CrossRef] [PubMed]
- Natale, G.; Bocci, G. Does metronomic chemotherapy induce tumor angiogenic dormancy? A review of available preclinical and clinical data. Cancer Lett. 2018, 432, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Kerbel, R.S. A Decade of Experience in Developing Preclinical Models of Advanced- or Early-Stage Spontaneous Metastasis to Study Antiangiogenic Drugs, Metronomic Chemotherapy, and the Tumor Microenvironment. Cancer J. 2015, 21, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Kerbel, R.S.; Grothey, A. Gastrointestinal cancer: Rationale for metronomic chemotherapy in phase III trials. Nat. Rev. Clin. Oncol. 2015, 12, 313–314. [Google Scholar] [CrossRef]
- Kerbel, R.S.; Guérin, E.; Francia, G.; Xu, P.; Lee, C.R.; Ebos, J.M.; Man, S. Preclinical recapitulation of antiangiogenic drug clinical efficacies using models of early or late stage breast cancer metastatis. Breast 2013, 22 (Suppl. 2), S57–S65. [Google Scholar] [CrossRef]
- Kerbel, R.S.; Shaked, Y. The potential clinical promise of ‘multimodality’ metronomic chemotherapy revealed by preclinical studies of metastatic disease. Cancer Lett. 2017, 400, 293–304. [Google Scholar] [CrossRef]
- Farooq, M.; El-Faham, A.; Khattab, S.N.; ElKayal, A.M.; Ibrahim, M.F.; Abu Taha, N.; Baabbad, A.; Wadaan, M.A.; Hamed, E.A. Biological Screening of Novel Derivatives of Valproic Acid for Anticancer and Antiangiogenic Properties. Asian Pac. J. Cancer Prev. 2014, 15, 7785–7792. [Google Scholar] [CrossRef]
- Huang, J.; Zhao, D.; Liu, Z.; Liu, F. Repurposing psychiatric drugs as anti-cancer agents. Cancer Lett. 2018, 419, 257–265. [Google Scholar] [CrossRef]
- Kourti, M.; Westwell, A.; Jiang, W.; Cai, J. Repurposing old carbon monoxide-releasing molecules towards the anti-angiogenic therapy of triple-negative breast cancer. Oncotarget 2019, 10, 1132–1148. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef] [PubMed]
- Deroanne, C.F.; Bonjean, K.; Servotte, S.; Devy, L.; Colige, A.; Clausse, N.; Blacher, S.; Verdin, E.; Foidart, J.-M.; Nusgens, B.V.; et al. Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene 2002, 21, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Kwon, H.J.; Lee, Y.M.; Baek, J.H.; Jang, J.-E.; Lee, S.-W.; Moon, E.-J.; Kim, H.-S.; Lee, S.-K.; Chung, H.Y.; et al. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat. Med. 2001, 7, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Huang, L. Targeting histone deacetylases for the treatment of cancer and inflammatory diseases. J. Cell. Physiol. 2006, 209, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef]
- Zecchin, A.; Pattarini, L.; Gutierrez, M.I.; Mano, M.; Mai, A.; Valente, S.; Myers, M.P.; Pantano, S.; Giacca, M. Reversible acetylation regulates vascular endothelial growth factor receptor-2 activity. J. Mol. Cell Biol. 2014, 6, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Mottamal, M.; Zheng, S.; Huang, T.L.; Wang, G. Histone Deacetylase Inhibitors in Clinical Studies as Templates for New Anticancer Agents. Molecules 2015, 20, 3898–3941. [Google Scholar] [CrossRef]
- Eyre, T.A.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. A phase 1 study to assess the safety, tolerability, and pharmacokinetics of CXD101 in patients with advanced cancer. Cancer 2019, 125, 99–108. [Google Scholar] [CrossRef]
- Mohammad, H.P.; Barbash, O.; Creasy, C.L. Targeting epigenetic modifications in cancer therapy: Erasing the roadmap to cancer. Nat. Med. 2019, 25, 403–418. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Nakashima, H.; Chiocca, E.A. Molecular responses to immune checkpoint blockade in glioblastoma. Nat. Med. 2019, 25, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. Immune Checkpoint Targeting in Cancer Therapy: Toward Combination Strategies with Curative Potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Pallin, D.J.; Baugh, C.W.; Postow, M.A.; Caterino, J.M.; Erickson, T.B.; Lyman, G.H. Immune-related Adverse Events in Cancer Patients. Acad. Emerg. Med. 2018, 25, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; López-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inogés, S.; De Andrea, C.; De Cerio, A.L.-D.; Tejada, S.; et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar] [CrossRef]
- Gunawan, F.; George, E.; Roberts, A. Combination immune checkpoint inhibitor therapy nivolumab and ipilimumab associated with multiple endocrinopathies. Endocrinol. Diabetes Metab. Case Rep. 2018, 2018, 17-0146. [Google Scholar] [CrossRef]
- Postow, M.A.; Hellmann, M.D.; Jhaveri, K.D.; Perazella, M.A.; Akturk, H.K.; Michels, A.W.; Kumar, B.; Ballas, Z.; Tervaert, J.-W.C.; Ye, C.; et al. Adverse Events Associated with Immune Checkpoint Blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef]
- Huang, A.C.; Postow, M.A.; Orlowski, R.J.; Mick, R.; Bengsch, B.; Manne, S.; Xu, W.; Harmon, S.; Giles, J.R.; Wenz, B.; et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017, 545, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Sandhu, S.K.; Workman, P.; De Bono, J.S. Envisioning the future of early anticancer drug development. Nat. Rev. Cancer 2010, 10, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Masucci, G.V.; Cesano, A.; Eggermont, A.; Fox, B.A.; Wang, E.; Marincola, F.M.; Ciliberto, G.; Dobbin, K.; Puzanov, I.; Taube, J.; et al. The need for a network to establish and validate predictive biomarkers in cancer immunotherapy. J. Transl. Med. 2017, 15, 223. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| I. Biologicals | ||||
| Antiangiogenic agent | Trade name | Class | Target | Indication * |
| Bevacizumab | Avastin (Mvasi) | MAb | VEGF-A isoforms | m/rCC, mCRC, rGB, m/rNSCLC, rOEC, rFTC, rPPC, mRCC |
| Ziv-Aflibercept | Cyramza | Recombinant fusion protein | VEGF-A, PlGF, VEGF-B, processed VEGF-C processed VEGF-D | mCRC |
| Ramucirumab | Zaltrap | MAb | VEGFR-2 | mCRC, mNSCLC, a/mGAC or GEJAC |
| II. Small molecule multi kinase inhibitors (MKIs) | ||||
| Sunitinib malate | Sutent | MKI | VEGFRs, PDGFRb, KIT, FLT-3 (CD135), CSF1R, RET | GIST, p/a/mPC, a/rRCC |
| Sorafenib tosylate | Nexavar | MKI | VEGFRs, PDGFRs, KIT, FLT-3 (CD135), CSF1R, RET, Raf | HCC, aRCC,p/r/mTC |
| Pazopanib hydrochloride | Votrient | MKI | VEGFRs, FGFRs, KIT | aRCC, aSTC |
| Axitinib | Inlyta | MKI | VEGFR1-3 | aRCC |
| Regorafenib | Stivarga | MKI | VEGFRs, PDGFRs, FGFRs, KIT, TIE2, Raf | mCRC, mGIST, HCC |
| Cabozantinib-S-malate | Cometriq | MKI | VEGFRs, RET, MET, TIE2, FLT-3 | HCC, p/mMTC, aRCC |
| Vandetanib | Caprelsa | MKI | VEGFRs, EGFR, RET, TIE2 | a/mMTC |
| III. Other molecules | ||||
| Everolimus | Afinitor | S/ThKI | mTOR | BC, a/mPC, aRC, SEGCA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jászai, J.; Schmidt, M.H.H. Trends and Challenges in Tumor Anti-Angiogenic Therapies. Cells 2019, 8, 1102. https://doi.org/10.3390/cells8091102
Jászai J, Schmidt MHH. Trends and Challenges in Tumor Anti-Angiogenic Therapies. Cells. 2019; 8(9):1102. https://doi.org/10.3390/cells8091102
Chicago/Turabian StyleJászai, József, and Mirko H.H. Schmidt. 2019. "Trends and Challenges in Tumor Anti-Angiogenic Therapies" Cells 8, no. 9: 1102. https://doi.org/10.3390/cells8091102
APA StyleJászai, J., & Schmidt, M. H. H. (2019). Trends and Challenges in Tumor Anti-Angiogenic Therapies. Cells, 8(9), 1102. https://doi.org/10.3390/cells8091102